
Abstract
In the past 10 years, large-scale genotyping has led to discoveries of sequence variants that confer the risk of many common and complex diseases. Due to pioneering work done, in large part, at deCODE genetics in Reykjavik, discoveries from Iceland have contributed substantially to key advances in population genetics. In cardiovascular medicine, a number of discoveries have been made, uncovering sequence variants that are associated with disorders such as coronary artery disease, atrial fibrillation, sick sinus syndrome, peripheral vascular disease, aortic aneurysm, and ischemic stroke. Thus, a wealth of genetic data has been accumulated in cardiology and has enhanced our understanding of a number of diseases. In many cases, these findings offer new mechanistic clues into the pathophysiology of complex cardiovascular diseases and may point toward novel therapeutic approaches in drug therapy. The next important step is to begin to transform these findings into practical clinical knowledge with the aim of improving the delivery of cardiovascular health care.
Introduction
In the past decade, large-scale genotyping has led to discoveries of sequence variants that confer the risk of common and complex diseases. Within cardiovascular medicine, genome-wide association studies (GWAS) have led to the discovery of a number of common sequence variants associated with many disorders, including coronary artery disease (CAD), atrial fibrillation (AF), peripheral vascular disease, aortic aneurysm, and ischemic stroke.[Citation1–6]
More recently, an increasing number of studies based on whole-genome sequencing (WGS) and whole-exome sequencing (WES) have uncovered rare variants associated with common heart diseases, including sick sinus syndrome and AF.[Citation7–9] This transition to studies based on rare variants identified by WGS and WES provides exciting opportunities to further explore the relationship between sequence variants and common diseases.
A major challenge, however, is to begin to transform these findings into practical clinical knowledge. The possibility to individualize risk of disease and tailor therapeutic interventions options based on genomic information is closer than ever. In the near future, large-scale genetic databases may provide a powerful tool to help guide clinical medicine.
Iceland’s role in genetic research
In the past 15–20 years, discoveries originating from Iceland have contributed substantially to key advances in population genetics. In large extent, this is due to the pioneering work done at deCODE genetics, founded in 1996 by Dr. Kari Stefansson.
The key goal of deCODE genetics was to find human genes associated with common diseases using population-based studies, and to apply the knowledge gained to uncover mechanisms of common diseases to guide drug development. So far, deCODE has discovered a number of genes believed to be involved in cardiovascular disease, cancer, dementia, diabetes, chronic kidney disease, and schizophrenia, among other diseases (http://www.ebi.ac.uk/gwas).
Despite having a small population of 330.000, which was relatively isolated for many centuries, it is not inbred by any measure and the incidence of autosomal recessive disorders, common in inbred communities, is low in Iceland. The genetic makeup of the nation is a northern European mix, with approximately 75% of the males of Scandinavian origin and 66% of the women of Gaelic origin.[Citation10] This supports the theory, put forward by historians, that the majority of females in the Icelandic founding population had Gaelic ancestry, whereas the majority of the males had Scandinavian ancestry. Icelanders are therefore by no means a genetic outlier in Europe. Iceland has an extensive genealogy database, enabling the makings of exact family pedigrees and in many cases inhabitants can trace their family lines back to the first settler who came to the island around the year 874 AD. The existence of this genealogy database has greatly facilitated genetic research.
The majority of Icelanders have supported the efforts of deCODE and participated in their research projects. To date, about 150 000 Icelanders have been genotyped with DNA microarrays and more than 20 000 of those have also had their whole-genome sequenced. Given the large fraction of the Icelandic population that has been chip-typed, precise long-range phasing is available genome-wide for all chip-typed Icelanders. This allows accurate imputation of sequence variants from whole-genome sequencing into chip-typed individuals, imputation referring to the statistical inference of unobserved genotypes. Additionally, in part made possible by the extensive genealogy database in Iceland, genotypes can be imputed into close relatives of chip typed individuals, creating in silico genotypes for a large number of Icelanders. The availability of genetic information for a large part of the nation to study creates unique opportunities for scientific and clinical advancement.
Genetic studies of cardiac arrhythmias
In population-based studies, both from Iceland and The Framingham Heart Study.[Citation11,Citation12], AF has been shown to segregate in families. In a study of more than 5000 individuals with AF in Iceland, the risk of developing AF was almost 2-fold in those who had a close relative with the arrhythmia and almost 5-fold if a close relative had been diagnosed with AF before the age of 60.[Citation11] This suggested a clear genetic predisposition, especially in those who were diagnosed at a relatively young age.
The development of microarrays for genotyping heralded a new era in genomic medicine, facilitating studies of associations of common sequence variants and common complex diseases. In GWAS approach, the principle is to genotype thousands to millions of sequence variants and to compare the allele distribution between individuals who have a particular disease and controls. The findings from such studies have usually shown a relatively small to modest effects of the genetic associations uncovered, generally in the range of 1.2- to 2-fold risk. However, even small to moderate effects can offer biologic insight and thus be of value in understanding the pathophysiology of common diseases. Most investigators, on the other hand, acknowledge the likelihood that the genetic effects on complex diseases are the results of multiple variants and their interactions. In addition, possible interactions with the lifestyle of the individual and a number of environmental factors are of significant importance. Many of the discoveries from GWAS have also underscored the complexity of genetic findings where the relationship between associated variant and disease has not always been immediately apparent.
As an example, two common variants found on 4q25 that associates with AF are in a noncoding area of the chromosome.[Citation3] The stronger of the two variants on 4q25 is present in 35% of individuals of Northern European descent and increases the risk of AF by 72%. We proposed that the variants in the noncoding area of 4q25 might have a role in regulatory function on a nearby gene. An interesting candidate is the most proximal gene, PITX2, a transcription factor that regulates right-left asymmetry of the heart during development and determines the occurrence of pulmonary myocardium, that is, atrial tissue which can be found in the pulmonary veins in mice.[Citation13] Both phenomena may be important in the pathogenesis of atrial fibrillation since ectopic beats and from the myocardial tissue in the pulmonary veins may be the triggers for atrial fibrillation.[Citation14]
Another example demonstrating the complexity of genetic findings associating with AF is a sequence variant on chromosome 16q22.[Citation5] Although there is a gene at this locus, its functional relationship to AF was not initially clear. This is a tumor suppressor gene, ZFHX3, which is associated with regulation of growth and differentiation of several tissues, including neurons and skeletal muscle. More recently, this gene has been associated with the transcription factor RUNX3 that translocates with transforming growth factor beta signaling and is an important mediator of fibrosis.[Citation15] Given the potential importance of fibrosis in the pathogenesis and prognosis of AF, it is conceivable that ZFHX3 increases susceptibility to the arrhythmia by modulating such processes.[Citation16]
A third locus for AF discovered by GWAS is the calcium-activated potassium channel KCNN3 on chromosome 1q21.[Citation17] Furthermore, by meta-analysis of GWAS data, six additional susceptibility loci for atrial fibrillation have been discovered.[Citation18]
We have used an electrocardiographic database in Iceland to study how sequence variants may influence key electrophysiologic parameters of the heart.[Citation19] To search for sequence variants that modulate heart rate, PR interval, and QRS duration, a GWAS was performed in more than 10 000 individuals. Several significant associations were identified, including a locus for heart rate (MYH6), four loci for PR interval (TBX5, SCN10A, CAV1, and ARHGAP24) and four for QRS duration (TBX5, SCN10A, 6p21, and 10q21). By testing for associations between these loci and subjects with selected arrhythmias, correlations were observed between TBX5 and CAV1 and AF, between TBX5 and advanced atrioventricular block, and between SCN10A and pacemaker implantation. Two simultaneously published studies demonstrated the association between sequence variants and cardiac conduction. One showed an association between SCNA10 and PR interval,[Citation20] whereas the other study revealed nine loci, including TBX5, SCN10A, CAV1, and ARHGAP24 affecting the PR interval.[Citation21]
The multiple common variants uncovered by GWAS have begun to unravel the genetic architecture of complex human traits, yielding robust associations with hundreds of genomic variants. By design, the GWAS methods capture a large fraction of the common variants of the human genome. However, it has become apparent that despite these successes, a substantial fraction of heritability of most complex traits remains unaccounted for. Common variants with large effects are very rare and rare variants with large effect are usually associated with Mendelian disorders. Therefore, it is unlikely that rare variants with large effects contribute importantly to the heritability of complex traits. On the other hand, it is more likely that a number of rare variants with low to modest effect contribute to the heritability of complex diseases although this may be hard to detect because of power issues ().
Figure 1. Feasibility of identifying genetic variants by risk allele frequency and strength of genetic effect (odds ratio). Adapted from Manolio T, et al., Nature 2009;461(7265):747–753, with permission.
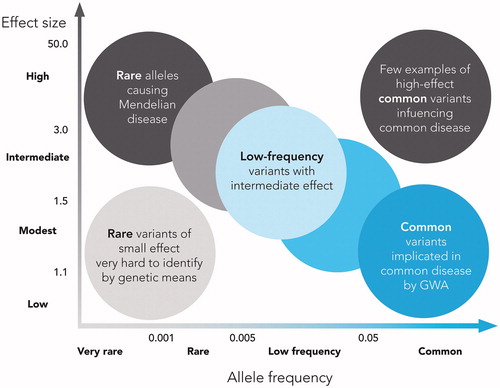
WGS is the most comprehensive method for analyzing the genome and reveals the complete DNA make-up of an organism. This method is expected to yield rare variants with moderate effect and copy number variants that GWAS may have missed. The cost has rapidly decreased from when this method was first available and is now only a fraction of the initial expense.
One of the first studies performed at deCODE using WGS identified a missense variant in MYH6, coding alpha heavy chain subunit of cardiac myosin, which was associated with a high risk (OR 12.5) of sick sinus syndrome (SSS).[Citation7] SSS is a common disease characterized by sinus bradycardia, sinus pauses, and chronotropic insufficiency and can lead to syncope among other symptoms. This was the first time that a sarcomere gene, which is usually thought of in the context of myocardial contraction, was linked to a conduction disease. The carriers of the MYH6 mutation have almost a 50% lifetime risk of SSS compared to a noncarrier lifetime risk of 6%. The MYH6 mutations also confers risk of AF, even after exclusion of known cases of SSS (OR 2.03).[Citation7]
In another study utilizing WGS data, we searched for variants conferring risk of early onset AF. A rare truncating frameshift deletion in the myosin encoding MYL4 gene was found to associate with early-onset AF under a recessive mode of inheritance.[Citation22] Eight homozygous carriers of the mutation were found in the Icelandic population, all of whom had been diagnosed with the arrhythmia at a very early age. Six of the homozygous carriers were diagnosed with AF by the age of 30 years, three of the homozygous carriers had received pacemaker implantations before the age of 50 years and three had suffered an ischemic stroke at a young age despite having a low-risk CHA2DS2-VASc score.
The association of both MYH6 and MYL4 with AF strongly implies that variants encoding sarcomer genes may be involved in the development of this common arrhythmia. These findings may offer new mechanistic clues into the pathophysiology of this complex arrhythmia and perhaps point toward novel therapeutic approaches in drug therapy.
Genetics of coronary artery disease
Coronary heart disease remains a leading cause of death and disability worldwide. Family clustering of acute myocardial infarction (AMI) suggests a strong genetic factor contributing to the development of this common disease. It has long been realized that coronary heart disease is the archetypal complex disease, involving multiple genes, environmental factors, and complex gene–gene and gene–environmental interactions.[Citation23] Additionally, several established risk factors of coronary heart disease have their own genetic complexity. Diabetes, hypertension, hyper- or dyslipidemias, and obesity suffice as examples.[Citation24–26] Thus, a thorough understanding of the genetics of coronary heart disease is of a major interest. However, the complexity has also raised doubts about the feasibility of detecting, through genetic studies, molecular mechanisms that would be useful targets for therapeutic or preventive interventions. Also, it has been confirmed that many, even very high numbers of genetic signals underly the common complex diseases, most having a relatively modest effect (OR <1.5).[Citation23]
A large international consortium with Icelandic participation [Citation27] had by the end of 2013 identified 46 loci with genome-wide significant association with CAD and myocardial infarction. Of these loci, 40% overlapped with classic risk factors, lipids (24%), hypertension (10%), diabetes (2%), or a combination of these risk factors (4%). However, 35 loci were in genomic regions not associated with known risk factors, raising the possibility of uncovering previously unknown molecular mechanisms contributing to atherogenesis, perhaps even a new fundamental understanding.
The first sequence variant identified was on chromosome 9p21 and confirmed by numerous studies in different cohorts globally.[Citation1,Citation2,Citation28] This common variant, present in homozygous form in 21% of the population, carried a 1.64-fold risk of developing AMI compared to noncarriers. The variants on chromosome 9p21 are located in an area containing the tumor suppressor genes CDKN2A and CDKN2B, encoding proteins that regulate cell proliferation, cell aging, senescence, and apoptosis.
The relationship between sequence variants and clinical phenotypes in CAD has been thoroughly pursued.[Citation29] Quantifying the extent of atherosclerosis is an example of detailed phenotyping that may serve as a method to take the genetic findings one step further toward a pathobiological understanding as well as translation into clinical utilization. The only genetic variants that have been shown to associate with the burden of coronary atherosclerosis are lipoprotein (a) and the variant on 9p21. The cumulative effect of known genetic risk variants for CAD on the development and progression of coronary atherosclerosis was studied in a cohort of 8.600 Icelandic patients with angiographically defined CAD.[Citation30] The effect of all known risk variants of CAD on the angiographic severity of the disease was quantified by plotting the number of affected vessels per risk allele against their effect on CAD risk (). With rising genetic risk, there was an increase in the number of significantly stenosed coronary arteries in both the Icelandic cohort and the control cohort from Atlanta, Georgia in the United States (US). Not surprisingly, the strongest effects were from the LPA-risk alleles and 9p21 that drive this association between genetic CAD risk and the burden of the atherosclerotic disease. When compared to the atherogenic impact of classical risk factors, the genetic risk, combining the effects of all the risk alleles, had higher odds ratio than male sex, lipid disorders, or diabetes on the extent of coronary atherosclerosis. Hypertension and smoking did not affect the distribution of atherosclerosis in either study population.
Figure 2. The effects of 50 single-nucleotide polymorphisms (SNPs) on the extent of coronary artery disease (CAD), expressed as the increase in number of diseased coronary arteries (with at least 50% stenosis) per SNP risk allele, plotted against their respective effect on CAD risk (odds ratio), previously reported in meta-analyses of genome-wide association studies. Combined effect sizes in the Icelandic and Emory Biobank samples are presented where available. The solid line denotes best linear fit, and the dashed lines indicate 95% confidence limits. Adapted from ref. Citation32.
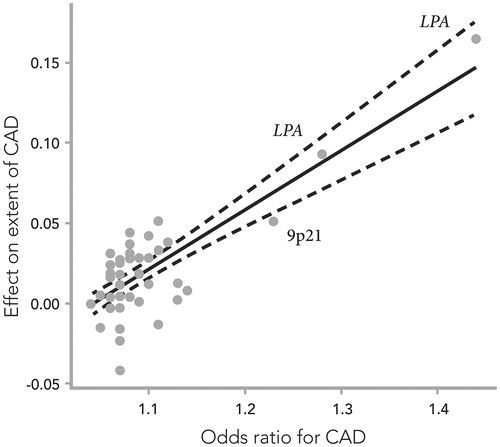
Many associations with classical risk factors of CAD have been discovered, including over 200 variants associated with blood lipids, over 10 with blood pressure and over 50 associated with diabetes (http://www.ebi.ac.uk/gwas). Many variants that affect serum levels of LDL or non-HDL cholesterol also affect the risk of CAD, strongly supporting a causal relationship.[Citation28] Most recently, a rare loss-of-function mutation in ASGR1, encoding subunit 1 of the asialoglycoprotein receptor, was found to lower non-HDL cholesterol and protect against CAD.[Citation31] On average, the ASGR1 mutation associated with a 0.40 mmol/L reduction in non-HDL cholesterol and a 34% reduction in risk of CAD. Interestingly, this reduction in coronary risk is considerably larger than would be expected from the lowering of non-HDL cholesterol alone.
Recently, the large Icelandic dataset was used to search for rare and low-frequency variants that affect lipid fractions and CAD,[Citation32] resulting in the discovery of 13 variants with large effects and confirming 14 previously reported variants. Five variants, in PCSK9, APOA1, ANGPLL4, and LDLR, were found to associate with CAD. The strongest association with CAD was found for the non-HDL genetic risk score. Non-HDL cholesterol includes not only LDL-cholesterol, but also the cholesterol contained in all atherogenic lipoproteins containing apo B, including VLDL and VLDL-remnants, intermediate density lipoprotein (IDL), lipoprotein(a), chylomicrons, and chylomicron remnants. After accounting for non-HDL cholesterol, no other risk score contributed independently to CAD risk. Conversely, non-HDL cholesterol significantly added to the risk attributable to LDL-cholesterol, underscoring the added atherogenic importance of remnant cholesterol.
Taken together, all the susceptibility loci that have been identified for CAD only explain around 10% of the heritability of the disease.[Citation23,Citation33] However, these recent papers, capitalizing on the powers of whole-genome sequencing, underscore the wealth of information to be anticipated in this new era of genomics, not only on rare and low-frequency polymorphisms with association to important phenotypes, but also providing clinically valuable mechanistic and pathobiologic insights.
Potential clinical use of genetic information
The term personalized or precision medicine has been introduced to refer to the ability to integrate clinical, molecular, and environmental markers of disease risk and to predict the response of an individual to a particular therapy. Until recently, this concept has perhaps found its greatest application in oncology, but identifying specific prognostic biomarkers in other fields of medicine, including cardiology, is also becoming feasible.
There has been a renewed interest in personalized or precision medicine. At the 2015 State of the Union address in the United States Congress, President Obama launched a precision medicine initiative.[Citation34] This ambitious plan involves genotyping up to one million individuals who are willing to participate in this program. Similar initiatives are underway in other countries including the United Kingdom and Finland. Iceland may, however, have a head start with the wealth of genetic and clinical information already available for the whole nation. Nonetheless, a constructive utilization of these data requires an orchestrated effort that still awaits the necessary initiative.
One clear example of the potential impact of genetics in clinical medicine involves mutations in the BRCA1 and BRCA2 genes and the associated high risk of malignancy, particularly breast cancer and ovarian cancer in women and prostate cancer in men.[Citation35] The overall lifetime risk of developing any cancer is increased 4.6-fold in the carriers of a five base deletion out of exon 9 in BRCA2, which is the predominant mutation type in Iceland. Moreover, the life expectancy of these BRCA2 mutation carriers is decreased by 11 years for women and 7 years for men.[Citation36]
Although some of the BRCA2 mutation carriers in Iceland were identified due to testing on clinical indications, the majority were discovered through research efforts at deCODE genetics. As the participants have only consented for participation in research, contacting them to warn them about their risk would be controversial.
Most healthcare providers within the cancer field spend the majority of their time in the area of diagnosis, treatment, and terminal care. Limited resources are spent on early detection and even less on prevention. In our opinion, the current knowledge of carriers of the BRCA2 mutation in Iceland provides a unique opportunity for early detection of cancer, even prevention although admittedly this opportunity carries with it numerous challenges.
In cardiovascular medicine, there may also be important opportunities to use genetic information to impact prognosis. This could be done using already existing genotypic information to try to influence outcomes in high-risk individuals. Sudden cardiac death (SCD), a major cause of mortality in Western countries, may be an interesting field to consider in this regard. Although majority of cases of arrhythmic death occur in elderly individuals and are more often than not associated with CAD, SCD in individuals under the age of 40 years is not a rare occurrence. The sudden death of a young and apparently healthy person is among the more challenging scenarios in clinical medicine, not least because of the perceived risk of the same fate in surviving relatives.
Most of the SCD in the young occur as a consequence of inherited arrhythmogenic disorders (IADs).[Citation37] IADs can be divided in two groups: cardiomyopathies which are disorders of abnormal structure and function of cardiac muscle, and channelopathies that are not structural abnormalities of the heart, but predispose to life-threatening arrhythmias by disrupting the electrical activity of myocyte membranes. Among these disorders are the long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, short QT syndrome, arrhythmogenic right ventricular cardiomyopathy, and hypertrophic cardiomyopathy.[Citation37] IADs are relatively uncommon diseases with a large number of known causative mutations. However, the clinical picture of IADs can be highly variable, even within the same family, due to the influence of environmental and genetic modifiers. Many individuals who carry the mutations associated with IADs can be completely asymptomatic. Likewise, the electrocardiogram may also be normal, either consistently or intermittently, and their disorder therefore in many individuals concealed.
The extensive genetic database at deCODE genetics, along with broad access to nationwide health care information and easy identification of subjects via a national identification number, offers a unique opportunity to further study individuals with the most common known genetic risk variants for IADs and sudden cardiac death. Such a study could yield unprecedented information on genetic epidemiology, genotype–phenotype relationship, risk of serious adverse cardiac events, and the possibility of using the genetic information for clinical risk stratification.
A key goal would be to investigate how the use of genetics might help in designing a precision approach to the prevention of SCD by integrating clinical and molecular data. This approach is the reverse of the classic paradigm in genetic research. Instead of searching for sequence variants to match and possibly account for phenotypic information, the emphasis is on phenotypic characterization of patients with a known risk genotype.
Such a study could also help to determine if and how initiating preventative measures in a group such as this is feasible. Preventive initiatives may include lifestyle modifications, lists with which drugs to avoid in LQTS, the use of prophylactic medications such as a beta blocker when appropriate, or even devices such as an implantable defibrillator in certain instances. There are, of course, many practical and ethical issues that need to be considered, but this type of approach is nevertheless within the realm of possibility.
In conclusion, a wealth of genetic data has been accumulated in cardiology and has enhanced our understanding of a number of diseases. The next important step is to use this information in a careful manner to improve our understanding of diseases processes and the delivery of health care. Iceland has a unique opportunity to take a leading role in this critical initiative which could reshape the practice of medicine.
Disclosure statement
David O. Arnar is a part-time employee of deCODE genetics. Other authors report no conflict of interest.
References
- McPherson R, Pertsemlidis A, Kavaslar N, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491.
- Helgadottir A, Thorleifsson G, Manolescu A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493.
- Gudbjartsson DF, Arnar DO, Helgadottir A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;488:353–357.
- Tucker NR, Clauss S, Ellinor PT. Common variation in atrial fibrillation: navigating the path from genetic association to mechanism. Cardiovasc Res. 2016;109:493–501.
- Gudbjartsson DF, Holm H, Gretarsdottir S, et al. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet. 2009;41:876–888.
- Helgadottir A, Thorleifsson G, Magnusson KP, et al. The same sequence variant on 9q21 associates myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–224.
- Holm H, Gudbjartsson DF, Sulem P, et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet. 2011;43:316–320.
- Gudbjartsson DF, Helgason H, Gudjonsson SA, et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet. 2015;47:435–444.
- Sulem P, Helgason H, Oddsson A, et al. Identification of a large set of rare complete human knockouts. Nat Genet. 2015;47:448–452.
- Helgason A, Sigurdardottir S, Gulcher J, et al. mtDNA and the origin of the icelanders: deciphering signals of recent population history. Am J Hum Genet. 2000;66:999–1016.
- Arnar DO, Thorvaldsson S, Manolio T, et al. Familial aggregation of atrial fibrillation in Iceland. Eur Heart J. 2006;27:708–712.
- Fox CS, Parise H, D’Agostino RB, et al. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291:2851–2855.
- Mommersteeg MT, Brown NA, Prall OW, et al. PITX2c and NKX2-5 are required for the formation and identity of the pulmonary myocardium. Circ Res. 2007;101:902–909.
- Haïssaguerre M, Jaïs P, Shah DC, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–666.
- Mabuchi M, Kataoka H, Miura Y, et al. Tumor suppressor, AT motif binding factor 1 (ATBF1), translocates to the nucleus with runt domain transcription factor 3 (RUNX3) in response to TGF-beta signal transduction. Biochem Biophys Res Commun. 2010;398:321–325.
- Darbar D, Roden DM. Genetic mechanisms of atrial fibrillation: impact on response to treatment. Nat Rev Cardiol. 2013;10:317–329.
- Ellinor P, Lunetta KL, Glazer NL, et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet. 2010;42:240–244.
- Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–675.
- Holm H, Gudbjartsson DF, Arnar DO, et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117–122.
- Chambers JC, Zhao J, Terracciano CMN, et al. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42:149–152.
- Pfeufer A, van Noord C, Marciante KD, et al. Genome-wide association study of PR interval. Nat Genet. 2010;42:153–159.
- Gudbjartsson DF, Holm H, Sulem P, et al. A frameshift mutation in the sarcomere gene MYL4 causes early onset familial atrial fibrillation. Eur Heart J. 2016. [Epub ahead of print]. doi: 10.1093/eurheartj/ehw379.
- Björkegren JLM, Kovacic JC, Dudley JT, et al. Genome-wide significant loci: how important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J Am Coll Cardiol. 2015;65:830–845.
- Levy D, Ehret GB, Rice K, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–687.
- Willer CJ, Sanna S, Jackson AU, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–169.
- Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206.
- CardiogramplusC4D Consortium, Delouskas P, Kanoni S, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33.
- Samani NJ, Endman J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453.
- Helgadottir A, Gretarsdottir S, Thorleifsson G, et al. Apolipoprotein(a) genetic sequence variants associated with systemic atherosclerosis and coronary atherosclerotic burden but not with venous thromboembolism. J Am Coll Cardiol. 2012;60:722–729.
- Bjornsson E, Gudbjartsson DF, Helgadottir A, et al. Common sequence variants associated with coronary artery disease associate with the extent of coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:1526–1531.
- Nioi P, Sigurdsson A, Thorleifsson H, et al. Variant ASGR1 associated with a reduced risk of coronary artery disease. N Engl J Med. 2016;374:2131–2141.
- Helgadottir A, Gretarsdottir S, Thorleifsson G, et al. Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat Genet. 2016;48:634–639.
- Roberts R. Genetics of coronary artery disease. Circ Res. 2014;114:1890–1903.
- Collins FS, Varmus H. A new initative on precision medicine. N Eng J Med. 2015;379:793–795.
- King MC, Levy-Lahad E, Lahad A. Population-based screening for BRCA1 and BRCA2: 2014 Lasker Award. JAMA. 2014;312:1091–1092.
- Editorial. Letters from Iceland. Nat Genet. 2015;47:425.
- Wilde AA, Behr ER. Genetic testing for inherited cardiac disease. Nat Rev Cardiol. 2013;10:571–583.