Abstract
Myopathies are frequently not confined to the skeletal muscles but also involve other organs or tissues. One of the most frequently affected organ in addition to the skeletal muscle is the heart (cardiac involvement, CI). CI manifests as impulse generation or conduction defects, focal or diffuse myocardial thickening, dilation of the cardiac cavities, relaxation abnormality, hypertrophic, dilated, restrictive cardiomyopathy, apical form of hypertrophic cardiomyopathy, noncompaction, Takotsubo phenomenon, secondary valve insufficiency, intra-cardiac thrombus formation, or heart failure with systolic or diastolic dysfunction. CI occurs in dystrophinopathies, Emery-Dreifuss muscular dystrophy, facioscapulohumeral muscular dystrophy, limb girdle muscular dystrophies, laminopathies, congenital muscular dystrophies, myotonic dystrophies, congenital myopathies, metabolic myopathies, desminopathies, myofibrillar myopathy, Barth syndrome, McLeod syndrome, Senger's syndrome, and Bethlem myopathy. Patients with myopathy should be cardiologically investigated as soon as their neurological diagnosis is established, since supportive cardiac therapy is available, which markedly influences prognosis and outcome of CI in these patients.
One of the organs most frequently affected in addition to the skeletal muscle in myopathies is the heart (cardiac involvement, CI) Citation1. Since the first recognition of muscular dystrophies in the second half of the 19th century CI has been described in various other primary myopathies and is even the dominant feature in some of them () Citation2. CI in myopathies comprises impulse generation or impulse propagation defects, myocardial abnormalities, secondary valve insufficiency, intracardial thrombus formation, or heart failure with systolic or diastolic dysfunction (). Early recognition of CI is warranted, since appropriate, supportive treatment greatly influences management and thus outcome and prognosis of these patients.
Table I. Primary myopathies with cardiac involvement.
Table II. Cardiac abnormalities in primary myopathies.
Diagnosing myopathies
The diagnosis of myopathies relies upon a comprehensive individual and family history, clinical neurologic examination, blood chemical investigations at rest or during exercise, nerve conduction studies and electromyography, muscle biopsy with histological, immunehistological or biochemical investigations, and genetic studies. Special attention should be directed towards the family history, particularly if other family members are also affected and present with the same or different manifestations.
Symptoms patients with myopathy report comprise those attributable to the skeletal muscle and those attributable to extra-cardiac manifestations. Symptoms of muscle affection are easy fatigability, exercise intolerance, spontaneous or induced muscle cramps, muscle aching after slight exercise, myalgias, muscle stiffness, double vision, drooping eyelid, fasciculations, transient or fixed weakness and wasting of limb, trunc, or facial muscles, scapular winging, dysphagia, dysarthria, gait disturbance, difficulties to get up from a chair or the floor, difficulties to carry the head, falls, or dark urine.
Signs of muscle affection on clinical neurologic examination include double vision, ptosis, ophthalmoparesis, myopathic face, tongue atrophy, focal or generalized transient or fixed, symmetrical or asymmetrical weakness, scapular winging, reduced tendon reflexes, muscle hypotonia, clinical myotonia, fasciculations, myokymia, pseudohypertrophy (calves, masseter, tongue), a positive Gower sign, gait disturbance (waddling gait, steppage gait), joint contractures, thorax deformities, or spinal deformities.
Cardiac manifestations of myopathies
CI in myopathies manifests as impulse generation or conduction abnormalities, myocardial abnormalities, secondary valve insufficiencies, or thrombogenic states. There are no pathognomonic electrocardiographic (ECG) abnormalities typical for CI in myopathies. All types of blocking, bradycardious or tachycardious supraventricular or ventricular ECG abnormalities can be observed. However, particular patterns are more frequent with the one than the other disorder. Atrio-ventricular (AV) block I can be found in two thirds of the patients with myotonic dystrophy type I. Atrial fibrillation occurs in almost all patients with laminopathies and in many of those with mitochondrial disorders. Complete AV-block is a frequent cardiac feature of Emery-Dreifuss muscular dystrophy (EDMD). Myocardial manifestations of CI are listed in , Citation3. In case of dilation of the cardiac cavities dilation of the anulus may result in secondary valve insufficiency. Atrial fibrillation and severe dilation may be associated with hypercoagulability.
Table III. Myopathies associated with hypertrophic, dilative, restrictive cardiomyopathy or with LVHT.
Frequency of CI
The prevalence of CI in various myopathies is listed in . It is highest in Barth syndrome, laminopathies, and dystrophinopathies Citation4–6. Bundle branch block can be found in one third and decreased systolic function in 22% of the patients with myotonic dystrophy I. Death is due to cardiac dysfunction in 10% of the DMD-patients.
Table IV. Frequency of cardiac involvement in primary myopathies.
Assessment of cardiac involvement
To diagnose and monitor CI in myopathies, the same cardiac methods and techniques are applied as for diagnosing cardiac disease of other causes. Diagnosing CI in myopathies requires a comprehensive cardiac investigation, including history, clinical cardiologic examination, surface ECG, ambulatory 24h-ECG, and echocardiography (). Patients with myopathy should undergo these basic cardiac investigations as soon as the neurological diagnosis has been established. If these investigations provide evidence for an impulse generation or conduction abnormality without clarifying its origin further invasive electrophysiological investigations are indicated (). If the basic cardiologic investigations provide evidence of a myocardial affection more sophisticated methods for its assessment may be applied, such as cardiac magnetic resonance imaging (MRI), stress echocardiography, tissue Doppler imaging, cardiac PET, myocardial scintigraphy with Tc-99m methoxy-isobutyl-isonitrile, I-123-β-methyl-p-iodophenylpentadecanoic acid or I-123 meta-idobenzyl-guanidine or myocardial biopsy. To exclude coronary heart disease as the cause of the cardiac abnormalities or to confirm an additional vascular pathology coronary angiography or SPECT may be necessary (). Particularly, if the onset of the neurological manifestations follows the onset of CI, CI may be easily overlooked or misinterpreted. Subclinical CI in dystrophinopathies may be detected only upon cardiac MRI Citation7, tissue Doppler imaging, or cardiac positron emission tomography (PET). After decease autopsy of the heart is obligatory ().
Figure 1. Flow-chart according to which patients with myopathies can be assessed for cardiac involvement
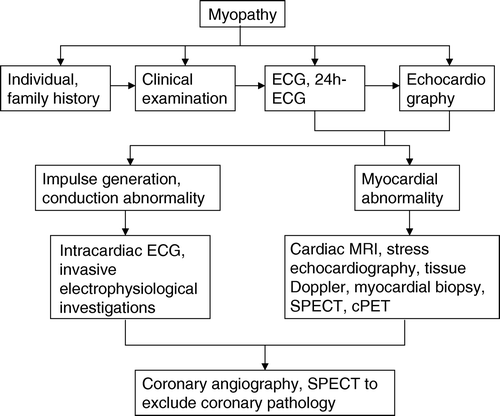
Figure 2. Cardiac autopsy specimen of a patient with suspected encephalomyopathy shows marked left ventricular hypertrabeculation distally to the papillary muscles
Myopathies with cardiac involvement
There is growing evidence that the prevalence of CI in myopathies is higher than previously thought (). In addition to muscular dystrophies, CI has been reported in metabolic myopathies, congenital myopathies, disorders of the contractile proteins, and some unclassified myopathies. CI usually increases with age and may precede, follow, or accompany the onset of skeletal muscle disease.
Muscular dystrophies
Dystrophinopathies
Dystrophinopathies are X-linked disorders due to deletions, insertions, or point mutations in the dystrophin gene on chromosome Xq21 (). Clinically, they manifest as DMD (out-of–frame mutation) or BMD (in-frame mutation). DMD is characterized by general weakness and wasting, with onset in the pelvic girdle and progression towards the distal and respiratory muscles. BMD has a similar phenotype, but with later onset and much slower progression and only late involvement of the respiratory muscles.
CI is an important feature of DMD, BMD patients and carriers, and comprises supraventricular or ventricular arrhythmias, abnormal Q-waves, ST-segment depression, prolonged QT-interval, hypertrophy signs, regional wall motion abnormalities, dilation, dilated cardiomyopathy (), secondary valve insufficiency, or heart failure with systolic or diastolic dysfunction Citation8, necessitating heart transplantation in selected cases. Most of these abnormalities are attributed to replacement of cardiomyocytes by connective tissue or fat, as assessed by gadolinium-enhanced cardiac MRI. In several DMD cases, reduction of the coronary vasodilated reserve, possibly due to involvement of the vascular smooth muscle, has been described. In two patients BMD was associated with isolated LVHT. In the early stages of the disease CI may be subclinical and only detected by instrumental investigations, such as cardiac MRI or MRI tagging. In single patients CI may worsen during general anesthesia. Besides these cardiac manifestations, there is some evidence for hypercoagulability in dystrophinopathies. Generally, the genotype/phenotype correlation for CI in dystrophinopathies is poor.
Both, DMD and BMD female carriers may be symptomatic from the mutation. The degree of CI in female carriers is, according to the Lyon-hypothesis, depended on the amount of X-chromosome inactivation. Accordingly, DMD-carriers can be asymptomatic or severely affected and can even develop dilation or heart failure, necessitating biventricular pacing or even heart transplantation ().
X-linked Emery-Dreifuss muscular dystrophy (XL-EDMD)
XL-EDMD is characterized by slowly progressive muscle weakness and wasting with humero-peroneal distribution and early onset contractures of the elbow joints, Achilles tendons, and posterior-cervical muscles Citation9. XL-EDMD is due to a mutation in the Emerin gene on chromosome Xq28 (). Emerin is a type II inner nuclear membrane protein, which regulates the flux of β-catenin, a transcription co-activator. Wild-type emerin restricts access of β-catenin to the nucleus, whereas mutated emerin dominantly stimulates β-catenin activity.
CI in XL-EDMD includes conduction defects, such as sinus bradycardia, AV-block I, or may cause sudden death without prior symptoms, unless treated with an early inserted pacemaker. In single cases heart failure, necessitating heart transplantation may occur. In a study on 18 XL-EDMD patients 10 required a pacemaker for bradyarrhythmias. Atrial fibrillation or flutter developed in 11 of them and atrial standstill in 5 of the 11. In single cases decreased accumulation of I-123 meta-idobenzyl-guanidine may predict left ventricular dysfunction. CI may precede the skeletal muscle affection by years.
Laminopathies
Laminopathies are due to mutations in the lamin A/C gene on chromosome 1q11–23 (), which encodes for lamin A/C, a component of the nuclear envelope. Lamins A and C are two different proteins produced by alternative splicing from the same gene. Lamin A/C mutations manifest at least as seven different disorders in the skeletal muscle, myocardium, peripheral nerve, or adipose tissue () Citation10. These include autosomal dominant EDMD (AD-EDMD), limb girdle muscular dystrophy (LGMD) 1B, characterized by proximal muscle weakness and wasting, AV-block, and dilated cardiomyopathy () Citation10, familial partial lipodystrophy, dilated cardiomyopathy with conduction system disease, autosomal recessive type 2 Charcot-Marie-Tooth disease, Hutchinson-Gilford progeria, and mandibulo-sacral dysplasia.
Table V. Phenotypic heterogeneity of mutations causing myopathy with cardiac involvement.
CI in laminopathies is characterized by conduction system or rhythm disturbances, such as supraventricular arrhythmias (sinustachycardia, atrial fibrillation, atrial tachycardia, or atrial standstill), AV-block, or ventricular arrhythmias, which occur early during the disease course, followed by dilation, dilated cardiomyopathy (), restrictive cardiomyopathy (), or systolic dysfunction Citation10, Citation11. Cardiac abnormalities may precede the onset of the skeletal muscle weakness by years Citation12. Rhythm abnormalities may be life threatening and often responsible for sudden cardiac death Citation10. CI in AD-EDMD occurs in adulthood and comprises bradycardia, AV-block I, atrial tachycardia, atrial fibrillation, or ventricular conduction blocks, ventricular arrhythmias, non-dilated cardiomyopathy, dilated cardiomyopathy (), or restrictive cardiomyopathy () Citation11. In single cases the heart may be subclinically affected and CI only detected upon tissue Doppler imaging or cardiac MRI. CI in LGMD1B may require implantation of an implantable cardioverter defibrillator (ICD) or, in single cases, heart transplantation Citation10, Citation12. In a family of 72 members with quadriceps myopathy due to a lamin A/C mutation, CI was found in 15%. Two thirds of the patients had dilated cardiomyopathy or ventricular arrhythmias, three quarters had bundle branch block, but all had atrial fibrillation.
Facioscapulohumeral muscular dystrophy (FSH)
FSH affects the facial, shoulder girdle, and, sometimes the peroneal muscles. Some cases with minor facial involvement but predominant affection of the limbs may mimic scapulo-peroneal syndrome. In addition to the muscle, the retina or the ears (sensorineural hearing loss) may be affected. FSH is caused by a deletion of an integral number of 3.3kb tandem repeats from the subtelomeric region on chromosome 4q35.
There is controversy concerning CI in FSH. Previous studies found no or hardly any clinically significant cardiac disease in FSH Citation13. More recent studies, however, provide increasing evidence that the conduction system and the myocardium are clinically or subclinically affected at least in some patients with FSH. In a multi-center study on 83 FSH patients, of whom, 62 were without cardiovascular risk factors, 10 developed clinical or subclinical CI (12%) Citation6. Five reported frequent palpitations as the equivalent of supraventricular paroxysmal tachycardia and 5 had asymptomatic supraventricular tachycardia Citation6. In a study on 24 patients with FSH the systo-diastolic variation of the integrated backscatter signal was reduced and the QT-dispersion increased Citation14. In 9 patients ventricular late potentials were recorded. QT and QTc-dispersion were inversely related to the integrated backscatter signal of the septum and posterior wall. In none of these patients CI manifested clinically Citation14. Studies on single cases or small case series additionally found sinus node dysfunction, manifesting as transient P-wave inversion, prolonged P-P-interval, or tall P-waves, premature atrial contractions, supraventricular arrhythmias, AV-block, intraventricular conduction delay, tall T-waves, ST-elevation, long-QT syndrome, ventricular tachycardia, or reduced Tl-201 uptake on scintigraphy Citation15. In single cases myocardial thickening has been described Citation15. Rarely, intractable heart failure may be the cause of death in FSH.
Limb girdle muscular dystrophies (LGMD)
LGMDs are a genetically heterogeneous group of autosomal dominant (6 entities) or autosomal recessive (10 entities) myopathies, clinically characterized by weakness and wasting of the shoulder and pelvic girdle muscles, calf hypertrophy and reduced tendon reflexes. Some patients are severely affected and resemble DMD (β-sarcoglycanopathy). The phenotype is due to mutations in genes, which encode for muscle membrane bound sarcoglycanes or the proteins such as myotilin, lamin A/C, caveolin-3, calpain, dysferlin, telethonin, E3-ubiquitine ligase, fukutin-related protein, or titin (). Caveolin-3 is the muscle-specific product of the caveolin gene family and integral membrane component of caveolae Citation17. Mutations in the caveolin-3 gene underlie not only LGMD1C but also rippling muscle disease, sporadic and familial forms of hyper-CK-emia, and distal myopathy () Citation17. A caveolin-3 mutation may cause all four distinct phenotypes within the same family Citation17. In all these patients the caveolin-3 expression in the myocardium was reduced to 60% of normal Citation17.
CI in LGMDs comprise incomplete right bundle branch block, tall R-waves in V1-2, left anterior hemiblock, shortened QT-interval, ST-segment elevation, abnormal Q-waves Citation16, reduction of the coronary vasodilated reserve, dilation of the cardiac cavities, dilated cardiomyopathy () Citation18, wall motion abnormalities, or heart failure Citation19, requiring heart transplantation in selected cases Citation18. A mutation in one of the sarcoglycan genes may disrupt the entire sarcoglycan complex in both the skeletal and cardiac muscle Citation20. Furthermore, there are indications that disruption of the sarcospan/sarcoglycan complex in vascular smooth muscles perturbates vascular function and may promote the development of cardiomyopathy Citation21. In a patient with dysferlinopathy (LGMD2B) echocardiography revealed left ventricular dilation and diffuse hypokinesia Citation22. LGMD2B may also go along with severe ECG abnormalities and subclinical affection of the right ventricular myocardium, as assessed by tissue Doppler-imaging Citation23. A study on 10 patients with γ-sarcoglycanopathy reported ECG abnormalities and myocardial relaxation abnormalities in four of them Citation23. In a study on 6 patients with LGMD2E half presented with CI and one of them with dilated cardiomyopathy. CI has been also reported in 8 of 9 patients with LGMD2I Citation24, manifesting as reduced systolic function, dilation of the cardiac cavities, or even dilated cardiomyopathy () Citation25. Overall, CI is assumed to occur in up to 20% of the patients with sarcoglycanopathies () Citation16.
Congenital muscular dystrophies
Congenital muscular dystrophies (CMD) constitute a clinically and genetically heterogeneous group of autosomal recessive myopathies, characterized by marked weakness, generalized hypotonia, joint contractures, and dystrophic changes on muscle biopsy, manifesting in early infancy. Mutations in the laminin α2 (LAMA2), fukutin (FCMD), integrin α7, O-mannose β-1,2-N-acetylglucosaminyl transferase, O-mannosyl transferase 1, fukutin-related protein, selenoprotein N1 (SEPM1), like-glykosyl-transferase, or collagen (COLA2, COLA3) genes have been identified as causative Citation26. According to histological findings they are subdivided into merosin-positive and merosin-negative forms. The function of most of these genes is unknown. For fukutin, however, there are indications that it is involved in the glycozilation of α-dystroglycan Citation27, Citation28.
CI has been predominantly reported in the Fukuyama type congenital muscular dystrophy (FCMD). The most frequently reported cardiac abnormalities in these patients are myocardial fibrosis or heart failure. In a study on 34 patients with FCMD 47% had a fractional shortening (FS) of <28% or reduced mean velocity of the circumferential fiber shortening. The FS decreased with age. Dilated cardiomyopathy requiring heart transplantation may be a rare feature of FCMD Citation28. Five patients died from heart failure. In a study on 10 merosin-positive patients with non-specified CMD three showed reduced systolic function and one dilation of the cardiac cavities. In a study on 42 patients with merosin-positive CMD left ventricular function was decreased. Among 6 children with merosin-negative CMD (LAMA2) one presented with cardiomyopathy and heart failure. In another study on six merosin-deficient children (LAMA2) two presented with dilated cardiomyopathy () Citation29. Cardiac abnormalities reported together with rigid spine syndrome (SEPM1) were, incomplete left bundle branch block, complete AV-block, heart failure, or a sinus venous defect () Citation30.
Myotonic dystrophy type 1 (MD1)
MD1 is an autosomal dominant multi-system disorder, usually involving the skeletal muscle, brain, heart, eyes, intestines, or endocrinium Citation31. Clinically, MD1 presents with myotonia, diffuse weakness and wasting, frontal baldness, diabetes, megacolon, cataracts, or infertility. MD1 is caused by an expanding trinucleotid CTG-repeat in the 3′ prime untranslated region of a serine-threonine kinase gene on chromosome 19q13.3 () Citation31. The mutation results in nuclear entrapment of mutant RNAs, which extensively interact with RNA-binding proteins in ribonuclear inclusions Citation32.
The most frequent cardiac abnormalities in MD1 are impulse generation or propagation defects, like AV-block I in 64%, prolonged QT-interval, QRS-prolongation in 32%, Torsades de pointes, myocardial thickening Citation33, or heart failure Citation34. There are also altered electro-anatomic patterns in the right cardiac chambers of MD1 patients as confirmed by electro-anatomic mapping Citation35. The most common sustained arrhythmia is reported to be atrial flutter. In a study on 37 patients the PQ-interval increased with age. In a study on 79 MD1 patients the QRS duration increased with disease progression Citation36. There is some evidence that the smooth muscle of the coronary arteries is affected. In single cases myocardial thickening and LVHT have been reported Citation37. Decreased systolic or diastolic function can be found in 22 and 30% of the patients respectively. Subclinical CI may be documented by reduced myocardial velocities on tissue Doppler echocardiography Citation38, which may also indicate early left ventricular systolic or diastolic dysfunction. There are also indications that there is parasympathetic autonomic dysfunction in some of these patients Citation39. The genotype/phenotype correlation between the CTG-repeat expansion and CI is generally poor Citation40. Factors other than the CTG-expansion size appear to determine severity and progression of CI in MD1 Citation41.
Myotonic dystrophy type 2 (MD2)
MD2 is an autosomal dominantly inherited myopathy, which presents with clinical features similar to MD1. Contrary to MD1, the MD2 phenotype is less severe, presents additionally with pseudohypertrophy of the calves, hyperhidrosis, or, less frequently than MD1, with dementia, hypersomnia, diabetes, or hypogonadism. MD2 is due to mutations in the ZNF9 gene on chromosome 3q21 and shows anticipation less frequently than MD1.
Cardiac abnormalities described in MD2 include sinus-bradycardia, supraventricular tachycardia, atrial fibrillation, supraventricular bigeminia, abnormal T-waves, right bundle branch block, ventricular ectopy, sustained monomorphic ventricular tachycardia, heart failure, or myocardial infarction Citation42. Atrial flutter may be provoked by electrophysiologic examination. Ventricular arrhythmias may necessitate implantation of an ICD or may cause sudden cardiac death. In a study on 379 DM2 patients CI was detected in 19% of them () Citation43.
Congenital (protein aggregation) myopathies
So far, congenital myopathies were diagnosed upon structural abnormalities on enzyme histochemistry, immunehistochemistry, or electron microscopy Citation44. With the elucidation of the underlying genetic defects the morphological classification of congenital myopathies has changed to the concept of protein aggregation myopathies, comprising disorders due to mutations in genes encoding for desmin, αB-crystallin, selenoprotein, myotilin, actin, myosin, tropomyosin, troponin T, nebulin, dynamin, myotubularin, cypher, or the ryanodine receptor Citation44. Since this new heuristic classification is still not fully established the old morphological classification is still in general use.
Nemaline myopathy
Nemaline myopathy is a rare, genetically heterogeneous, autosomal dominant or recessive myopathy, characterized by severe, generalized weakness and hypotonia from birth or infancy Citation45, Citation46. Nemaline myopathy is due to mutations in the α-actin, α-tropomyosin, β-tropomyosin, slow troponin T, or nebulin gene ().
CI in nemaline myopathy comprises myocardial thickening, hypertrophic cardiomyopathy (), dilation of the cardiac cavities, or heart failure Citation47. Hypertrophic cardiomyopathy has been recently described for the first time in a 2-year-old child carrying an α-actin mutation Citation47. A patient with nemaline myopathy also developed dilated cardiomyopathy (), requiring heart transplantation Citation48. Additionally, myocardial relaxation may be impaired and may result in diastolic dysfunction Citation49. In a report on two patients with congenital fiber-type dysproportion myopathy atrial fibrillation, AV-block, dilation of the cardiac cavities, and heart failure, necessitating heart transplantation, were described. Another patient presented with dilated cardiomyopathy and complete AV-block () Citation50.
Central core disease
Central core disease is due to mutations in the ryanodine receptor gene and clinically characterized by general hypotonia, muscle cramps and predominantly proximal lower limb weakness at birth Citation51. Also congenital dislocation of the hip, pes cavus, pes planus, shortening of the Achilles tendon, extensive lumbar lordosis or kyphoscoliosis may be present. Most cases, however, are only mildly disabled. CI in central core disease presents with myocardial dilation, or heart failure, necessitating heart transplantation in single cases Citation52.
Centronuclear myopathy
Centronuclear myopathy is a rare congenital myopathy due to mutations in the dynamin 2 gene on chromosome 19p13.2 () Citation52. Rarely CI has been described, manifesting as myocardial thickening, dilated cardiomyopathy (), heart failure, or sudden cardiac death Citation52. A single patient required heart transplantation so far Citation52.
Desmin myopathy
Desmin is the main intermediate filament protein in the skeletal muscle and myocardium and important as part of the cytoskeleton Citation53. The desmin filament lattice surrounds the Z-discs, interconnects them to each other, interlinks myofibrils, and links the contractile apparatus to the sarcolemma, cytoskeleton, cytoplasmic organelles, and the nucleus Citation53. In mildly affected cases, distal limb and axial weakness, and, in severely affected patients, general weakness are the predominant neurological abnormalities Citation54. Histologically, desmin myopathy 2 is characterized by abnormal aggregates of desmin-type intermediate filaments in the skeletal, cardiac, or rarely, the intestinal smooth muscle Citation54. Desminopathy due to mutations in the desmin gene present with progressive weakness of the limb muscles and desmin-reactive granular aggregates in myofibers. Lack of desmin results in susceptibility of myofibers and cardiomyocytes to damage Citation53.
CI in desmin myopathy comprises ECG abnormalities, such as atrial flutter, AV-block, or ventricular tachycardia. Additionally, there may be myocardial thickening, dilation, or heart failure with systolic or diastolic dysfunction Citation1. In some cases, predominantly right ventricular dysfunction has been described Citation1. Generally, it is assumed that rather the absence of an intact desmin filament system than the accumulation of the protein is responsible for skeletal muscle and cardiac abnormalities.
Myofibrillar myopathy
Myofibrillar myopathy is morphologically characterized by desintegration of the Z-disk and myofibrils, followed by abnormal accumulation of various proteins Citation55. Myofibrillar myopathy is genetically heterogeneous due to mutations in the Z-disk-related proteins desmin, αB-crystallin, myotilin, myotubularin (myotubular myopathy), LMNA, myosin heavy chain IIa (myopathy with contractures, ophthalmoplegia and rimmed vacuoles, also termed inclusion body myopathy 3), or selenoprotein N1 (multiminicore disease with external ophthalmoplegia) Citation55. Recently, myofibrillar myopathy due to mutations in the Z-band alternatively spliced PDZ motif-containing protein (cypher/ZASP (Z-band alternatively spliced PDZ-motif containing protein), zaspopathy) has been reported Citation56. In a study on 54 patients with myofibrillar myopathy Z-band alternatively spliced PDZ-motif containing protein mutations were detected in 11 Citation55.
CI in myofibrillar myopathy includes dilated cardiomyopathy or LVHT Citation55, Citation56. In a Qatary family with myofibrillar myopathy one presented with restrictive cardiomyopathy, one with hypertrophic cardiomyopathy necessitating heart transplantation, and one with complete heart block necessitating implantation of a pacemaker Citation57.
Distal myopathies
Distal myopathy with rimmed vacuoles (hereditary inclusion body myopathy 2)
Distal myopathy with rimmed vacuoles has been recently shown to be the same disorder as hereditary inclusion body myopathy and is due to mutations in the tile UDP-GlcNAc2-epimerase gene, which encodes for a bifunctional protein with activities of the UDP-GlcNAc2-epimerase and the ManNAc-kinase Citation58. The UDP-GlcNAc2-epimerase catalyzes the rate-limiting step in the sialic acid biosynthesis and ManNAc-kinase catalyzes the next step. CI occurs in 18% of the patients (Table IV) amd often manifests as sudden cardiac death (58). CI has not been described in Myoshi myopathy (dysferlin), tibial muscular dystrophy (titin), or autosomal dominant distal myopathy also known as hyaline myopathy or myosin storage disease (myosin heavy chain 7).
Metabolic myopathies
Glycogenoses
Glycogenoses are a heterogeneous group of system disorders, due to impaired activity of enzymes involved in the glucose metabolism. Clinically, these patients present with proximal weakness, hypotonia (floppy infant), fatigue, respiratory failure, seizures, cardiac or liver involvement. Histologically, glycogenoses show intracellular glycogen accumulations. Depending on the affected enzyme, 11 different types have been described so far Citation59.
CI has been described in glycogenosis type IIa (M. Pompe), IIb (Danon's disease) III (Forbes disease), IV (McArdle's disease), VII (M. Tarui), and glycogenosis type IX so far. Most well known is CI in type II glycogenosis (acid-α glucosidase-deficiency, Pompe's disease) manifesting as premature ectopic beats, severe myocardial thickening, or sudden death Citation60. In type III glycogenosis ECG abnormalities can be observed in up to 90% of the patients. Myocardial thickening occurs in up to 20% of these cases. In a family with type IV glycogenosis myocardial thickening, heart failure, and heart transplantation were reported Citation61. In a patient with Tarui's disease CI manifested as anginal chest pain for years, myocardial thickening, and abnormal myocardial texture Citation62. In two infants with type IX glycogenosis large QRS complexes, short PQ-interval and myocardial thickening were reported Citation63.
Danon's disease, a lysosomal glycogen storage disease, is characterized by mental retardation, vacuolar myopathy with mild general weakness, and fatal hypertrophic cardiomyopathy in males before 20 years Citation64. Single patients may additionally present with Wolff-Parkinson-White syndrome Citation65. Females are without CNS or PNS abnormalities but may develop cardiomyopathy in adulthood Citation66. Danon's disease is due to mutations in the lysosomal associated protein 2 (LAMP-2) gene on chromosome Xq25-q25, which encodes for a lysosome-associated glycoprotein Citation67. Typically, autophagic vacuoles accumulate in the myocardium and skeletal muscle. On immunehistochemistry LAMP2 is completely absent in these patients Citation67. Autophagic vacuoles are likely to be causal for the depressed contractile function resulting in an attenuated left ventricular pump reserve. Severe cardiac muscle involvement may lead to early death already at infancy Citation66.
Fatty acid oxidation disorders
Mitochondrial β-oxidation of long-chain fatty acids provides energy for heart and muscle during prolonged aerobic exercise and for hepatic ketogenesis during long-term fasting Citation68. Oxidation of long-chain fatty acids may be disturbed at various enzymatic levels. To gain access to the β-oxidation enzymes, long-chain fatty acids need to be transferred via the inner mitochondrial membrane Citation68. The shuttle is dependent on various enzymes and carnitine. Carnitine is essential for esterification of long-chain fatty acids. Carnitine is transported into the intermembrane space via the organic cation/carnitine transporter. Carnitine is conjugated with long-chain fatty acids by the carnitine palmitoyl transferase 1 (CPT1). Long-chain fatty acids are transferred across the inner plasma membrane by the carnitine-acylcarnitine translocase (CACT), and conjugated back to coenzyme A for subsequent β-oxidation by the carnitine palmitoyl transferase 2 (CPT2). The clinical presentation of fatty acid oxidation disorders varies significantly according to the underlying enzymatic defect Citation69. They may present as isolated cardiomyopathy, myopathy, or hepatopathy Citation70.
Deficiency of the organic cation/carnitine transporter, which has been recently localized to mitochondria Citation71, or carnitine uptake defect cause primary carnitine deficiency (PCD). PCD is characterized by increased loss of carnitine to the urine and decreased carnitine accumulation in various tissues. Plasma carnitine levels are very low Citation72. Patients may present with neonatal metabolic acidosis, hypoketotic hypoglycemia, hepatic encephalopathy, epilepsy, recurrent infections, hepatomegaly, hyperammonemia, skeletal myopathy, and hypertrophic or dilated cardiomyopathy with lipid accumulation Citation70, Citation71. Most of the disease manifestations of PCD respond well to carnitine supplementation. Defects in the liver isoform of CPT1 present with recurrent attacks of fasting hypoketotic hypoglycemia. The heart and the skeletal muscles, which express a genetically distinct form of CPT1, are usually unaffected. These patients can have elevated levels of plasma carnitine. CACT deficiency presents in most cases in the neonatal period with hypoglycemia, hyperammonemia, cardiomyopathy, or arrhythmias leading to cardiac arrest. Plasma carnitine levels are extremely low. Patients with the myopathic form of CPT2 deficiency present with muscle pain, cramps, rhabdomyolysis or myoglobinuria triggered by prolonged exercise. In these patients in-vivo oxidation of long-chain fatty acids is severely impaired during prolonged low-intensity exercise Citation73. More severe variants of CPT2 deficiency present in the neonatal period together with multiple congenital anomalies. Among 107 patients with inherited fatty acid oxidation disorders arrhythmias were recorded in 22% Citation68.
Very-long chain acetyl CoA dehydrogenase deficiency and long chain 3-hydroxy-acetyl CoA dehydrogenase deficiency, are also disorders of the mitochondrial long-chain fatty-acid oxidation, and may present with hypoketotic hypoglycemia, rhabdomyolysis, exercise-induced episodes of intense generalized muscle pain, weakness, or myoglobinuria Citation74. A frequent cardiac manifestation of very-long chain acetyl CoA dehydrogenase deficiency and long-chain 3-hydroxy acetyl-CoA dehydrogenase deficiency is hypertrophic cardiomyopathy () Citation74, Citation75.
Respiratory chain disorders (RDCs)
Respiratory chain disorders are a group of genotypically or phenotypically heterogeneous disorders, which are associated with impaired function of the respiratory chain, Krebs cycle, β-oxidation, transport proteins, assembly proteins or structural proteins. The most frequent and important of these are disorders due to abnormal respiratory chain function, leading to insufficient ATP production via the oxidative phosphorylation system. RCDs are predominantly multisystem diseases with great phenotypic variability, why they are easily overlooked or misdiagnosed for years. RCDs are due to mutations in mitochondrial desoxy-ribonucleic acid (mtDNA) or nuclear DNA (nDNA) located genes, encoding for components of the respiratory chain complexes I-V, t-RNAs, or rRNAs Citation76.
Most respiratory chain disorders are multi-system diseases affecting a variety of organs or tissues, including the heart. However, CI may also occur isolated, even as the presenting feature. CI in respiratory chain disorders comprises ECG abnormalities, focal or diffuse myocardial thickening, hypertrophic cardiomyopathy Citation77, dilation of the cardiac cavities, dilated cardiomyopathy Citation77, LVHT () Citation78 or heart failure Citation79, Citation80. Heart failure may also respond to biventricular pacing Citation81. Intractable heart failure requires heart transplantation in a growing number of patients Citation82.
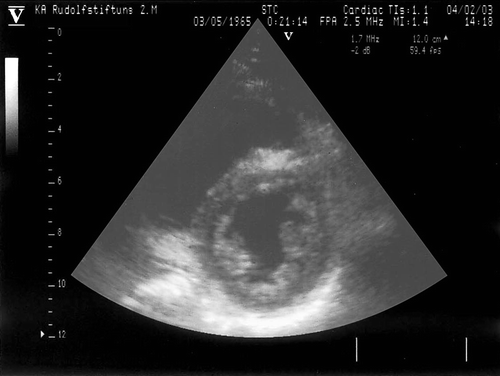
Figure 3. Echocardiographic parasternal short-axis view of a patient with MELAS syndrome shows extensive hypertrabeculation of the lateral and posterior wall
Myoadenylate deaminase (MADA) deficiency
MADA deficiency is characterized by childhood onset muscle cramps, stiffness, post-exercise myalgia, or weakness. Myocardial thickening seems to be a rare cardiac manifestation of MADA deficiency Citation83. In a single case MADA deficiency manifested as recurrent muscle cramps and exercise intolerance and was associated with myocardial thickening and LVHT Citation3.
CI has not been reported in glycogenosis type X (muscle phosphoglycerate mutase), glycogenosis type XI, enolase deficiency (β-enolase), or carnitine palmitoyl-transferase deficiency 1 (CPT1) so far.
Unclassified myopathies
Senger's syndrome
Senger's syndrome is an autosomal recessive condition, characterized by congenital heart defects, and morphologically abnormal mitochondria of the skeletal muscle, lactacidosis, and cataracts Citation84. Senger's syndrome is due to depletion of the adenine nucleotide translocator, presumably following a yet unidentified adenine nucleotide translocator-specific transcriptional or translational error. Interestingly, the gene encoding for the adenine nucleotide translocator is not only involved in the pathogenesis of Senger's syndrome but also that of autosomal dominant chronic progressive external ophthalmoplegia (CPEO) and FSH, in the latter by its depression through the subtelomeric deletion of certain tandem repeats on chromosome 4q35 Citation84. In single cases Senger's syndrome is associated with hypertrophic cardiomyopathy Citation85.
Barth syndrome
Barth syndrome is a rare X-linked recessive disorder of infancy characterized by myopathy, cardiomyopathy, cyclic neutropenia, short stature, low cholesterol, and mitochondrial abnormalities Citation86. Barth syndrome is due to mutations in the taffazine gene on chromosome Xq28. In addition to Barth syndrome taffazin mutations are also responsible for X-linked endocardial fibroelastosis, X-linked fatal infantile dilated cardiomyopathy (CMD3A), or familial isolated noncompaction of left ventricular myocardium () Citation87. The exact function of taffazine is unknown but there are indications that it is directly involved in the metabolism of cardiolipin localized in the inner mitochondrial membrane Citation88. Cardiolipin not only plays a role in the maintenance of the membrane potential and architecture but also provides essential structural and functional support to proteins involved in mitochondrial bioenergetics Citation88.
CI in Barth syndrome comprises endocardial fibroelastosis, increased number of mitochondria in cardiomyocytes Citation86, myocardial thickening in the majority of the cases, or LVHT Citation86. In a study on 34 patients with Barth syndrome 90%, had cardiomyopathy and 50% LVHT () Citation89. Ventricular arrhythmias were found in 43% of the cases Citation89. Ventricular arrhythmias appear to be a dominant feature of CI, which often require the implantation of an ICD Citation89.
Bethlem myopathy (Ullrich syndrome)
Bethlem myopathy is an autosomal dominant, early onset, slowly progressive limb-girdle myopathy with contractures of the fingers, the elbow and the Achilles tendon Citation90. The disease is due to mutations in each of the three type VI collagen subunit genes. The interaction of type VI and type IV collagen within the basal lamina provides a possible molecular mechanism for the pathogenesis of this disorder Citation13, Citation91.
Usually, the heart is not severely affected in Bethlem myopathy Citation92. In a study on 50 patients with Bethlem myopathy CI was found in none of them Citation92. Only in single cases myocardial thickening has been described Citation13.
McLeod syndrome
McLeod syndrome is a multi-system disorder including the CNS (chorea, epilepsy), the PNS (axonal polyneuropathy), and the blood cells (acanthocytosis of the erythrocytes). Mild myopathy is also a common manifestation in most cases Citation93. Rarely, life-threatening rhabdomyolysis may occur Citation93. Often, patients present with mild, asymptomatic hyperCK-emia Citation94. The absence of the Kell Kx membrane protein seems to be causative Citation95. CI has been rarely observed and is described as progressive heart disease in these patients Citation95.
CI has not been described so far in oculo-pharyngeal muscular dystrophy (polyA-binding protein), epidermiolysis bullosa simplex (plectin), Schwartz-Jampel syndrome (perlecan), or Brody disease (calcium ATPase). In a single patient rippling muscle disease (caveolin-3) was associated with arrhythmias and cardiomyopathy Citation96.
Therapy of cardiac abnormalities in primary myopathies
General
Therapy of cardiac abnormalities due to CI in primary myopathies is not at variance to therapy of cardiac disease due to other causes. If atrial fibrillation is tachycardious, digitalis, amiodarone or β-blockers should be added. Electrical cardioversion may be tried, if atrial fibrillation lasts <1 year and if the left atrial diameter is <50mm. In case of bradycardia, intractable to drugs, or AV-block III, a pacemaker is indicated. In case of severe ventricular arrhythmias, an ICD may be required. Supraventricular re-entry tachycardia may be treated by high-frequency catheter ablation. In cases of intracardiac thrombus formation, anticoagulation is essential. Systolic dysfunction may be treated with high-dose angiotensin-converting enzyme inhibitors (ACEI), diuretics, or in case of tachycardia, β-blockers in low dosages. Diastolic dysfunction may respond to ACEI as well. Intractable heart failure may necessitate implantation of a biventricular pacemaker, or even heart transplantation.
Diagnosis-sensitive
In respiratory chain disorders all drugs should be discontinued, which could impair the respiratory chain function, such as acetyl-salicylic acid, valproic acid, barbiturates, biguanides, tetracyclines, chloramphenicol, zidovudin, some local anesthetics, or phenothiazines Citation97. In single DMD patients with end stage cardiac dysfunction continuous intravenous infusion of milrinone or dobutamine resulted in long-term improvement of cardiac function for up to 30 months Citation98. In a retrospective study on 111 DMD patients, of whom 48 received steroids, the decline in cardiac function was significantly lower as compared to those not receiving steroids Citation99, Citation100. In a study on 62 DMD patients and 7 BMD patients ACEI or β-blockers were given after the first abnormal echocardiography in these patients Citation101. β-blockers were added if there was no ventricular remodeling upon ACEI after three months Citation101. In a study on 25 DMD patients, 2 with FCMD and one with EDMD application of the β-blocker carvedilol resulted in significant improvement of left ventricular systolic function, whereas ACEI were ineffective. Recent studies have also shown that ACEI have a beneficial effect directly on muscle force in patients with myopathy Citation102. L-carnitine is not only beneficial to extracardiac manifestations of PCD, but also beneficial to hypertrophic or dilated cardiomyopathy Citation71. A beneficial effect in patients with CPT1, CPT2, and CACT was reported for a low-fat diet supplemented with medium chain triglycerides, which are metabolized by mitochondria independently from carnitine, carnitine supplements, or avoidance of fasting and sustained exercise Citation103. Hypertrophic cardiomyopathy in patients with Pompe's disease responds well to enzyme replacement therapy with recombinant human acid-α-glucosidase Citation104. Particularly, the QT-interval decreases and hypertrophy signs decline Citation105. Also left ventricular function improves Citation104. The earlier affected subjects receive this therapy, the better their outcome. Aerobic conditioning in eight patients with McArdle's disease increased the cardiac output in 15% of these patients Citation106.
Experimental approaches
The tri-block poloxamers, specifically poloxamer 188 (P188), are able to stabilize the membranes of dystrophic myocardium in animal models and may offer a new therapeutic approach for cardiac disease in DMD Citation107. There are also indications, at least in a mouse model, that L-carnitine together with activators of the peroxisome proliferator-activated receptors resolve lipotoxic, hypertrophic cardiomyopathy in PCD Citation72. In juvenile visceral steatosis mice, an animal model of PCD, low lipid diet markedly attenuated the development of hypertrophic cardiomyopathy Citation108.
Conclusion
CI in primary myopathies occurs more frequently than previously thought. Because of the high prevalence of CI, patients with myopathies should be cardiologically investigated as soon as their neurological diagnosis is established, since sufficient cardiac therapy improves CI in these patients and since management of these patients and disease course are strongly influenced by the degree of CI. Future studies should be directed towards the detection of subclinical CI, if early treatment of CI in the subclinical stage improves the outcome of these patients, if prophylactic cardiac therapy attenuates progression of CI, and if CI develops or becomes symptomatic with progression of the muscular manifestations in myopathies, in which CI has not been described so far.
References
- Finsterer J, Stollberger C. Cardiac involvement in primary myopathies. Cardiology. 2000; 94: 1–11
- Funakoshi M, Tsuchiya Y, Arahata K. Emerin and cardiomyopathy in Emery-Dreifuss muscular dystrophy. Neuromusc Disord. 1999; 9: 108–14
- Finsterer J, Stollberger C, Blazek G. Neuromuscular implications in left ventricular hypertrabeculation/noncompaction. Int J Cardiol. 2006; 110: 288–300
- Srinivasan R, Hornyak JE, Badenhop DT, Koch LG. Cardiac rehabilitation after heart transplantation in a patient with Becker's muscular dystrophy: A case report. Arch Phys Med Rehabil. 2005; 86: 2059–61
- Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, et al. Myotonic dystrophy type 2: Molecular, diagnostic and clinical spectrum. Neurology. 2003; 60: 657–64
- Trevisan CP, Pastorello E, Armani M, Angelini C, Nante G, Tomelleri G, et al. Facioscapulohumeral muscular dystrophy and occurrence of heart arrhythmia. Eur Neurol. 2006; 56: 1–5
- Mavrogeni S, Tzelepis GE, Athanasopoulos G, Maounis T, Douskou M, Papavasiliou A, et al. Cardiac and sternocleidomastoid muscle involvement in Duchenne muscular dystrophy: An MRI study. Chest. 2005; 127: 143–8
- Cox GF, Kunkel LM. Dystrophies and heart disease. Curr Opin Cardiol. 1997; 12: 329–43
- Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Hrenneaux M, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction system disease. N Engl J Med. 1999; 341: 1715–24
- Ben Yaou R, Gueneau L, Demay L, Sotra S, Chikhaoui K, Richard P, et al. Heart involvement in lamin A/C related diseases. Arch Mal Coeur Vaiss. 2006; 99: 848–55
- Sanna T, Dello Russo A, Toniolo D, Vytopil M, Pelargonio G, De Martino G, et al. Cardiac features of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutations. Eur Heart J. 2003; 24: 2227–36
- Mercuri E, Brown SC, Nihoyannopoulos P, Poulton J, Kinali M, Richard P, et al. Extreme variability of skeletal and cardiac muscle involvement in patients with mutations in exon 11 of the lamin A/C gene. Muscle Nerve. 2005; 31: 602–9
- deVisser M, deVoogt WG, laRiviere GV. The heart in Becker muscular dystrophy, facioscapulohumeral dystrophy, and Bethlem myopathy. Muscle Nerve. 1992; 15: 591–6
- Galetta F, Franzoni F, Sposito R, Plantinga Y, Femia FR, Galluzzi F, et al. Subclinical cardiac involvement in patients with facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2005; 15: 403–8
- Finsterer J, Stollberger C, Meng G. Cardiac involvement in facioscapulohumeral muscular dystrophy. Cardiology. 2005; 103: 81–3
- Melacini P, Fanin M, Duggan DJ, Freda MP, Berardinelli A, Danieli GA, et al. Heart involvement in muscular dystrophies due to sarcoglycan gene mutations. Muscle Nerve. 1999; 22: 473–9
- Cagliani R, Bresolin N, Prelle A, Gallanti A, Fortunato F, Sironi M, et al. A CAV3 microdeletion differentially affects skeletal muscle and myocardium. Neurology. 2003; 61: 1513–9
- Boito CA, Melacini P, Vianello A, Prandini P, Gavassini BF, Bagattin A, et al. Clinical and molecular characterization of patients with limb-girdle muscular dystrophy type 2I. Arch Neurol. 2005; 62: 1894–9
- Poppe M, Bourke J, Eagle M, Frosk P, Wrogemann K, Greenberg C, et al. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann Neurol. 2004; 56: 738–41
- Barresi R, DiBlasi C, Negri T, Brugnoni R, Vitali A, Felisari G, et al. Disruption of heart sarcoglycan complex and severe cardiomyopathy caused by beta sarcoglycan mutations. J Med Genet. 2000; 37: 102–7
- Coral-Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, et al. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscle dystrophy. Cell. 1999; 98: 465–74
- Kuru S, Yasuma F, Wakayama T, Kimura S, Konagaya M, Aoki M, et al. A patient with limb girdle muscular dystrophy type 2B (LGMD2B) manifesting cardiomyopathy. Rinsho Shinkeigaku. 2004; 44: 375–8
- Calvo F, Teijeira S, Fernandez JM, Teijeiro A, Fernandez-Hojas R, Fernandez-Lopez XA, et al. Evaluation of heart involvement in gamma-sarcoglycanopathy (LGMD2C). A study of ten patients. Neuromuscul Disord. 2000; 10: 560–6
- Gaul C, Deschauer M, Tempelmann C, Vielhaber S, Klein HU, Heinze HJ, et al. Cardiac involvement in limb-girdle muscular dystrophy 2I: Conventional cardiac diagnostic and cardiovascular magnetic resonance. J Neurol. 2006; 253: 1317–22
- Muller T, Krasnianski M, Witthaut R, Deschauer M, Zierz S. Dilated cardiomyopathy may be an early sign of the C826A Fukutin-related protein mutation. Neuromuscul Disord. 2005; 15: 372–6
- Louhichi N, Triki C, Quijano-Roy S, Richard P, Makri S, Meziou M, et al. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities. Identification of a founder mutation in Tunisian families. Neurogenetics. 2004; 5: 27–34
- Toda T, Kobayashi K, Kondo-Iida E, Sasaki J, Nakamura Y. The Fukuyama congenital muscular dystrophy story. Neuromusc Disord. 2000; 10: 153–9
- Murakami T, Hayashi YK, Noguchi S, Ogawa M, Nonaka I, Tanabe Y, et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann Neurol. 2006; 60: 597–602
- Spyrou N, Philpot J, Foale R, Camici PG, Muntoni F. Evidence of left ventricular dysfunction in children with merosin-deficient congenital muscular dystrophy. Am Heart J. 1998; 136: 474–6
- Pethig K, Manz F, Kuhn H. Hypertrophic non-obstructive cardiomyopathy in rigid spine syndrome. Z Kardiol. 1995; 84: 490–5
- Thornton C. The myotonic dystrophies. Sem Neurol. 1999; 19: 25–33
- Mahadevan MS, Yadava RS, Yu Q, Balijepalli S, Frenzel-McCardell CD, Bourne TD, et al. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat Genet. 2006; 38: 1066–70
- Nakada T, Yonesaka S. Hypertrophic cardiomyopathy’ in a girl with adult-type myotonic dystrophy. Pediatr Int. 1999; 41: 586–8
- Phillips MF, Harper PS. Cardiac disease in myotonic dystrophy. Cardiovasc Res. 1997; 33: 13–22
- Dello Russo A, Pelargonio G, Parisi Q, Santamaria M, Messano L, Sanna T, et al. Widespread electroanatomic alterations of right cardiac chambers in patients with myotonic dystrophy type 1. J Cardiovasc Electrophysiol. 2006; 17: 34–40
- Merlevede K, Vermander D, Theys P, Legius E, Ector H, Robberecht W. Cardiac involvement and CTG expansion in myotonic dystrophy. J Neurol. 2002; 249: 693–8
- Finsterer J, Stollberger C, Wegmann R, et al. Left ventricular hypertrabeculation in myotonic dystrophy type 1. Herz. 2001; 26: 287–90
- Vinereanu D, Bajaj BP, Fenton-May J, Rogers MT, Madler CF, Fraser AG. Subclinical cardiac involvement in myotonic dystrophy manifesting as decreased myocardial Doppler velocities. Neuromuscul Disord. 2004; 14: 188–94
- Di Leo R, Rodolico C, De Gregorio C, Recupero A, Coglitore S, Annesi G, et al. Cardiovascular autonomic control in myotonic dystrophy type 1: A correlative study with clinical and genetic data. Neuromuscul Disord. 2004; 14: 136–41
- Finsterer J, Gharehbaghi-Schnell E, Stollberger C, et al. Relation of cardiac abnormalities and CTG-repeat size in myotonic dystrophy. Clin Genet. 2001; 59: 350–5
- Groh WJ, Lowe MR, Simmons Z, Bhakta D, Pascuzzi RM. Familial clustering of muscular and cardiac involvement in myotonic dystrophy type 1. Muscle Nerve 2005; 31: 719–24
- Day JW, Roelofs R, Leroy B, Pech I, Benzow K, Ranum LPW. Clinical and genetic characteristics of a five-generation family with a novel form of myotonic dystrophy (DM2). Neuromusc Disord. 1999; 9: 19–27
- Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, et al. Myotonic dystrophy type 2: Molecular, diagnostic and clinical spectrum. Neurology. 2003; 60: 657–64
- Goebel HH. Congenital myopathies in the new millennium. J Child Neurol. 2005; 20: 94–101
- Michele DE, Albaya FP, Metzger JM. A nemaline myopathy mutation in alpha-tropomyosin causes defective regulation of striated muscle force production. J Clin Invest. 1999; 104: 1575–81
- Nowak KJ, Wattanasirichaigoon D, Goebel HH, Wilce M, Pelin K, Donner K, et al. Mutations in the skeletal muscle (-actin gene in patients with actin myopathy and nemaline myopathy. Nature Genetics. 1999; 23: 208–13
- D'Amico A, Graziano C, Pacileo G, Petrini S, Nowak KJ, Boldrini R, et al. Fatal hypertrophic cardiomyopathy and nemaline myopathy associated with ACTA1 K336E mutation. Neuromuscul Disord. 2006; 16: 548–52
- Muller-Hocker J, Schafer S, Mendel B, Lochmuller H, Pongratz D. Nemaline cardiomyopathy in a young adult: An ultraimmunohistochemical study and review of the literature. Ultrastruct Pathol. 2000; 24: 407–16
- Michele DE, Coutu P, Metzger JM. Divergent abnormal muscle relaxation by hypertrophic cardiomyopathy and nemaline myopathy mutant tropomyosins. Physiol Genomics. 2002; 9: 103–11
- Fujita K, Nakano S, Yamamoto H, Ito H, Goto Y, Kusaka H. An adult case of congenital fiber type disproportion (CFTD) with cardiomyopathy. Rinsho Shinkeigaku. 2005; 45: 380–2
- Benkusky NA, Farrell EF, Valdivia HH. Ryanodine receptor channelopathies. Biochem Biophys Res Commun. 2004; 322: 1280–5
- Al-Ruwaishid A, Vajsar J, Tein I, Benson L, Jay V. Centronuclear myopathy and cardiomyopathy requiring heart transplant. Brain Dev. 2003; 25: 62–6
- Carlsson L, Thornell LE. Desmin-related myopathies in mice and man. Acta Physiol Scand. 2001; 171: 341–8
- Arbustini E, Morbini P, Grasso M, Fasani R, Verga L, Bellini O, et al. Restrictive cardiomyopathy, atrioventricular block and to subclinical myopathy in patients with desmin-immunoreactive material deposits. J Am Coll Cardiol. 1998; 31: 645–53
- Selcen D, Engel AG. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol. 2005; 57: 269–76
- Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003; 42: 2014–27
- El-Menyar AA, Al-Suwaidi J, Gehani AA, Bener A. Clinical and histologic studies of a Qatari family with myofibrillar myopathy. Saudi Med J. 2004; 25: 1723–6
- Nishino I, Malicdan MC, Murayama K, Nonaka I, Hayashi YK, Noguchi S. Molecular pathomechanism of distal myopathy with rimmed vacuoles. Acta Myol. 2005; 24: 80–3
- Kaplan J-C, Fontain B. Neuromuscular disorders: Gene location. Neuromusc Disord. 2006; 16: 64–90
- Stöllberger C, Finsterer J, Bittner RE. Angina for 14 years. Lancet. 1997; 349: 1292
- Regalado JJ, Rodriguez MM, Ferrer PL. Infantile hypertrophic cardiomyopathy of glycogenosis type IX: Isolated cardiac phosphorylase kinase deficiency. Pediatr Cardiol. 1999; 20: 304–7
- Metzl JD, Elias ER, Berul CI. An interesting case of infant sudden death: Severe hypertrophic cardiomyopathy in Pompe's disease. Pacing Clin Electrophysiol. 1999; 22: 821–2
- Ewert R, Gulijew A, Wensel R, Dandel M, Hummel M, Vogel M, et al. Glycogenosis type IV as a seldom cause of cardiomyopathy – report about a successful heart transplantation. Z Kardiol. 1999; 88: 850–6
- Fanin M, Nascimbeni AC, Fulizio L, Spinazzi M, Melacini P, Angelini C, et al. Generalized lysosome-associated membrane protein-2 defect explains multisystem clinical involvement and allows leukocyte diagnostic screening in Danon disease. Am J Pathol. 2006; 168: 1309–20
- Balmer C, Ballhausen D, Bosshard NU, Steinmann B, Boltshauser E, Bauersfeld U, et al. Familial X-linked cardiomyopathy (Danon disease): diagnostic confirmation by mutation analysis of the LAMP2gene. Eur J Pediatr. 2005; 164: 509–14
- Morisawa Y, Fujieda M, Murakami N, Naruse K, Okada T, Morita H, et al. Lysosomal glycogen storage disease with normal acid maltase with early fatal outcome. J Neurol Sci. 1998; 160: 175–9
- Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med 2006; 27: 495–502
- Stanley CA. Carnitine deficiency disorders in children. Ann N Y Acad Sci. 2004; 1033: 42–51
- Bonnet, D, Martin, D, Pascale, De Lonlay, Villain, E, Jouvet, P, Rabier, D, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100:2248–53.
- Lamhonwah AM, Tein I. Novel localization of OCTN1, an organic cation/carnitine transporter, to mammalian mitochondria. Biochem Biophys Res Commun. 2006; 345: 1315–25
- Asai T, Okumura K, Takahashi R, Matsui H, Numaguchi Y, Murakami H, et al. Combined therapy with PPARalpha agonist and L-carnitine rescues lipotoxic cardiomyopathy due to systemic carnitine deficiency. Cardiovasc Res. 2006; 70: 566–77
- Hou JW. Primary systemic carnitine deficiency presenting as recurrent Reye-like syndrome and dilated cardiomyopathy. Chang Gung Med J. 2002; 25: 832–7
- Orngreen MC, Duno M, Ejstrup R, Christensen E, Schwartz M, Sacchetti M, et al. Fuel utilization in subjects with carnitine palmitoyltransferase 2 gene mutations. Ann Neurol. 2005; 57: 60–6
- Oey NA, den Boer ME, Wijburg FA, Vekemans M, Auge J, Steiner C, et al. Long-chain fatty acid oxidation during early human development. Pediatr Res. 2005; 57: 755–9
- Mathur A, Sims HF, Gopalakrishnan D, Gibson B, Rinaldo P, Vockley J, et al. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation. 1999; 99: 1337–43
- Leonard JV, Shapira AHV. Mitochondrial respiratory chain disorders I: Mitochondrial DNA defects. Lancet. 2000; 355: 299–304
- Bonnet D, Rustin P, Rotig A, Le Bidois J, Munnich A, Vouhe P, et al. Heart transplantation in children with mitochondrial cardiomyopathy. Heart. 2001; 86: 570–3
- Finsterer J, Stöllberger C. Hypertrabeculated left ventricle in mitochondriopathy. Heart. 1998; 80: 632
- Houstek J, Klement P, Floryk D, Antonicka H, Hermanska J, Kalous M, et al. A novel deficiency of mitochondrial ATPase of nuclear origin. Hum Mol Genet. 1999; 8: 1967–74
- Wallace DC. Mitochondrial defects in cardiomyopathy and neuromuscular disease. Am Heart J. 2000; 139: 70–85
- Stöllberger, C, Finsterer, J. Disappearance of left ventricular hypertrabeculation/noncompaction after biventricular pacing in a patient with neuromuscular disorder. J Card Fail. 2007; (in press).
- Ozawa E, Noguchi S, Mizuno Y, Hagiwara Y, Yoshida M. From dystrophinopathy to sarcoglycanopathy: Evolution of a concept of muscular dystrophy. Muscle Nerve. 1998; 21: 421–38
- Skyllouriotis ML, Marx M, Bittner RE, Skyllouriotis P, Gross M, Wimmer M. Myoadenylate deaminase deficiency, hypertrophic cardiomyopathy and gigantism syndrome. Pediatr Neurol. 1997; 17: 61–6
- Sharer JD. The adenine nucleotide translocase type 1 (ANT1): A new factor in mitochondrial disease. IUBMB Life. 2005; 57: 607–14
- Atiq M, Iqbal S, Ibrahim S. Sengers disease: A rare association of hypertrophic cardiomyopathy and congenital cataracts. Indian J Pediatr. 2004; 71: 437–40
- Bleyl SB, Mumford BR, Brown-Harrison MC, Pagotto LT, Carey JC, Pysher TJ, et al. Xq28-linked noncompaction of the left ventricular myocardium: Prenatal diagnosis and pathologic analysis of affected individuals. Am J Med Genet. 1997; 72: 257–65
- Brady AN, Shehata BM, Fernhoff PM. X-linked fetal cardiomyopathy caused by a novel mutation in the TAZ gene. Prenat Diagn. 2006; 26: 462–5
- Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. 2007; 292: C33–C44
- Spencer CT, Bryant RM, Day J, Gonzalez IL, Colan SD, Thompson WR, et al. Cardiac and clinical phenotype in Barth syndrome. Pediatrics. ee346 2006; 118: 337
- Merlini L, Villanova M, Sabatelli P, Malandrini A, Maraldi NM. Decreaed expression of laminin beta 1 in chromosome 21-linked Bethlem myopathy. Neuromusc Disord. 1999; 9: 326–9
- Sewry CA, Muntoni F. Inherited disorders of the extracellular matrix. Curr Opin Neurol. 1999; 12: 519–26
- van der Kooi AJ, de Voogt WG, Bertini E, Merlini L, Talim FB, Ben Yaou R, et al. Cardiac and pulmonary investigations in Bethlem myopathy. Arch Neurol. 2006; 63: 1617–21
- Jung HH, Brandner S. Malignant McLeod myopathy. Muscle Nerve. 2002; 26: 424–7
- Danek A, Rubio JP, Rampoldi L, Ho M, Dobson-Stone C, Tison F, et al. McLeod neuroacanthocytosis: Genotype and phenotype. Ann Neurol. 2001; 50: 755–64
- Witt TN, Danek A, Reiter M, Heim MU, Dirschinger J, Olsen EG, et al. McLeod syndrome: A distinct form of neuroacanthocytosis. Report of two cases and literature review with emphasis on neuromuscular manifestations. J Neurol. 1992; 239: 302–6
- Koul RL, Chand RP, Chacko A, Ali M, Brown KM, Bushnarmuth SR, et al. Severe autosomal recessive rippling muscle disease. Muscle Nerve. 2001; 24: 1542–7
- Finsterer J. Central nervous system manifestations of mitochondrial disorders. Acta Neurol Scand. 2006; 114: 217–38
- Cripe LH, Barber BJ, Spicer RL, Wong BL, Weidner N, Benson DW, et al. Outpatient continuous inotrope infusion as an adjunct to heart failure therapy in Duchenne muscular dystrophy. Neuromuscul Disord. 2006; 16: 745–8
- Biggar WD, Harris VA, Eliasoph L, Alman B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord. 2006; 16: 249–55
- Markham LW, Spicer RL, Khoury PR, Wong BL, Mathews KD, Cripe LH, et al. Steroid therapy and cardiac function in Duchenne muscular dystrophy. Pediatr Cardiol. 2005; 26: 768–71
- Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, et al. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005; 112: 2799–804
- Onder G, Vedova CD, Pahor M. Effects of ACE inhibitors on skeletal muscle. Curr Pharm Des. 2006; 12: 2057–64
- Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006; 142: 77–85
- Klinge L, Straub V, Neudorf U, Schaper J, Bosbach T, Gorlinger K, et al. Safety and efficacy of recombinant acid alpha-glucosidase (rhGAA) in patients with classical infantile Pompe disease: Results of a phase II clinical trial. Neuromuscul Disord. 2005; 15: 24–31
- Ansong AK, Li JS, Nozik-Grayck E, Ing R, Kravitz RM, Idriss SF, et al. Electrocardiographic response to enzyme replacement therapy for Pompe disease. Genet Med. 2006; 8: 297–301
- Haller RG, Wyrick P, Taivassalo T, Vissing J. Aerobic conditioning: an effective therapy in McArdle's disease. Ann Neurol. 2006; 59: 922–8
- Townsend D, Yasuda S, Metzger J. Cardiomyopathy of Duchenne muscular dystrophy: Pathogenesis and prospect of membrane sealants as a new therapeutic approach. Expert Rev Cardiovasc Ther. 2007; 5: 99–109
- Jalil MA, Horiuchi M, Wakamatsu M, Li MX, Begum L, Suzuki K, et al. Attenuation of cardiac hypertrophy in carnitine-deficient juvenile visceral steatosis (JVS) mice achieved by lowering dietary lipid. J Biochem (Tokyo) 2006; 139: 263–70
- Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. American College of Cardiology/American Heart Association Task Force; European Society of Cardiology Committee for Practice Guidelines; European Heart Rhythm Association and the Heart Rhythm Society. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death–executive summary: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Eur Heart J. 2006; 27: 2099–140
- Park OY, Ahn Y, Park WS, Lim JH, Park HW, Kim JH, et al. Rapid progression from hypertrophic cardiomyopathy to heart failure in a patient with Becker's muscular dystrophy. Eur J Heart Fail. 2005; 7: 684–8
- Muller JS, Piko H, Schoser BG, Schlotter-Weigel B, Reilich P, Gurster S, et al. Novel splice site mutation in the caveolin-3 gene leading to autosomal recessive limb girdle muscular dystrophy. Neuromuscul Disord. 2006; 16: 432–6
- Ohkubo M, Ino T, Shimazaki S, Yabuto K, Okada R, Sato T. Multicore myopathy associated with multiple pterygium syndrome and hypertrophic cardiomyopathy. Pediatr Cardiol. 1996; 17: 53–6
- Sekijima Y, Ikeda S, Katai S, Matsuda M, Hashimoto T, Haruta S, et al. Cytoplasmic body myopathy with hypertrophic cardiomyopathy. Intern Med. 1995; 34: 166–70
- Case LE, Kishnani PS. Physical therapy management of Pompe disease. Genet Med. 2006; 8: 318–27
- Scaglia F, Towbin JA, Craigen WJ, Belmont JW, Smith EO, Neish SR, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004; 114: 925–31
- Schroder JM, May R, Shin YS, Sigmund M, Nase-Huppmeier S. Juvenile hereditary polyglucosan body disease with complete branching enzyme deficiency (type IV glycogenosis). Acta Neuropathol (Berl) 1993; 85: 419–30
- DeAdamo P, Fassone L, Gedeon A, Janssen EAM, Bione S, Bolhuis PA, et al. The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet. 1997; 61: 862–7
- Willemsen MA, van Oort AM, ter Laak HJ, Sengers RC, Gabreels FJ. Multicore myopathy with restrictive cardiomyopathy. Acta Paediatr. 1997; 86: 1271–4
- Ishiwata S, Nishiyama S, Seki A, Kojima S. Restrictive cardiomyopathy with complete atrioventricular block and distal myopathy with rimmed vacuoles. Jpn Circ J. 1993; 57: 928–33
- Campos Y, Martin MA, Caballero C, Rubio JC, de la Cruz F, Tunon T, et al. Single large-scale mitochondrial DNA deletion in a patient with encephalopathy, cardiomyopathy, and prominent intestinal pseudo-obstruction. Neuromuscul Disord. 2000; 10: 56–8
- Berger PB, Duffy J, Reeder GS, Karon BL, Edwards WD. Restrictive cardiomyopathy associated with the eosinophilia-myalgia syndrome. Mayo Clin Proc. 1994; 69: 162–5
- Finsterer J, Stollberger C, Gaismayer K, Janssen B. Acquired noncompaction in Duchenne muscular dystrophy. Int J Cardiol. 2006; 106: 420–1
- Ichida F, Tsubata S, Bowles KR, Hanenda N, Uese K, Miyawaki T. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation. 2001; 103: 1256–63
- Chebel S, Ben Hamda K, Boughammoura A, Frih Ayed M, Ben Farhat MH. Cardiac involvement in Steinert's myotonic dystrophy. Rev Neurol (Paris) 2005; 161: 932–9