
Abstract
Aims
This study aimed to assess the practicality of using a stepwise pedigree-based approach to differentiate between familial and sporadic Dilated Cardiomyopathy (DCM), while also considering timing of the genetic analysis. The analysis includes an examination of the extent to which complete family investigations were conducted in real-world scenarios as well as the length of the investigation.
Methods
The stepwise pedigree approach involved conducting a comprehensive family history spanning 3 to 4 generations, reviewing medical records of relatives, and conducting clinical screening using echocardiography and electrocardiogram on first-degree relatives. Familial DCM was diagnosed when at least 2 family members were found to have DCM, and genetic analysis was considered as an option. This study involved a manual review of all DCM investigations conducted at the Centre of Cardiovascular Genetics at Umeå University Hospital, where the stepwise pedigree approach has been employed since 2007.
Results
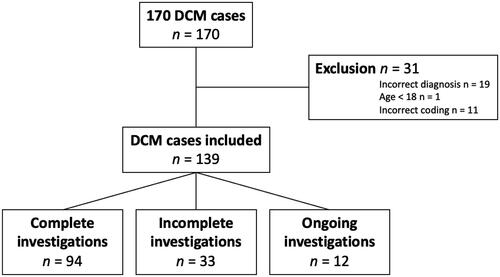
The investigation process had a mean duration of 643 days (95% CI 560.5–724.9). Of the investigations preformed, 94 (68%) were complete, 12 (9%) were ongoing, and 33 (24%) were prematurely terminated and thus incomplete. At the conclusion of the investigations, 55 cases (43%) were classified as familial DCM, 50 (39%) as sporadic DCM, and 22 (18%) remained unassessed due to incomplete pedigrees. Among the familial cases, genetic verification was achieved in 40%.
Conclusion
The stepwise pedigree approach is time consuming, and the investigations are often incomplete which may suggest that a more direct approach to genetic analysis, may be warranted.
Introduction
Dilated cardiomyopathy (DCM) is a heart muscle disease characterized by left ventricular dilatation with contractile dysfunction, in the absence of ischaemic heart disease, or pressure or volume overload [Citation1]. After ischaemic heart disease, DCM is the second most common cause of heart failure, having an estimated prevalence of >0.4% in the general population [Citation2]. Approximately, 30% of DCM cases are familial DCM, which is diagnosed when the patient of interest (proband) meets the criteria for DCM and has two or more family members diagnosed with DCM, or has a first-degree relative who experienced sudden cardiac death at <35 years of age [Citation2,Citation3].
Genetic studies have revealed that up to 40% of familial DCM cases can be genetically verified, with pronounced genetic heterogeneity, including >30 genes identified as encoding different components of the myocardium structure [Citation4–10]. The most common genetic causes of familial DCM are variants of the TTN gene, which account for 10–20% of familial DCM, and the second most common disease-associated gene is the LMNA gene, which accounts for 4–8% of familial DCM [Citation6–9,Citation11]. TTN-associated DCM usually exhibits a less severe disease form, whereas variants in the LMNA gene are associated with a more aggressive disease form with an early onset and high risk of malignant arrhythmias [Citation4–9].
It is a clinical challenge to separate sporadic and familial DCM. Previous studies have demonstrated that approximately 5% of cases can be identified as familial based on family history, whereas up to 50% of cases can be identified as familial through cardiovascular screening of family members with echocardiography [Citation12–15]. Several clinical guidelines emphasize the importance of a thorough three-generation family history, and physical examination (including echocardiography) of first-degree family members of the affected patient.
There is ongoing debate regarding the timing and frequency of genetic analyses in DCM patients, with several positional statements recommending genetic testing of any index patient with familial accumulation or a characteristic phenotype, e.g. cardiac conduction disturbances [Citation1,Citation16–18]. However, the restriction of genetic analyses to this selected group of patients has been questioned, since some minor studies have demonstrated similar frequencies of disease-associated variants in familial and non-familial cases, and due to the recognition of de novo variants [Citation6,Citation19,Citation20]. Additionally, patients often lack knowledge about family history, and it can be difficult to perform a complete family screening. Moreover, the investigations are time-consuming and place great demands on the index patient who must act as the contact person between the genetic centre and the family.
Recently published ESC guidelines for the management of cardiomyopathies include a class I recommendation for a thorough 3- to 4-generation family history, including clinical screening (with imaging and ECG) for all first-degree relatives, which is in agreement with previously published position statements and consensus documents [Citation1,Citation16–18]. These guidelines also include a class I recommendation for genetic testing of patients fulfilling the diagnostic criteria for cardiomyopathy, when such testing will enable diagnosis, prognostication, therapeutic stratification, reproductive management, and/or cascade genetic testing [Citation1]. This is a new recommendation of genetic analysis, which makes no clear distinction between familial and sporadic cardiomyopathies, and the guidelines highlight the difficulties in distinguishing sporadic from familial cases. Notably, the issue genetic analysis timing is not addressed. The ESC guideline recommendations regarding genetic analysis are in line with the EHRA, HRS, APHRS, LAHRS consensus statement, which also recommends the genetic analysis of all probands regardless of family history [Citation21].
The Umeå University hospital Centre for cardiovascular Genetics (CCG) is responsible for family investigations of DCM in Northern Sweden and has applied a stepwise pedigree approach. The approach to perform genetic analysis only when the disease is classified as familial was based on previously published guidelines and position statements [Citation1,Citation16–18,Citation22]. The CCG unit comprises cardiologists, genetic counsellors, and clinical geneticists, who work together as a cardiomyopathy team. In Sweden, genetic testing for DCM is fully financed by the public healthcare system.
In the present study, we retrospectively analysed the work of the CCG. Our aims were to examine the applicability of the stepwise pedigree approach, the proportion of investigations that have been completed, and the percentage of investigations that lead to a genetic diagnosis.
Materials and methods
Upon referral to Umeå University hospital CCG, each patient is contacted by a genetic counsellor, and asked to complete a family history form, and to get consent forms signed to enable retrieval and review of relatives’ medical records. After their medical records are reviewed, first-degree relatives are offered cardiac screening with echocardiography and ECG. Based on these results, the disease is classified as either sporadic or familial. Patients considered to have familial disease are offered genetic analysis after completing genetic counselling. In a minority of cases—when there is suspicion of a more aggressive disease, such as LMNA-associated DCM, usually in the presence of atrioventricular (AV) block or in cases with a history of ventricular arrythmias—upfront genetic analysis was performed.
From 2007 to 2022, a total of 170 family investigations regarding DCM have been performed at the CCG unit. We performed a thorough manual journal review, and constructed a dataset containing information regarding patients’ age, sex, phenotype, duration of the investigation, and investigational findings. We excluded 31 cases from this analysis (19 due to incorrect diagnosis, 1 who was <18 years of age, and 11 due to incorrect coding).
When the index patient—despite repeated reminders by the CCG coordinator—did not return the family form and/or consent forms, the investigation was terminated and classified as incomplete. Regarding ECG abnormalities, the presence or absence of AV-block was noted. Regarding symptoms, patients with both dyspnoea and reduced physical energy were classified as dyspnoeic. If a history of ventricular arrythmias was reported at referral, the CCG formulated a second opinion regarding to whether these arrythmias were documented and correctly assessed. If so, the cardiologist at the CCG made the decision of whether an upfront genetic analysis should be performed.
Statistics
Statistical analyses were performed using the IBM Statistical Package for the Social Sciences (SPSS) version 28. A p value <0.05 was considered as the level of significance. Differences between groups were analysed using the chi2 test and Student’s t-test when appropriate. Normal distribution was assumed based on cohort size and the central limit theorem.
Ethics
This study was conducted in compliance with the Declaration of Helsinki and was approved by the Swedish Ethical Review Authority (Dnr: 2023-03307-01).
Results
Our analysis included a total of 139 cases. Among these patients, 49 (35%) were women, and the mean age at referral was 53.4 years (95% CI 51.1–55.7 years). The majority of patients had a left ventricular ejection fraction of ≤35% (), and the mean left ventricular end-diastolic diameter was 66 mm (95% CI 63.9–67.2 mm) at the time of diagnosis.
Table 1. Symptoms and basic phenotypic features of the index patients.
The mean duration of investigation was 643 days (95% CI 560.5–724.9). illustrates the distribution of the duration time. Among the analysed investigations, 94 (68%) were complete investigations, 12 (9%) were still ongoing, and 33 (24%) were prematurely terminated and therefore incomplete (). The ongoing investigations were not excluded from the analysis of duration time.
Figure 1. Scatterplot of the distribution of investigation duration in days.
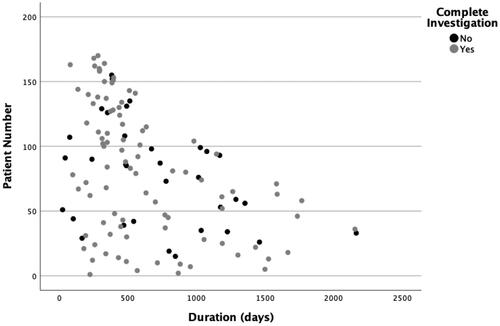
Figure 2. Flow-chart of DCM patients.
At the end of the investigation, 55 cases (43%) were classified as familial DCM, 50 (39%) as sporadic DCM, and 22 (18%) were not assessed due to incomplete pedigrees. Additionally, 12 ongoing investigations had not yet been assessed as familial or sporadic. Among the familial cases, 38% were assessed based on family screening. In the remaining 62%, review of the relative’s medical records and clinical screening of first-degree relatives did not contribute any additional information, except to confirm the family history ().
Table 2. Final assessment of investigated cases.
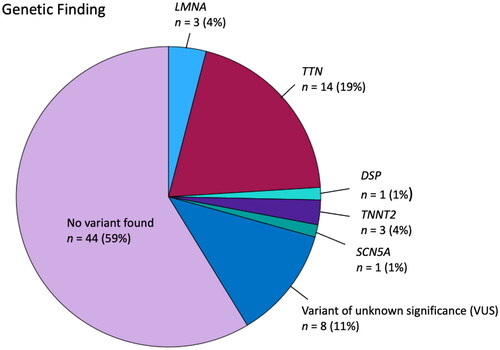
A total of 75 genetic analyses were performed, of which 23 (31%) revealed a genetic variant classified as pathogenic or likely pathogenic. Among these identified genetic variants, 19% were in the TTN gene, 4% in the LMNA gene, and 4% in the TNNT2 gene (). A complete list of the pathogenic or likely pathogenic variants are found in the Supplementary Material.
Figure 3. Pie chart of the genetic findings.
Among the 23 patients with a history of ventricular arrythmias, 10 cases were considered to be familial. Genetic analyses were performed in 14 of these cases, of which 4 revealed a pathogenic or likely pathogenic variant.
Compared to male patients, female patients were younger (52.1 vs. 54.1 years, p < 0.001). Classification as familial was based on anamnesis in a higher proportion of female patients compared to male patients (71 vs. 55%, p = 0.022). Additionally, the proportion of completed investigations was higher among female patients than male patients (80 vs. 70%, p = 0.009). Male and female patients did not significantly differ regarding symptoms, ECG, and echocardiographic findings.
A total of 55 patients were considered to have familial DCM, of which 40% of cases were genetically verified. The genetically verified patients were somewhat younger than the non-genetically verified patients (54 vs. 58 years; p = 0.034). These two groups did not significantly differ regarding symptoms, ECG, or echocardiographic findings.
Discussion
Our present analysis demonstrated that the stepwise pedigree approach—including a review of medical records, obtaining a 3-generation family history, screening first-degree relatives, and performing genetic analysis—is a time-consuming approach, with a mean duration of almost 2 years and a high proportion of incomplete investigations. The long duration likely reflects the complexity of this stepwise pedigree approach. Gathering medical records from multiple hospitals in different regions is a time-consuming task, as is the screening of family member. The referral of a DCM patient’s relative for a screening echocardiography is usually a screening of an asymptomatic person, which is often a low-priority investigation in actual clinical practice. Furthermore, the genetic analysis itself is a time-consuming procedure that usually takes several months.
The high rate of incomplete investigations highlights a gap in knowledge relating to the experience and burden of the investigation. We suspect that the investigations might be too demanding and time-consuming for the index patient, who must function as the link between their relatives and the healthcare system.
Compared to male patients, the female patients were younger and fewer in number. This may be an indirect sign that physicians are more prone to refer men to CCG, with women mainly being referred when they are younger and there is a higher suspicion of familial or more aggressive disease. We also noticed that women were more commonly assessed as having familial DCM based on only family history, which could indicate that women are more informed about their relatives and their medical history.
Surprisingly, we found that a high degree of cases could be classified as “familial disease” based on family history alone, which is in contrast to previously published data [Citation12]. The proportion and the distribution of the genetic findings in this cohort were consistent with previously published data [Citation7,Citation23,Citation24]. This was the first study of genetic findings in cases of DCM in northern Sweden. Since CCG is responsible for all family investigations in this area—including performing and interpreting the genetic analyses—we were able to analyse the complete investigations, from referral to final assessment.
One limitation of this study is the relatively small cohort size compared to previously published data. When the unit was founded, only a few cases per year were referred to CCG. The number of referred cases has substantially increased over the following years. Notably, we analysed investigations performed over a lengthy time period, and the analysed gene panels have changed during these years. We have not addressed these changes in this project. Furthermore, CCG attendance is dependent on referrals, and different hospitals may have different routines for referrals. Since there is likely a significant selection bias at the level of referral, this study cannot analyse the proportions of familial compared to sporadic DCM.
The findings of this investigation highlight the downside of the pedigree approach, notably including the high proportion of incomplete investigations. This is problematic because it can lead to poor identification of people at risk or in the early stages of serious heart disease. Additionally, the duration of almost 2 years to complete a full family investigation is probably not a pragmatic approach in a healthcare system with limited resources.
It may seem appealing to use an alternative approach, with genetic analysis of all DCM patients, in parallel with family history and screening of first-degree relatives. This alternative approach would be consistent with the guidelines, as they do not mark a clear difference in the recommendations for genetic investigation between sporadic and familial forms of DCM. Further studies are needed to evaluate whether such a strategy would enable shorter investigation time, a higher proportion of complete investigations, and reasonable genetic yield. Notably, since inherited cardiomyopathies are characterized by variable penetrance, even a complete pedigree would not reveal all cases of familial disease, which strengthens the argument in favour of upfront genetic analysis, regardless of family history and screening results.
In conclusion, this study demonstrated that it is feasible to follow the ESC guidelines regarding DCM diagnostics and family screening, even at a relatively small referral centre. The proportion of positive genetic findings was comparable to that found in larger centres. However, the stepwise pedigree approach is time-consuming, and the investigations are often incomplete, which may suggest the utility of a more direct approach to genetic analysis.
Revised manuscript Clean Copy.docx
Download MS Word (88.3 KB)supplemantary file 1.docx
Download MS Word (15.4 KB)Acknowledgements
We thank Karin Karlsson and Stellan Mörner at the Center for Cardiovascular Genetics at Umea University hospital for their support and wise comments during the study. The authors also acknowledge Clinical Genomics Umea for scientific guidance regarding the genetic analyses performed.
Disclosure statement
TA received speaker’s honoraria from Actelion Ltd, Vifor Pharma, Astra Zeneca, Pharmacosmos, and Boehringer Ingelheim. No potential conflict of interest was reported by the author(s)
Additional information
Funding
References
- Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44(37):3503–3626. doi: 10.1093/eurheartj/ehad194.
- Rosenbaum AN, Agre KE, Pereira NL. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat Rev Cardiol. 2020;17(5):286–297. doi: 10.1038/s41569-019-0284-0.
- Sweet M, Taylor MR, Mestroni L. Diagnosis, prevalence, and screening of familial dilated cardiomyopathy. Expert Opin Orphan Drugs. 2015;3(8):869–876. doi: 10.1517/21678707.2015.1057498.
- Tayal U, Ware JS, Lakdawala NK, et al. Understanding the genetics of adult-onset dilated cardiomyopathy: what a clinician needs to know. Eur Heart J. 2021;42(24):2384–2396. doi: 10.1093/eurheartj/ehab286.
- Merlo M, Cannatà A, Gobbo M, et al. Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail. 2018;20(2):228–239. doi: 10.1002/ejhf.1103.
- Morales A, Hershberger RE. The rationale and timing of molecular genetic testing for dilated cardiomyopathy. Can J Cardiol. 2015;31(11):1309–1312. doi: 10.1016/j.cjca.2015.06.034.
- Pugh TJ, Kelly MA, Gowrisankar S, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16(8):601–608. doi: 10.1038/gim.2013.204.
- Peters S, Johnson R, Birch S, et al. Familial dilated cardiomyopathy. Heart Lung Circ. 2020;29(4):566–574. doi: 10.1016/j.hlc.2019.11.018.
- Wilsbacher LD. Clinical implications of the genetic architecture of dilated cardiomyopathy. Curr Cardiol Rep. 2020;22(12):170. doi: 10.1007/s11886-020-01423-w.
- Stava TT, Leren TP, Bogsrud MP. Molecular genetics in 4408 cardiomyopathy probands and 3008 relatives in Norway: 17 years of genetic testing in a national laboratory. Eur J Prev Cardiol. 2022;29(13):1789–1799. doi: 10.1093/eurjpc/zwac102.
- Goidescu CM. Dilated cardiomyopathy produced by lamin A/C gene mutations. Clujul Med. 2013;86(4):309–312.
- Michels VV, Moll PP, Miller FA, et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326(2):77–82. doi: 10.1056/NEJM199201093260201.
- Grünig E, Tasman JA, Kücherer H, et al. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol. 1998;31(1):186–194. doi: 10.1016/s0735-1097(97)00434-8.
- Baig MK, Goldman JH, Caforio AL, et al. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol. 1998;31(1):195–201. doi: 10.1016/s0735-1097(97)00433-6.
- McKenna CJ, Codd MB, McCann HA, et al. Idiopathic dilated cardiomyopathy: familial prevalence and HLA distribution. Heart. 1997;77(6):549–552. doi: 10.1136/hrt.77.6.549.
- Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8(8):1308–1339. doi: 10.1016/j.hrthm.2011.05.020.
- Charron P, Arad M, Arbustini E, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010;31(22):2715–2726. doi: 10.1093/eurheartj/ehq271.
- Musunuru K, Hershberger RE, Day SM, et al. Genetic testing for inherited cardiovascular diseases: a scientific statement From the American Heart Association. Circ Genom Precis Med. 2020;13(4):e000067. 20200723. doi: 10.1161/hcg.0000000000000067.
- Hershberger RE, Parks SB, Kushner JD, et al. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1(1):21–26. doi: 10.1111/j.1752-8062.2008.00017.x.
- Hershberger RE, Norton N, Morales A, et al. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3(2):155–161. doi: 10.1161/CIRCGENETICS.109.912345.
- Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases. Heart Rhythm. 2022;19(7):e1–e60. doi: 10.1016/j.hrthm.2022.03.1225.
- Hershberger RE, Lindenfeld J, Mestroni L, et al. Genetic evaluation of cardiomyopathy–a Heart Failure Society of America practice guideline. J Card Fail. 2009;15(2):83–97. doi: 10.1016/j.cardfail.2009.01.006.
- Parks SB, Kushner JD, Nauman D, et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008;156(1):161–169. doi: 10.1016/j.ahj.2008.01.026.
- Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619–628. doi: 10.1056/NEJMoa1110186.