
Abstract
Objective: Evaluate the safety and efficacy of golimumab through week 120 in Japanese patients with active rheumatoid arthritis (RA) previously treated with DMARDs.
Methods: Japanese patients with active RA despite prior DMARDs were randomized to placebo (Group 1, n = 105), golimumab 50 mg (Group 2, n = 101), or golimumab 100 mg (Group 3, n = 102). At week 16, Group 1 patients crossed over to golimumab 50mg; after week 52, a one-time golimumab dose reduction from 100 to 50 mg was permitted. Assessments included ACR20/50/70 responses and good/moderate DAS28-ESR responses. Radiographic progression was assessed with the van der Heijde-modified Sharp (vdH-S) score. Safety and efficacy were assessed through week 120.
Results: ACR20 response rates at week 52 in Group 1, Group 2, and Group 3 were 70.6%, 71.4%, and 81.9%, respectively, and maintained through week 104 (87.2%, 85.1%, 88.9%, respectively) and week 120 (86.1%, 87.0%, 89.5%, respectively). Similar trends were observed for ACR50, ACR 70, and DAS28-ESR. Median change in total vdH-S at weeks 52, 104, and 120 ranged from 0.0 to 1.5 across treatment groups. Through week 120, 93.8%/97.1% had an AE with golimumab 50 mg/100 mg, respectively, and 19.7%/11.8% had an SAE. Infections were the most common AE.
Conclusion: Clinical response to golimumab 50 mg and 100 mg was maintained over 2 years in Japanese patients with active RA despite prior DMARDs.
Introduction
Rheumatoid arthritis (RA) is estimated to affect up to 1.0% of the Japanese population [Citation1]. The joint destruction that is characteristic of RA can lead to a substantial loss of physical function [Citation2]. Treatment recommendations generally support the goal of achieving low disease activity or remission [Citation3], which may prevent long-term radiographic damage and disability [Citation4–6].
Golimumab is a human monoclonal anti-tumor necrosis factor α (TNFα), and is approved in Japan for use as monotherapy or in combination with methotrexate (MTX) for adults with moderate to severe RA. The safety and efficacy of subcutaneous (SC) golimumab with and without MTX were evaluated in three large global trials of patients with RA who were MTX-naïve (GO-BEFORE) [Citation7], MTX-inadequate responders (GO-FORWARD) [Citation8], or anti-TNF-experienced (GO-AFTER)[Citation9] as well as in two Japanese studies in MTX-inadequate responder patients with MTX (GO-FORTH) [Citation10] and in disease-modifying antirheumatic drug (DMARD)-inadequate responders (GO-MONO) [Citation11]. In the GO-MONO trial, significantly greater proportions of patients receiving golimumab monotherapy had improvements in the signs and symptoms of RA at week 14 compared with those receiving placebo [Citation11]. Long-term control of disease activity with golimumab monotherapy is clinically relevant, especially for patients who are intolerant to MTX. Here we report the final safety and efficacy results from the GO-MONO trial through week 120.
Patients and methods
Patients
Patient eligibility criteria have been previously described in detail [Citation11]. Briefly, Japanese patients aged 20–75 years with active RA despite prior DMARD therapy were eligible. Active RA was defined as at least six swollen joints (0–66) and at least six tender joints (0–68) and at least two of the following: (1) C-reactive protein (CRP) ≥ 2.0 mg/dL or erythrocyte sedimentation rate (ESR) by Westergren method ≥28 mm/h, (2) morning stiffness ≥30 minutes, (3) investigator-documented radiographic evidence of bone erosion, or (4) positive test for anti-cyclic citrullinated peptide antibody or rheumatoid factor.
Study design
GO-MONO was a Phase 2/3, multicenter, randomized, placebo-controlled trial [Citation11]. Patients were randomly assigned (1:1:1) to receive SC injections of placebo (Group 1), golimumab 50 mg (Group 2), or golimumab 100 mg (Group 3) at baseline and every 4 weeks thereafter. At week 16, patients in Group 1 crossed over to golimumab 50 mg. After week 52, patients in Group 3 could have their dose reduced to 50 mg at the investigator’s discretion. The final golimumab administration was at week 116. All DMARD therapy was discontinued at least 4 weeks before the first study agent administration. After week 24, concomitant DMARD use, including MTX, was permitted at the investigator’s discretion for patients who did not achieve ≥20% improvement in the American College of Rheumatology criteria (ACR20) [Citation12]. Concomitant use of stable doses of oral corticosteroids (≤10 mg of prednisolone/day) was permitted, and changes in the dose and/or discontinuation were allowed after week 16. Concomitant use of MTX and prednisolone (time to initiation and dose) was examined in a post-hoc analysis. Additionally, baseline characteristics were compared between patients who did and did not initiate concomitant MTX therapy during the trial.
The trial was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the institutional review board at each site. All the patients were required to give written informed consent before any study-related procedures.
Assessments
The primary endpoint was the proportion of patients with an ACR20 response [Citation12] at week 14. Efficacy was assessed using the ACR criteria, the ACR Index of Improvement (ACR-N), and the 28-joint count Disease Activity Score using ESR (DAS28-ESR). The proportion of patients with ACR20, ACR50, and ACR70 responses, a DAS28-ESR good or moderate response [Citation13], and DAS28-ESR remission (<2.6) were determined. The Health Assessment Questionnaire-Disability Index (HAQ-DI) [Citation14] was used to assess improvements in physical function, with a minimal clinically important difference defined as an improvement greater than 0.25 units. In a post hoc analysis, remission rates were determined through week 120 using the clinical disease activity index (CDAI, ≤ 2.8) [Citation15], the simplified disease activity index (SDAI, ≤ 3.3) [Citation16], and the Boolean criteria [Citation16]. Efficacy assessments were collected through week 120.
In the current analysis, radiographs of the hands and feet were collected at weeks 0, 52, 104, and 120 and were scored using the van der Heijde modification of the Sharp (vdH-S) methodology [Citation17]. Readers of the radiographs were blinded to patient identity, treatment group, and time point at which the radiographs were obtained.
Safety assessments were performed through week 120 and included adverse event (AE) reporting and laboratory tests. Serum samples were collected through week 120 for evaluation of the presence of antibodies to golimumab [Citation18]. On days when study agent was administered, serum samples were collected before the administration of golimumab.
Statistical analysis
Clinical efficacy and radiographic results from week 52 to week 120 were summarized using descriptive statistics according to the randomized treatment group. Observed data were reported with no imputation for missing data. Only patients who had at least one radiograph after week 52 were included in the radiographic analyses. No statistical comparisons were performed among the treatment groups after week 16 when all the patients were receiving golimumab.
Adverse events were reported using descriptive statistics for all randomized and treated patients grouped according to actual treatment received. Because of changes in treatment that were permitted under the protocol (i.e. placebo crossover and golimumab dose reduction in Group 3), some patients may have been included in more than one group.
Results
Baseline characteristics and patient disposition
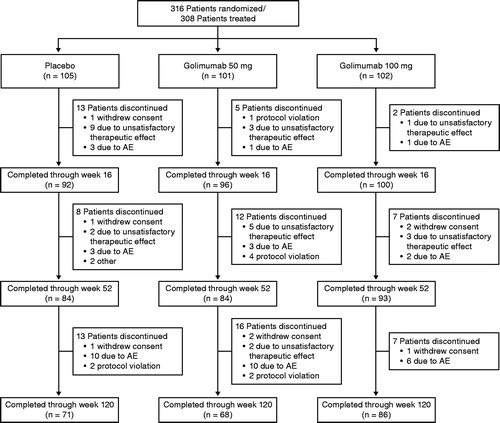
Baseline demographics and disease characteristics were well balanced among the treatment groups [Citation11]. A total of 316 patients were randomized; eight withdrew consent before any study treatment was administered. Therefore, 308 patients (Group 1, n = 105; Group 2, n = 101; Group 3, n = 102) were treated and received at least one dose of study agent. Patient disposition through week 24 has been previously reported in detail [Citation11]. A total of 225 patients completed assessments through week 120 (Group 1, n = 71, 67.6%; Group 2, n = 68, 67.3%; Group 3, n = 86, 84.3%). Eighty-three patients (Group 1, n = 34; Group 2, n = 33; Group 3, n = 16) discontinued golimumab therapy through week 120, with the most common reasons being AEs (n = 39 [12.7%]) and unsatisfactory therapeutic effect (n = 25 [8.1%]) (). Seventeen patients in Group 3 had a golimumab dose reduction from 100 mg to 50 mg after week 52 at the investigator’s discretion per protocol.
Figure 1. Patient disposition through week 120. AE: adverse event.
Efficacy
At week 24, significantly greater proportions of patients in Group 2 and Group 3 achieved an ACR20 response when compared with Group 1 (46.5% and 69.6% vs. 17.1%; p < .0001 for both) [Citation11]. The proportions of patients with an ACR50 and ACR70 response at week 24 were also significantly greater in Group 2 and Group 3 than in Group 1 [Citation11]. Following crossover from placebo to golimumab at week 16, the proportions of patients in Group 1 with an ACR20, ACR50, and ACR70 response increased over time and were similar to those in Group 2 by week 20 (). At week 52, 70.6% of patients in Group 1, 71.4% of patients in Group 2, and 81.9% of patients in Group 3 had an ACR20 response. These proportions were maintained or improved over time through week 120, when more than 85% of patients in each treatment group had an ACR20 response (). Similar trends were observed for ACR50 and ACR70 responses at week 120, with 69.4% to 74.4% of patients across the treatment groups having an ACR50 response and 44.2% to 56.5% having an ACR70 response ().
Figure 2. The proportions of patients with an ACR20 (a), ACR50 (b), and ACR70 (c) response through week 120. All placebo patients crossed over to golimumab at week 16 (dashed line). ACR20/50/70, ≥ 20%/50%/70% improvement in American College of Rheumatology criteria.
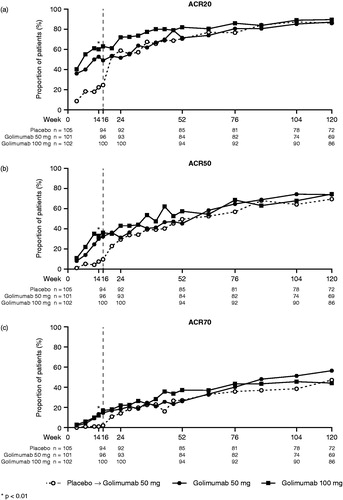
Table 1. Clinical efficacy results at weeks 52, 104, and 120.
Mean ACR-N scores were numerically lower in Group 1 (44.9) and Group 2 (44.1) compared with Group 3 (52.2) at week 52 (). Mean scores increased, and were similar across the treatment groups, at week 104 and week 120. The proportion of patients with a DAS28-ESR good response ranged from 36.9% to 44.7% across the treatment groups at week 52, with numerically lower proportions in Group 1 and Group 2 compared with Group 3 (); these rates continued to increase through week 120. A similar trend was observed with the proportion of patients achieving DAS28-ESR remission.
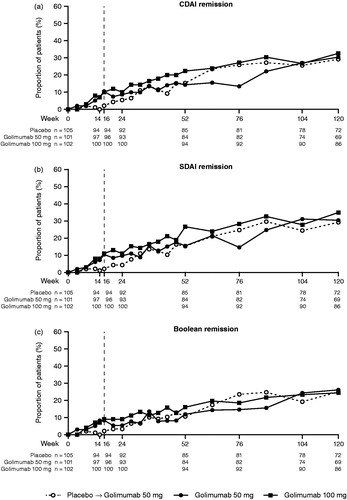
The proportions of patients with CDAI, SDAI, and Boolean remission were numerically greater in Groups 2 and 3 than in Group 1 through the placebo-controlled portion of the trial (). After week 24, the proportion of patients in Group 1 achieving remission began to increase and approach those in Groups 2 and 3.
Figure 3. The proportions of patients achieving remission by CDAI (a), SDAI (b), and Boolean (c) criteria through week 120. All placebo patients crossed over to golimumab at week 16 (dashed line). CDAI: clinical disease activity index; SDAI: simplified disease activity index.
At week 24, patients randomized to Groups 2 and 3 had greater mean improvements from baseline in HAQ-DI score when compared with those in Group 1 [Citation11]. Physical function continued to improve in all three groups after week 16 when all patients were receiving golimumab, and these improvements were maintained through weeks 52, 104, and 120. More than 60% of the patients in each treatment group achieved an improvement in HAQ-DI ≥ 0.25 at week 52, and this proportion was maintained or increased through week 120 ().
Mean changes from baseline in total vdH-S score were generally numerically lower in Group 3 than in Group 2 or Group 1 at weeks 52, 104, and 120 (); although no statistical comparisons were conducted after week 24. The median change from baseline at weeks 52, 104, and 120 ranged from 0.0 to 1.5 across the treatment groups; 30.6% to 44.9% of patients had a change in total vdH-S score ≤0 at week 104, and these proportions were maintained at week 120, suggesting that many patients experienced minimal radiographic progression. The proportions of patients in each treatment group who had a change from baseline in total vdH-S score greater than the smallest detectable change (SDC) were similar among the treatment groups at week 52 (range: 17.4–20.5%) and remained steady at weeks 104 and 120. Changes in total vdH-S scores at week 120 were comparable among the treatment groups (). The median changes from baseline in total vdH-S score at week 52 and week 120 in patients who initiated MTX were 4.0 and 3.0 in Group 2 and 2.0 and 3.0 in Group 3, respectively; the median changes at week 52 and week 120 for patients who did not initiate MTX during the study were 0.0 and 0.5 in Group 2 and −0.5 and 0.0 in Group 3, respectively. Among patients who initiated MTX during the study, numerically greater proportions of patients had a change in total vdH-S score > SDC at week 120 compared with those who had a change in total vdH-S score ≤ SDC in Group 1 (62.5% [n = 10/16] vs. 36.4% [n = 20/55]), Group 2 (66.7% [n = 10/15] vs. 35.8% [n = 19/53]), and Group 3 (47.4% [n = 9/19 vs. 36.4% [n = 24/66]).
Figure 4. Change in Total vdH-S score in each treatment group at week 120 for all patients (a), patients who initiated methotrexate (b), and patients who remained on monotherapy (c). vdH-S, van der Heijde modification of the Sharp score.
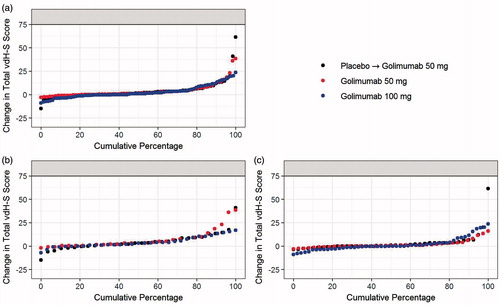
Table 2. Radiographic results through week 120.
Changes in concomitant medications
Changes in concomitant DMARD therapies were permitted after week 24, and approximately 40% of all the patients (Group1, n = 41 [39.0%]; Group 2, n = 42 [41.6%]; Group 3, n = 37 [36.3%]) initiated MTX therapy during the study (); mean ± SD initial doses of MTX were 6.0 ± 1.6 mg/week, 5.4 ± 1.3 mg/week, and 5.6 ± 1.2 mg/week, respectively. The proportions of patients who initiated MTX therapy between week 24 and week 52 were 20.0% (n = 21) in Group 1, 29.7% (n = 30) in Group 2, and 18.6% (n = 19) in Group 3; overall, the proportions of patients initiating MTX in Groups 1 and 2 gradually decreased over time (Supplemental Figure 1). Patients who initiated MTX during the trial generally had shorter disease duration, but higher DAS-ESR, DAS-CRP, HAQ-DI, and patient’s pain and global assessment scores, and higher ESR and CRP levels at baseline when compared with patients who did not receive concomitant MTX (Supplemental Table 1).
Table 3. Proportion of patients receiving concomitant MTX and/or prednisolone through week 120.
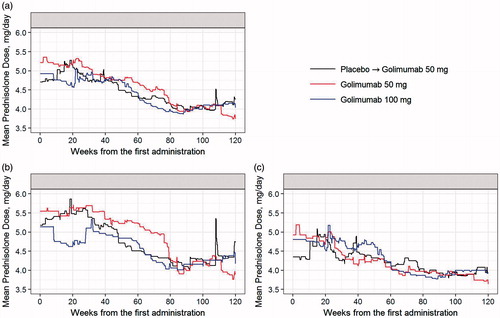
Before week 16, 206 patients used concomitant prednisolone (Group 1, n = 68; Group 2, n = 70; Group 3, n = 68), with a mean daily dose of 4.82 mg in Group 1, 5.17 mg in Group 2, and 4.87 mg in Group 3. After week 16, 225 patients used concomitant prednisolone (Group 1, n = 77; Group 2, n = 76; Group 3, n = 72), with a mean dose of 4.57 mg in Group 1, 4.74 mg in Group 2, and 4.22 mg in Group 3. In Groups 1 and 2, numerically greater proportions of patients received concomitant prednisolone together with MTX through the study when compared with the proportions of patients receiving prednisolone without MTX (). The trends of mean daily prednisolone dose were similar between all the groups and gradually decreased over the course of the study. The mean prednisolone dose at baseline for patients who initiated MTX therapy was more than 5.0 mg/day, and that of the patients without MTX was less than 5.0 mg/day (). The mean prednisolone dose used by patients who did not receive concomitant MTX remained low throughout the study ().
Figure 5. Mean prednisolone dose per day through week 120 for all the patients (a), patients who initiated methotrexate (b), and patients who remained on monotherapy (c). First administration refers to week 0 administration of placebo or golimumab.
Safety
AEs among patients receiving placebo (up to week 16) have been previously reported in detail [Citation11]. Through week 120, 193 patients received at least one administration of golimumab 50 mg and 102 patients received at least one administration of golimumab 100 mg; among these, 181 (93.8%) and 99 (97.1%), respectively, had at least one AE (). The most common AE was nasopharyngitis, with no apparent difference between the two golimumab doses.
Table 4. Adverse events through week 120.
Through week 120, 19.7% of patients receiving golimumab 50 mg and 11.8% of patients receiving golimumab 100 mg had at least one serious AE (SAE) (). Infections were the most common type of SAE: cellulitis (50 mg, n = 2; 100 mg, n = 2), herpes zoster (50 mg, n = 3), gastroenteritis (50 mg, n = 1; 100 mg, n = 1), pyelonephritis (50 mg, n = 2), pneumonia (50 mg, n = 1; 100 mg, n = 1), and dental pulpitis and intervertebral discitis each occurring in one patient receiving golimumab 50 mg, and sepsis, urosepsis, liver abscess, and viral enterocolitis each occurring in one patient receiving golimumab 100 mg. There were no cases of active tuberculosis. Five malignancies were reported: three among patients receiving the 50-mg dose (ovarian neoplasm, n = 2 and colon cancer, n = 1) and two among patients receiving the 100-mg dose (breast cancer, n = 1; pancreatic carcinoma, n = 1). Two deaths occurred during the trial (brain stem hemorrhage, 50 mg; pancreatic carcinoma, 100 mg).
Through week 120, a total of 42 patients had an injection site reaction (50 mg, n = 24 [12.4%]; 100 mg, n = 18 [17.6%]). All injection site reactions were considered to be mild, and none resulted in discontinuation of the study treatment. No anaphylactic reactions or serum sickness-like reactions occurred throughout the trial.
Immunogenicity
Through week 120, 295 patients (Group 1, n = 92; Group 2, n = 101; Group 3, n = 102) received at least one golimumab administration and had evaluable samples for the analysis of antibodies to golimumab. Fifteen patients (Group 1, n = 3; Group 2, n = 7; Group 3, n = 5) tested positive for antibodies to golimumab.
Discussion
In the GO-MONO trial of Japanese patients with active RA, patients who received SC golimumab monotherapy 50 mg or 100 mg from baseline (Groups 2 and 3) had significantly greater mean improvements in efficacy measures at week 14 compared with placebo [Citation11]. Patients who were randomized to receive placebo at baseline (Group 1) demonstrated improvements in clinical efficacy at week 20, with ACR 20/50/70 response rates approaching those in Groups 2 and 3 by week 24 [Citation11]. In all three groups, clinical response continued to improve from week 24 to week 52 and was maintained through week 120, with no apparent differences between the golimumab dose groups (50 mg and 100 mg).
Radiographic progression was minimal through week 120 as the majority of the patients did not have radiographic progression greater than the SDC. Median changes in total vdH-S score ranged from 1.0 to 1.5 across the three treatment groups. The median change in total vdH-S score was numerically higher in Group 1 than in Groups 2 and 3, which is consistent with the evidence that joint damage early in the RA disease process may not be reversible [Citation6,Citation19]. The change in total vdH-S score at week 120 was maintained for patients who initiated concomitant MTX during the trial and those who did not. Radiographic progression in the GO-MONO trial appeared to be slightly higher than that observed in the GO-FORTH trial (patients receiving golimumab 50 mg or 100 mg plus MTX). This may indicate that golimumab monotherapy may not be adequate to prevent radiographic progression in some patients; however, the comparison between the two trials may be scientifically inadequate given the different patient populations and study designs. In a post-hoc analysis of the GO-FORTH trial, among patients with a high level of disease activity at baseline, those who received golimumab 100 mg plus MTX had less radiographic progression than did those who received golimumab 50 mg plus MTX [Citation20].
It should be noted that approximately 40% of the patients initiated MTX therapy through week 120. Patients who initiated MTX during the trial tended to have more active disease at baseline compared with patients who remained on golimumab monotherapy. Although a majority of patients did not initiate concomitant MTX, golimumab monotherapy may not have provided sufficient control of radiographic progression for some patients. The number of patients receiving concomitant prednisolone increased from 206 before week 16 to 225 who initiated prednisolone after week 16; however, the mean prednisolone dose decreased over time in all three treatment groups. It should be noted that the use of MTX and prednisolone was at the discretion of the investigator; no data were collected regarding the reasons for initiation or discontinuation (other than AEs) of concomitant therapies.
The incidence and type of AEs observed in GO-MONO were consistent with previous phase 3 trials of SC golimumab in patients with rheumatic diseases [Citation21–25], including the GO-FORTH trial of Japanese patients with RA who received golimumab plus MTX [Citation26]. No apparent dose effect was observed. Infections were the most common type of AE. Of note, there were no cases of active tuberculosis. Through week 120, five malignancies were reported, and two deaths occurred.
The clinical efficacy results in GO-MONO are limited by the use of observed data after week 24. Because there was no imputation for missing data, clinical efficacy response rates through week 120 may have been overestimated. However, golimumab monotherapy 50 mg or 100 mg was efficacious overall through 2 years of therapy in this population of Japanese patients with active RA despite prior DMARD therapy. Some patients with more severe disease may benefit more from combination therapy with MTX.
Conflict of interest
TT has received research grants from Abbott, Astra Zeneca, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharma Corporation, Novartis, Takeda Pharmaceutical, and Wyeth.
MH has received research grants from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharma Corporation, Takeda Pharmaceutical, and Wyeth and received consultant fees from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Janssen Pharmaceutical, and Mitsubishi Tanabe Pharma Corporation.
YT has received research grants from Abbott, Astellas Pharmaceutical, Banyu Pharmaceutical, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharma Corporation, Pfizer, and Takeda Pharmaceutical.
HY has received research grants from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharmaceutical, Otsuka Pharmaceutical, Roche, Takeda Pharmaceutical, and Wyeth.
NI has received research grants from Astellas Pharmaceutical, Chugai Pharmaceutical, Eizai Pharmaceutical, and Mitsubishi Tanabe Pharma Corporation.
KY has received research grants from Astellas Pharmaceutical, Chugai Pharmaceutical, Eizai Pharmaceutical, Immunofuture Inc, Mitsubishi Tanabe Pharma Corporation, Santen Pharmaceutical, and Wyeth.
NM has no conflicts of interest.
TK has received research grants from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharma Corporation, Otsuka Pharmaceutical, Pfizer, Takeda Pharmaceutical, and Wyeth.
YU and YI are employees of Janssen Pharmaceutical K.K.
TY is an employee of Mitsubishi Tanabe Pharma Corporation.
DB is an employee of Janssen Research & Development, LLC.
Supplementary Table 1
Download MS Word (27.5 KB)Acknowledgements
The authors thank Miyo Ota, PhD, of Janssen Pharmaceutical K.K. for statistical support and Rebecca Clemente, PhD, of Janssen Scientific Affairs, LLC, for writing support.
Additional information
Funding
Related Research Data
References
- Yamanaka H, Sugiyama N, Inoue E, Taniguchi A, Momohara S. Estimates of the prevalence of and current treatment practices for rheumatoid arthritis in Japan using reimbursement data from health insurance societies and the IORRA cohort (I). Mod Rheumatol. 2014;24:33–40.
- Welsing PM, van Gestel AM, Swinkels HL, Kiemeney LALM, van Riel PL. The relationship between disease activity, joint destruction, and functional capacity over the course of rheumatoid arthritis. Arthritis Rheum. 2001;44:2009–17.
- Smolen JS, Landewe R, Breedveld FC, Buch M, Burmester G, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73:492–509.
- Baumgartner SW, Fleischmann RM, Moreland LW, Schiff MH, Markenson J, Whitmore JB. Etanercept (Enbrel) in patients with rheumatoid arthritis with recent onset versus established disease: improvement in disability. J Rheumatol. 2004;31:1532–7.
- Lillegraven S, van der Heijde D, Uhlig T, Kvien TK, Haavardsholm EA. What is the clinical relevance of erosions and joint space narrowing in RA? Nat Rev Rheumatol. 2012;8:117–20.
- Quinn MA, Emery P. Window of opportunity in early rheumatoid arthritis: possibility of altering the disease process with early intervention. Clin Exp Rheumatol. 2003;21:S154–S7.
- Emery P, Fleischmann RM, Moreland LW, Hsia EC, Strusberg I, Durez P, et al. Golimumab, a human anti-tumor necrosis factor α monoclonal antibody, injected subcutaneously every four weeks in methotrexate-naive patients with active rheumatoid arthritis: twenty-four-week results of a phase III, multicenter, randomized, double-blind, placebo-controlled study of golimumab before methotrexate as first-line therapy for early-onset rheumatoid arthritis. Arthritis Rheum. 2009;60:2272–83.
- Keystone EC, Genovese MC, Klareskog L, Hsia EC, Hall ST, Miranda PC, et al. Golimumab, a human antibody to tumour necrosis factor α given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate therapy: the GO-FORWARD Study. Ann Rheum Dis. 2009;68:789–96.
- Smolen JS, Kay J, Doyle MK, Landewe R, Matteson EL, Wollenhaupt J, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor α inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet. 2009;374:210–21.
- Tanaka Y, Harigai M, Takeuchi T, Yamanaka H, Ishiguro N, Yamamoto K, et al. Golimumab in combination with methotrexate in Japanese patients with active rheumatoid arthritis: results of the GO-FORTH study. Ann Rheum Dis. 2012;71:817–24.
- Takeuchi T, Harigai M, Tanaka Y, Yamanaka H, Ishiguro N, Yamamoto K, et al. Golimumab monotherapy in Japanese patients with active rheumatoid arthritis despite prior treatment with disease-modifying antirheumatic drugs: results of the phase 2/3, multicentre, randomised, double-blind, placebo-controlled GO-MONO study through 24 weeks. Ann Rheum Dis. 2013;72:1488–95.
- Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38:727–35.
- van Riel PLCM, van Gestel AM, Scott DL. EULAR Handbook of Clinical Assessments in Rheumatoid Arthritis. Alphen Aan Den Rijn, The Netherlands: Van Zuiden Communications, BV; 2000.
- Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23:137–45.
- Aletaha D, Nell VPK, Stamm T, Uffmann M, Pflugbeil S, Machold K, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther. 2005;7:R796–806.
- Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LH, Funovits J, et al. American College of Rheumatology/European League Against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Arthritis Rheum. 2011;63:573–86.
- van der Heijde DMFM, van Leeuwen MA, van Riel PL, Koster AM, van't Hof MA, van Rijswijk MH, et al. Biannual radiographic assessments of hands and feet in a three-year prospective followup of patients with early rheumatoid arthritis. Arthritis Rheum. 1992;35:26–34.
- Kay J, Matteson EL, Dasgupta B, Nash P, Durez P, Hall S, et al. Golimumab in patients with active rheumatoid arthritis despite treatment with methotrexate: a randomized, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum. 2008;58:964–75.
- van der Kooij SM, le Cessie S, Goekoop-Ruiterman YPM, de Vries-Bouwstra JK, van Zeben D, Kerstens PJ, et al. Clinical and radiological efficacy of initial vs delayed treatment with infliximab plus methotrexate in patients with early rheumatoid arthritis. Ann Rheum Dis. 2009;68:1153–8.
- Tanaka Y, Harigai M, Takeuchi T, Yamanaka H, Ishiguro N, Yamamoto K, et al. Prevention of joint destruction in patients with high disease activity or high C-reactive protein levels: Post hoc analysis of the GO-FORTH study. Mod Rheumatol. 2016;26:323–30.
- Braun J, Deodhar A, Inman RD, van der Heijde D, Mack M, Xu S, et al. Golimumab administered subcutaneously every 4 weeks in ankylosing spondylitis: 104-week results of the GO-RAISE study. Ann Rheum Dis. 2012;71:661–7.
- Emery P, Fleischmann RM, Doyle MK, Strusberg I, Durez P, Nash P, et al. Golimumab, a human anti-tumor necrosis factor monoclonal antibody, injected subcutaneously every 4 weeks in patients with active rheumatoid arthritis who had never taken methotrexate: 1-year and 2-year clinical, radiologic, and physical function findings of a phase III, multicenter, randomized, double-blind, placebo-controlled study. Arthritis Care Res (Hoboken). 2013;65:1732–42.
- Kavanaugh A, McInnes IB, Mease PJ, Krueger GG, Gladman DD, van der Heijde D, et al. Clinical efficacy, radiographic and safety findings through 2 years of golimumab treatment in patients with active psoriatic arthritis: results from a long-term extension of the randomised, placebo-controlled GO-REVEAL study. Ann Rheum Dis. 2013;72:1777–85.
- Keystone EC, Genovese MC, Hall S, Miranda PC, Bae SC, Palmer W, et al. Golimumab in patients with active rheumatoid arthritis despite methotrexate therapy: results through 2 years of the GO-FORWARD study extension. J Rheumatol. 2013;40:1097–103.
- Smolen JS, Kay J, Landewe RBM, Matteson EL, Gaylis N, Wollenhaupt J, et al. Golimumab in patients with active rheumatoid arthritis who have previous experience with tumour necrosis factor inhibitors: results of a long-term extension of the randomised, double-blind, placebo-controlled GO-AFTER study through week 160. Ann Rheum Dis. 2012;71:1671–9.
- Tanaka Y, Harigai M, Takeuchi T, Yamanaka H, Ishiguro N, Yamamoto K, et al. Clinical efficacy, radiographic progression, and safety through 156 weeks of therapy with subcutaneous golimumab in combination with methotrexate in Japanese patients with active rheumatoid arthritis despite prior methotrexate therapy: final results of the randomized GO-FORTH trial. Mod Rheumatol. 2016;26:481–90.