
Abstract
Objectives: The objective of this study is to evaluate the safety and effectiveness of subcutaneous tocilizumab (TCZ-SC) in a real-world clinical setting in Japan.
Methods: This single arm, 26-week prospective observational study enrolled patients with RA who were either TCZ naïve or switched from TCZ-IV to TCZ-SC (TCZ-IV-SC group) (UMIN Clinical Trials Registry UMIN000011102). All patients received TCZ-SC 162 mg every 2 weeks and data were collected until week 26 or discontinuation.
Results: Overall 784 (78.1%) were TCZ naïve and 219 (21.8%) were in the TCZ-IV-SC group. 70.9% received disease-modifying antirheumatic drugs at baseline. Adverse events (AEs) and serious AEs occurred in 28.2% and 4.9% of patients, respectively (TCZ-naïve: 29.5% and 5.2%; TCZ-IV-SC: 23.2% and 4.1%). Infections and infestations were the most common AEs (7.4%) and serious AEs (1.7%). Two TCZ-naïve patients died. TCZ-naïve patients had an improvement in median Clinical Disease Activity Index (CDAI) score and mean Disease Activity Score in 28 joints as measured by erythrocyte sedimentation rate (DAS28-ESR) from baseline to week 26. The TCZ-IV-SC group had similar median CDAI scores and mean DAS28-ESR over 26 weeks.
Conclusions: There were no unexpected safety signals with TCZ-SC. TCZ-SC was effective in reducing disease activity in TCZ-naïve patients and maintaining remission in TCZ-IV-SC patients.
Introduction
First-line treatment of rheumatoid arthritis (RA) frequently includes the use of conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) such as methotrexate. If these first-line treatments fail or are not tolerated, an approved biologic DMARD (bDMARD) with a different mechanism of action, such as an anti-tumor necrosis factor agent, an interleukin 6 (IL-6) receptor antagonist, a t-cell costimulation modulator or a Janus kinase inhibitor, may be used [Citation1,Citation2].
Tocilizumab (TCZ) is a recombinant humanized monoclonal antibody directed against the human IL-6 receptor [Citation3]. The use of intravenous TCZ (TCZ-IV) has been approved in over 100 countries, and TCZ-IV has demonstrated efficacy with a well-established safety profile as monotherapy or in combination with csDMARDs in patients with RA [Citation4–10]. Subcutaneous TCZ (TCZ-SC) was also approved internationally after it demonstrated efficacy in patients with RA, with a consistent safety profile to that of TCZ-IV in clinical trials [Citation11–13]. Although many studies have evaluated TCZ-IV in real-world clinical settings, the data for TCZ-SC have been limited [Citation14–16].
With the availability of a SC formulation, some patients may wish to switch from their current TCZ-IV regimen for the convenience of self-administration. In the phase III SUMMACTA study, the efficacy and safety profiles in patients who switched from TCZ-IV in combination with csDMARDs to TCZ-SC were comparable with those in patients who maintained TCZ-IV [Citation13,Citation17]. Similar results were observed with TCZ monotherapy in the phase III MUSASHI study, in which patients who received TCZ-IV monotherapy and switched to TCZ-SC monotherapy experienced efficacy and safety comparable with those in patients who received TCZ-IV monotherapy [Citation18]. However, data from these clinical trials may not fully represent the safety and effectiveness of TCZ-SC in patients in real-world clinical settings. To date, there has been one real-world study that evaluated TCZ effectiveness when switching from IV to SC administration; the investigators found that TCZ-SC maintained a favorable RA disease activity level previously achieved with TCZ-IV [Citation19]. In order to further understand the real-world safety and effectiveness data of TCZ-SC, including patients who may switch from TCZ-IV, the Japanese health authority requested a postmarketing surveillance (PMS) study.
The objective of this open-label, multicenter, prospective, observational PMS study was to evaluate the safety and effectiveness of TCZ-SC in real-world clinical settings in Japan.
Methods
Patients
All patients included in this PMS study met the American College of Rheumatology 1987 classification criteria for RA and had not previously received TCZ-SC. Patients who were TCZ naïve or receiving TCZ-IV were eligible for enrollment. The criteria for inclusion in the TCZ-SC switched group, hereafter referred to as the TCZ-IV-SC group, were: the period from the last dose of TCZ-IV to the first dose of TCZ-SC had to be ≤60 days; the last dose of TCZ-IV had to follow the last dose of another bDMARD; and the patients must have received TCZ-IV for ≥45 days. If these criteria were not met, the patients were considered TCZ naïve. Patients were excluded if they met any of the contraindications specified in the Japanese prescribing information [Citation20].
Protocol
Patients received TCZ-SC 162 mg every 2 weeks either as monotherapy or in combination with csDMARDs for 26 weeks. There were no restrictions for concomitant medication use; corticosteroid use was permitted. Safety data were collected from initiation of TCZ-SC to week 26 or until discontinuation of TCZ-SC. Effectiveness data were collected at weeks 12 and 26. The primary objective was to evaluate the incidence of adverse drug reactions and the changes in Disease Activity Score in 28 joints as measured by erythrocyte sedimentation rate (DAS28-ESR) and Clinical Disease Activity Index (CDAI) from baseline to weeks 12 and 26 in both groups.
Secondary objectives were to assess the remission rates as determined by DAS28-ESR and CDAI and the changes in DAS28-ESR and CDAI from baseline to weeks 12 and 26 in both groups. Anti-TCZ immunoglobulin E (IgE) antibodies were measured in patients who developed systemic injection reactions and injection site reactions. Since adverse drug reactions have a strict cause–effect drug relationship, they are presented here as part of the broader category of adverse events (AEs).
This study was a multi-institutional, prospective, non-interventional, observational study and approved by the Pharmaceuticals and Medical Devices Agency (PMDA) and was conducted in accordance with Good Post-marketing Study Practice (GPSP: Ministry of Health, Labour and Welfare ordinance). Documentation was in accordance with Good Vigilance Practice/GPSP. The study protocol was reviewed and approved in advance by PMDA. For this reason, no ethical review by the individual facilities participating in the study was needed. Because informed consent is not required for post-marketing observational studies that are conducted under the GPSP in Japan, the present study did not solicit informed consent from patients.
Statistical methods
AEs were coded and classified using system organ classes and preferred terms according to the Medical Dictionary for Regulatory Activities (version 18.1). Numerical values were reported for the incidence of AEs and the proportions of patients achieving DAS28-ESR and CDAI remission. Mean (SD) DAS28-ESR and median (IQR) and mean (SD) CDAI scores were reported. For assessment of changes from baseline to week 26, DAS28-ESR was described by using the mean (SD) because DAS28-ESR scores are normally distributed. CDAI was described by using the median (IQR) because CDAI scores are not normally distributed [Citation21–23]. Missing effectiveness data were replaced with the last available preceding data during the observational period according to the last observation carried forward method, as specified in the protocol. Data from ≥4 weeks after the last dose of TCZ-SC were excluded from analysis in patients who discontinued TCZ-SC.
Results
A total of 1036 patients were enrolled (). Case report forms could not be collected for 16 patients, excluding them from the analysis. Of the remaining 1020 patients, 17 patients were excluded from the safety analyses due to retrospective enrollment, which was considered a protocol deviation; a total of 1003 patients were then included in the safety analyses. Of these 1003 patients, 21.8% (219/1003 patients) were in the TCZ-IV-SC group and 78.1% (784/1003 patients) were TCZ naïve. Effectiveness analyses included 785 patients; 218 patients were excluded from the safety population due to unevaluable efficacy reports (missing data, 216 patients), a non-RA diagnosis (one patient), and concomitant treatment with TCZ-IV and TCZ-SC (one patient).
Figure 1. Patient disposition in the 26-week postmarketing surveillance program. CDAI: Clinical Disease Activity Index; CRF: case report form; DAS28-ESR: Disease Activity Score in 28 joints as measured by erythrocyte sedimentation rate; IV: intravenous; RA: rheumatoid arthritis; TCZ: tocilizumab. aPatient data up to time of discontinuation were available for DAS28-ESR and CDAI calculations.
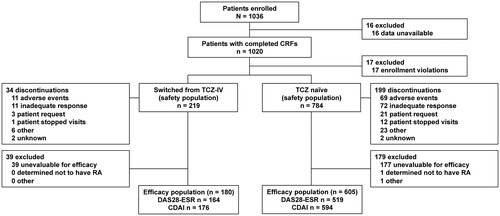
The proportion of patients who received TCZ-SC up to week 12 was 86.4% (848/981 patients); 76.2% (748/981 patients) received it until week 26. Of the 1003 patients, 22 were excluded due to transfer to another hospital (TCZ-IV-SC group: four patients, TCZ-naïve group: 18 patients). In total, 13.5% (133/981 patients) had discontinued TCZ-SC by week 12, and 23.7% (233/981 patients) had discontinued by week 26 (15.8% [34/215 patients] in the TCZ-IV-SC group and 25.9% [199/766 patients] in the TCZ-naïve group). At the end of the 26-week study period, the most common reasons for treatment discontinuation were inadequate response (35.6% [83/233 patients]) and AEs (34.3% [80/233 patients]). Two patients in the TCZ-naïve group and no patients in the TCZ-IV-SC group discontinued TCZ-SC after achieving DAS28-ESR and CDAI remission.
The baseline mean age, weight, and disease duration of the total population were 60.6 years, 53.4 kg, and 10.4 years, respectively, with 85.4% (857/1003 patients) being female (TCZ-naïve group: 60.9 years, 53.4 kg, 9.9 years, 84.6% [664/784 patients], respectively; TCZ-IV-SC group: 59.5 years, 53.6 kg, 11.9 years, 88.1% [193/219 patients], respectively; ). At baseline, 70.9% (712/1003 patients) of all patients received csDMARDs and 44.3% (445/1003 patients) received corticosteroids (TCZ-naïve group: 72.4% [568/784 patients], 46.9% [368/784 patients], respectively; TCZ-IV-SC group: 65.7% [144/219 patients], 35.1% [77/219 patients], respectively). Comorbidities were present in 59.2% (594/1003 patients) of all patients, respiratory disease in 11.3% (114/1003 patients) of all patients, and diabetes mellitus in 6.9% (70/1003) (TCZ-naïve group: 57.0% [447/784 patients], 10.7% [84/784 patients], and 6.8% [54/784 patients], respectively; TCZ-IV-SC group: 67.1% [147/219 patients], 13.6% [30/219 patients], and 7.3% [16/219 patients], respectively).
Table 1. Patient demographic and baseline characteristics.
Safety
AEs occurred in 28.2% (283/1003) of the safety population (23.2% [51/219 patients] in the TCZ-IV-SC group and 29.5% [232/784] in the TCZ-naïve group; ). The patient-year (PY)-adjusted AE rates in the TCZ-IV-SC group were numerically lower than those in the TCZ-naïve group (78.8 versus 98.9; ). Infections and infestations were the most common AE (4.5% [10/219 patients] in the TCZ-IV-SC group versus 8.2% [65/784 patients] in the TCZ-naïve group), with infection rates of 11.1 per 100 PY for the TCZ-IV-SC group and 21.7 for the TCZ-naïve group. The most common infection was nasopharyngitis (two events occurred in two patients in the TCZ-IV-SC group and 12 events occurred in eight patients in the TCZ-naïve group). The next most common AE was laboratory investigations (2.7% [6/219 patients] in the TCZ-IV-SC group versus 6.8% [54/784 patients] in the TCZ-naïve group), which occurred at a rate of 7.0 per 100 PY in the TCZ-IV-SC group and 18.8 per 100 PY in the TCZ-naïve group. Laboratory investigations mainly included white blood cell count decreased, blood triglycerides increased, and so on. Decreased white blood cell counts occurred in two patients in the TCZ-IV-SC group and 15 patients in the TCZ-naïve group. Of the 15 TCZ-naïve patients who had decreased white blood cell counts, 10 had an onset of the event within 12 weeks from the start of TCZ-SC. Increased blood triglycerides occurred in one patient in the TCZ-IV-SC group and in 17 patients in the TCZ-naïve group. Of the 17 TCZ-naïve patients who developed increased triglycerides, 13 developed them within 12 weeks after the start of TCZ-SC. Increased lipid levels occurred once in the TCZ-naïve group. One event of hyperlipidemia occurred in the TCZ-IV-SC group and 16 in the TCZ-naïve group.
Table 2. Adverse events and serious adverse events.
Table 3. Adverse events per 100 patient years.
Serious AEs (SAEs) rates were comparable between groups (4.1% [9/219 patients] in the TCZ-IV-SC group versus 5.2% [41/784 patients] in the TCZ-naïve group; ). The rate of serious infections was 5.8 per 100 PY in the TCZ-naïve group (2.2% [18/784 patients]), and no serious infections occurred in the TCZ-IV-SC group; ). The most common serious infections with ≥2 events were cellulitis, in three patients, and pneumonia and bacterial pneumonia, in two patients each. The most common SAEs in the TCZ-IV-SC group were nervous system disorders (1.3% [3/219 patients]), which occurred at a rate of 3.0 per 100 PY and included one patient each with dizziness, epilepsy, and myoclonic epilepsy. Six malignancies occurred: in the TCZ-IV-SC group (0.4% [1/219 patients]), one patient had gastric cancer, and in the TCZ-naïve group (0.5% [4/784 patients]), two patients had gastric cancer, one patient had a malignant lung neoplasm, and one patient had metastases to the lymph nodes and breast cancer. One SAE of diverticulitis perforation occurred in the TCZ-naïve group. There were two SAEs of decreased white blood cell counts (one patient in each group) and one SAE of decreased neutrophil counts in one patient in the TCZ-naïve group, as determined by the attending physician. Two patients in the TCZ-naïve group died, one due to pneumonia pneumococcal and the other due to cardiac failure.
Eight patients in the TCZ-IV-SC group and six patients in the TCZ-naïve group experienced injection site reactions, all non-serious. Anti-TCZ IgE antibodies were measured in four of the 14 patients who experienced injection site reactions, and one patient from the TCZ-naïve group tested positive. Systemic injection reactions, defined as an event occurring during or within 24 h after injection of TCZ-SC and classified under preferred terms known to be potentially associated with allergic reactions, were observed in 3.1% (32/1003 patients) of all patients (TCZ-IV-SC group: 5.0% [11/219 patients]; TCZ-naïve group: 2.6% [21/784 patients]); two events that occurred in the TCZ-IV-SC group were serious. Anti-TCZ IgE antibodies were measured in five of 32 patients who experienced systemic injection reactions, and one patient tested positive from the TCZ-naïve group; the patient had experienced both injection site reactions and systemic injection reactions. No anaphylaxis events occurred.
Effectiveness
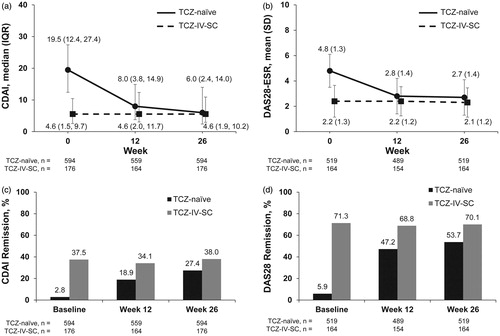
TCZ-naïve patients had improvements in median and mean CDAI scores from baseline to week 26 (median: 19.5 versus 6.0; mean: 21.1 versus 9.7), with 27.4% (163/594 patients) achieving CDAI remission at week 26 (CDAI ≤2.8 and 2.8% [17/594 patients] were in remission at baseline; and Supplemental Table 1). Patients in the TCZ-IV-SC group maintained median CDAI scores from baseline to week 26 (median: 4.6 versus 4.6; mean: 7.3 versus 6.8) and maintained CDAI remission at week 26 (37.5% [66/176 patients] versus 38.0% [67/176 patients]).
Figure 2. CDAI, DAS28-ESR, and remission by group over 26 weeks. Median (IQR) CDAI (a) and mean (SD) DAS28-ESR (b) from baseline to week 26 for patients in the TCZ-naïve and TCZ-IV-SC groups. CDAI (c) and DAS28-ESR (d) remission rates from baseline to week 26 in the TCZ-naïve and TCZ-IV-SC groups. CDAI: Clinical Disease Activity Index; DAS28-ESR: Disease Activity Score in 28 joints as measured by erythrocyte sedimentation rate; IQR: interquartile range; SD: standard deviation; TCZ: tocilizumab; TCZ-IV-SC: patients who switched from intravenous to subcutaneous tocilizumab.
When patients were stratified by baseline weight, the CDAI remission rates at week 26 were not significantly different between the body weight categories for both the TCZ-naïve and TCZ-IV-SC groups (TCZ-naïve: weight <40 kg, 16.6% [3/18 patients]; weight ≥40 to <60 kg, 27.0% [117/433 patients]; ≥60 kg, 29.1% [37/127 patients]; TCZ-IV-SC: weight <40 kg, 0.0% [0/2 patients]; weight ≥40 to <60 kg, 37.8% [50/132 patients]; ≥60 kg: 41.4% [17/41 patients]) (Supplemental Table 2). When patients were grouped by baseline body mass index (BMI), the proportion who achieved CDAI remission was not significantly different between groups (TCZ-naïve: BMI <18.5 kg/m2, 26.5% [13/49 patients]; ≥18.5 to <25 kg/m2, 26.6% [90/338 patients]; ≥25 to <30 kg/m2, 23.8% [16/67 patients]; ≥30 kg/m2, 16.6% [2/12 patients]; TCZ-IV-SC: BMI <18.5 kg/m2, 33.3% [5/15 patients]; ≥18.5 to <25 kg/m2, 40.5% [45/111 patients]; ≥25 to <30 kg/m2, 33.3% [8/24 patients]; ≥30 kg/m2, 33.3% [1/3 patients]). CDAI remission rates at week 26 were not significantly different for those with disease durations of <2 years, ≥2 to <5 years, ≥5 to <10 years, and ≥10 years (TCZ-naïve: 34.7% [41/118 patients], 33.6% [32/95 patients], 23.3% [29/124 patients] and 24.3% [45/185 patients], respectively; TCZ-IV-SC: 25.0% [2/8 patients], 48.0% [12/25 patients], 37.7% [17/45 patients], and 34.2% [25/73 patients], respectively). In the TCZ-naïve group, more patients who were bDMARD-naïve achieved CDAI remission at week 26 than those who had previously received a bDMARD (37.5% [93/248 patients] versus 20.2% [70/346 patients]). Patients who received a csDMARD at baseline in combination with TCZ-SC had a comparable CDAI remission rate with the TCZ-SC monotherapy group (TCZ-naïve: 26.7% [117/438 patients] and 29.4% [46/156 patients]; TCZ-IV-SC: 37.0% [43/116 patients] and 40.0% [24/60 patients]). The CDAI remission rate in patients without a comorbidity was 32.1% (82/255 patients) in the TCZ-naïve group and 54.5% (30/55 patients) in the TCZ-IV-SC group.
TCZ-naïve patients had improvements in mean DAS28-ESR scores from baseline to week 26 (4.8 versus 2.7), with 53.7% (279/519 patients) achieving DAS28-ESR remission at week 26 (DAS28-ESR <2.6 and 5.9% [31/519 patients] were in remission at baseline, and Supplemental Table 1)). The mean DAS28-ESR scores of the patients in the TCZ-IV-SC group were maintained from baseline to week 26 (2.2 versus 2.1) similar results were observed for DAS28-ESR remission rates (71.3% [117/164 patients] versus 70.1% [115/164 patients]).
When patients were stratified by baseline weight, the DAS28-ESR remission rate at week 26 was comparable between groups in both the TCZ-naïve and TCZ-IV-SC groups (TCZ-naïve: weight <40 kg, 50.0% [7/14 patients]; ≥40 to <60 kg, 54.4% [207/380 patients]; ≥60 kg, 52.2% [59/113 patients]; TCZ-IV-SC: weight <40 kg, 66.6% [2/3 patients]; ≥40 to <60 kg, 71.5% [88/123 patients]; ≥60 kg, 67.5% [25/37 patients]; Supplemental Table 3). When patients were grouped by baseline BMI, the DAS28-ESR remission rates at week 26 were not significantly different across the BMI categories (TCZ-naïve: BMI <18.5 kg/m2, 43.4% [20/46 patients]; ≥18.5 kg/m2 to <25 kg/m2, 55.1% [166/301 patients]; BMI ≥25 kg/m2 to <30 kg/m2, 45.0% [27/60 patients]; ≥30 kg/m2, 27.2% [3/11 patients]; TCZ-IV-SC: BMI <18.5 kg/m2, 81.2% [13/16 patients]; ≥18.5 kg/m2 to <25 kg/m2, 71.6% [76/106 patients]; BMI ≥25 kg/m2 to <30 kg/m2, 52.1% [12/23 patients]; ≥30 kg/m2, 50.0% [1/2 patients]). The proportion of patients who achieved DAS28-ESR remission at week 26 was numerically higher among those with shorter disease duration (TCZ-naïve: <2 years, 68.8% [73/106 patients], ≥2 to <5 years, 55.2% [47/85 patients], ≥5 to <10 years, 48.6% [54/111 patients], ≥10 years, 49.0% [81/165 patients]; TCZ-IV-SC, <2 years: 87.5% [7/8 patients]; ≥2 to <5 years, 80.0% [20/25 patients]; ≥5 to <10 years, 78.0% [32/41 patients]; ≥10 years, 61.6% [45/73 patients]). Patients who received a csDMARD at baseline in combination with TCZ-SC had a DAS28-ESR remission rate comparable with those who received TCZ-SC monotherapy (TCZ-naïve: 56.1% [216/385 patients] and 47.0% [63/134 patients]; TCZ-IV-SC: 71.2% [77/108 patients] and 67.8% [38/56 patients]). A numerically higher proportion of patients achieved DAS28-ESR remission at week 26 when the patients were bDMARD naïve (TCZ-naïve: 60.6% [131/216 patients] versus 48.8% [148/303 patients]; TCZ-IV-SC: 76.6% [46/60 patients] versus 66.3% [69/104 patients]) and had no comorbidities (TCZ-naïve: 64.1% [138/215 patients] versus 46.3% [141/304 patients]; TCZ-IV-SC: 85.4% [47/55 patients] versus 62.3% [68/109 patients]).
Discussion
This observational PMS study in patients with RA treated in a real-world clinical setting evaluated patients who either switched from TCZ-IV to TCZ-SC or were TCZ naïve and received TCZ-SC. Overall, TCZ-SC was effective and had a safety profile consistent to that observed with TCZ-IV in previous PMS studies and clinical trials [Citation4–10,Citation15,Citation16]. No new safety signals were observed. TCZ-SC potentially reduced the disease activity in TCZ-naïve patients and maintained remission in the TCZ-IV-SC group.
In the Japanese PMS study of patients with RA treated with TCZ-IV and in the clinical trials, the most common AEs and SAEs were infections, similar to those observed in our study [Citation11,Citation13,Citation15]. In our study, we also found that AEs of special interest (malignancies, GI perforations and cardiovascular dysfunction) occurred at rates similar to those in the TCZ-IV PMS study. Therefore, the results of our study do not raise new concerns.
In our study, TCZ-naïve patients experienced higher rates of AEs (29.5% versus 23.2%; 98.9 per 100 PY versus 78.8 per 100 PY) and SAEs (5.2% versus 4.1%; 15.0 per 100 PY versus 10.1 per 100 PY) than TCZ-IV-SC patients. All of the serious infections occurred in the TCZ-naïve group. Patients in the TCZ-IV-SC group may have experienced fewer AEs because those who had an AE or were intolerant to TCZ would have already discontinued TCZ prior to switching formulations. Patients in this study had received TCZ-IV prior to baseline for over 2 years (mean 884.7 days). In the 3-year extended PMS study of TCZ-IV, 9.9% of patients (3.67 per 100PY) developed a serious infection and 57.6% of that subset subsequently discontinued treatment [Citation24]. Also, in a cumulative analysis of global clinical trials: in the all-exposed patient population (patients who received ≥1 dose of TCZ), rates of AE and SAE were highest during the first 12 months of treatment and declined afterward [Citation25]. Laboratory abnormalities, although not always serious or leading to discontinuation, also occurred more frequently in the early phase of TCZ treatment. In this study, more events of hyperlipidemia, increased blood triglycerides, and decreased white blood cells were reported in the TCZ-naïve group than in the TCZ-IV-SC group. Again, patients previously receiving TCZ who experienced these AEs may have discontinued treatment prior to switching.
TCZ-SC could be effective in maintaining remission in patients who switched from IV to SC administration. Similar results were observed in the 84-week open-label extension period of the phase III MUSASHI study, as well as in a real-world comparison of DAS28-ESR clinical response in patients at baseline and 3 months after switching from TCZ-IV to TCZ-SC [Citation18,Citation19]. TCZ-SC potentially reduced disease activity in TCZ-naïve patients. In the 24-week, phase III, double-blind arm of the MUSASHI study, the DAS28-ESR and CDAI scores in the TCZ-SC and TCZ-IV groups improved similarly [Citation12]. These findings indicate that multiple patient populations, including TCZ-naïve patients and those who have previously received TCZ-IV, can benefit from the therapeutic effects of TCZ-SC.
In this study, the proportion of patients who achieved CDAI and/or DAS28-ESR remission was different for multiple baseline factors such as disease duration, comorbidities, and prior use of a bDMARD. Patients who had previously received a bDMARD had lower DAS28-ESR and CDAI remission rates than those who were bDMARD naïve; the prior bDMARD potentially failed to regulate the disease state. Patients who previously received TCZ-IV had a CDAI remission rate of 37.5% at baseline and then achieved a CDAI remission rate of 38.0% with TCZ-SC at week 26, supporting the argument that TCZ-SC was effective in managing RA in this patient group. As can be expected, a more severe disease state may be more difficult to manage, which can attenuate the likelihood of response to TCZ. Patients who have longer disease duration, have a comorbidity, or previously received a bDMARD may consequently have worse disease control, and these patients may not achieve remission.
A higher proportion of patients who had a low BMI (<25 kg/m2) achieved DAS28-ESR remission than those patients with higher BMIs. The small number of patients in the higher BMI groups may have accounted for the variation observed in the DAS28-ESR response rates. TCZ-SC was given at a fixed dose, and patients with a higher BMI may fail to maintain serum trough concentrations of TCZ required for a response. Previous studies showed that following a switch from TCZ-IV every 4 weeks to TCZ-SC every 2 weeks, patients with higher body weight had reduced TCZ serum trough concentrations and reduced efficacy [Citation18,Citation26].
CDAI and DAS28-ESR remission rates were similar between monotherapy and combination therapy. Comparable therapeutic responses with TCZ monotherapy and combination therapy have also been observed in clinical trials [Citation27,Citation28]. The CDAI remission rates were lower than the DAS28-ESR remission rates; this may be explained by the components included in the index and score calculation. TCZ influences ESR, thus affecting DAS28-ESR; the CDAI does not use acute phase reactants in its calculation [Citation29]. Changes in DAS28-ESR were affected by TCZ, presumably leading to favorable improvement in efficacy evaluations.
The study design of this PMS study was less restrictive than that of phase III studies and confounders within the study (e.g. BMI and concomitant medication) can cause bias in the signal-to-noise ratio [Citation30]. Also, this study did not have a control group; all patients received TCZ.
Conclusions
The results of this real-world study demonstrate that TCZ-SC is a tolerable and effective treatment for patients with RA. Safety results were consistent with the known safety profile of TCZ, and no new safety concerns were observed. TCZ-SC potentially reduced the disease activity in TCZ-naïve patients and maintained remission in the TCZ-IV-SC group. TCZ-SC has promising potential to play an important role in the treatment strategy for RA, either as monotherapy or in combination with csDMARDs such as methotrexate.
Conflicts of interest
T. A. reports personal fees from Chugai, Eli Lilly Japan K.K., GlaxoSmithKline K.K., Pfizer Inc., and UCB Japan Co. Ltd during the conduct of the study; grants and personal fees from Astellas, Takeda, Mitsubishi-Tanabe, Chugai, Pfizer Inc., Daiichi Sankyo, Eisai, and AbbVie; grants from Otsuka and personal fees from Bristol-Myers Squibb Co. outside the submitted work. K. F. reports personal fees from Chugai during the conduct of the study; grants and personal fees from Chugai and Bristol-Myers Squibb Co. and personal fees from Pfizer Inc. and Eli Lilly outside the submitted work. K. Y. reports personal fees from Chugai during the conduct of the study; grants and personal fees from Chugai and Mitsubishi-Tanabe and personal fees from Pfizer Inc., AbbVie, Bristol-Myers Squibb Co., Takeda, GlaxoSmithKline, Nippon Shinyaku, Eli Lilly, Janssen, Eisai, Astellas and Acetlion outside the submitted work. M. T. reports personal fees from Chugai during the conduct of the study. K. K. reports personal fees from Chugai during the conduct of the study. HK reports grants and personal fees from Chugai during the conduct of the study; grants and personal fees from AbbVie, Ayumi, Mitsubishi-Tanabe, Astellas, Takeda, and Eisai and personal fees from Novartis, Sanofi, Eli Lilly, GlaxoSmithKline, Bristol-Myers Squibb Co., Pfizer Inc., and Janssen outside the submitted work.
Supplementary Tables
Download MS Word (48.5 KB)Acknowledgements
The authors thank all investigators for their contributions to the implementation of our study. Support for third-party writing assistance for this manuscript, furnished by Denise Kenski, PhD, of Health Interactions, was provided by F. Hoffmann-La Roche, Ltd.
Additional information
Funding
Related Research Data
References
- Smolen JS, Landewe R, Breedveld FC, Buch M, Burmester G, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73(3):492–509.
- Singh JA, Saag KG, Bridges SL, Jr, Akl EA, Bannuru RR, Sullivan MC, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2016;68(1):1–26.
- Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford). 2012;51(Suppl 5):v3–11.
- Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67(11):1516–23.
- Genovese MC, McKay JD, Nasonov EL, Mysler EF, da Silva NA, Alecock E, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheum. 2008;58(10):2968–80.
- Jones G, Sebba A, Gu J, Lowenstein MB, Calvo A, Gomez-Reino JJ, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis. 2010;69(1):88–96.
- Kremer JM, Blanco R, Brzosko M, Burgos-Vargas R, Halland AM, Vernon E, et al. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate: results from the double-blind treatment phase of a randomized placebo-controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheum. 2011;63(3):609–21.
- Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371(9617):987–97.
- Nishimoto N, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Azuma J, et al. Study of active controlled tocilizumab monotherapy for rheumatoid arthritis patients with an inadequate response to methotrexate (SATORI): significant reduction in disease activity and serum vascular endothelial growth factor by IL-6 receptor inhibition therapy. Mod Rheumatol. 2009;19(1):12–19.
- Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and castleman disease. Blood. 2008;112(10):3959–64.
- Kivitz A, Olech E, Borofsky M, Zazueta BM, Navarro-Sarabia F, Radominski SC, et al. Subcutaneous tocilizumab versus placebo in combination with disease-modifying antirheumatic drugs in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66(11):1653–61.
- Ogata A, Tanimura K, Sugimoto T, Inoue H, Urata Y, Matsubara T, et al. Phase III study of the efficacy and safety of subcutaneous versus intravenous tocilizumab monotherapy in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66(3):344–54.
- Burmester GR, Rubbert-Roth A, Cantagrel A, Hall S, Leszczynski P, Feldman D, et al. A randomised, double-blind, parallel-group study of the safety and efficacy of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional disease-modifying antirheumatic drugs in patients with moderate to severe rheumatoid arthritis (SUMMACTA study). Ann Rheum Dis. 2014;73(1):69–74.
- Bykerk VP, Ostor AJ, Alvaro-Gracia J, Pavelka K, Ivorra JA, Graninger W, et al. Tocilizumab in patients with active rheumatoid arthritis and inadequate responses to DMARDs and/or TNF inhibitors: a large, open-label study close to clinical practice. Ann Rheum Dis. 2012;71(12):1950–4.
- Koike T, Harigai M, Inokuma S, Ishiguro N, Ryu J, Takeuchi T, et al. Effectiveness and safety of tocilizumab: postmarketing surveillance of 7901 patients with rheumatoid arthritis in Japan. J Rheumatol. 2014;41(1):15–23.
- Pappas D, John A, Curtis J, Reed G, Karki C, Magner R, et al. Dosing of intravenous tocilizumab in a real-world setting of rheumatoid arthritis: analyses from the Corrona registry. Rheumatol Ther. 2016;3:103–15.
- Burmester GR, Rubbert-Roth A, Cantagrel A, Hall S, Leszczynski P, Feldman D, et al. Efficacy and safety of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional DMARDs in patients with RA at week 97 (SUMMACTA). Ann Rheum Dis. 2016;75(1):68–74.
- Ogata A, Atsumi T, Fukuda T, Hirabayashi Y, Inaba M, Ishiguro N, et al. Sustainable efficacy of switching from intravenous to subcutaneous tocilizumab monotherapy in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2015;67(10):1354–62.
- Iwamoto N, Fukui S, Umeda M, Nishino A, Nakashima Y, Suzuki T, et al. Evaluation of switching from intravenous to subcutaneous formulation of tocilizumab in patients with rheumatoid arthritis. Mod Rheumatol. 2016;26(5):662–6.
- ACTEMRA (tocilizumab) injection, for intravenous use injection, for subcutaneous use [package insert]. South San Francisco, CA: Genentech, Inc; 2017.
- Aletaha D, Smolen J. The Simplified Disease Activity Index (SDAI) and the Clinical Disease Activity Index (CDAI): A review of their usefulness and validity in rheumatoid arthritis. Clin Exp Rheumatol. 2005;23(5Suppl39):S100–S8.
- van der Heijde DM, van’t Hof MA, van Riel PL, van Leeuwen MA, van Rijswijk MH, van de Putte LB. Validity of single variables and composite indices for measuring disease activity in rheumatoid arthritis. Ann Rheum Dis. 1992;51(2):177–81.
- Aletaha D, Nell VP, Stamm T, Uffmann M, Pflugbeil S, Machold K, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther. 2005;7(4):R796–806.
- Yamamoto K, Goto H, Hirao K, Nakajima A, Origasa H, Tanaka K, et al. Longterm safety of tocilizumab: results from 3 years of followup postmarketing surveillance of 5573 patients with rheumatoid arthritis in Japan. J Rheumatol. 2015;42(8):1368–75.
- Genovese MC, Rubbert-Roth A, Smolen JS, Kremer J, Khraishi M, Gomez-Reino J, et al. Longterm safety and efficacy of tocilizumab in patients with rheumatoid arthritis: a cumulative analysis of up to 4.6 years of exposure. J Rheumatol. 2013;40(6):768–80.
- Abdallah H, Hsu JC, Lu P, Fettner S, Zhang X, Douglass W, et al. Pharmacokinetic and pharmacodynamic analysis of subcutaneous tocilizumab in patients with rheumatoid arthritis from 2 randomized, controlled trials: SUMMACTA and BREVACTA. J Clin Pharmacol. 2017;57(4):459–68.
- Burmester GR, Rigby WF, van Vollenhoven RF, Kay J, Rubbert-Roth A, Kelman A, et al. Tocilizumab in early progressive rheumatoid arthritis: FUNCTION, a randomised controlled trial. Ann Rheum Dis. 2016;75(6):1081–91.
- Dougados M, Kissel K, Sheeran T, Tak PP, Conaghan PG, Mola EM, et al. Adding tocilizumab or switching to tocilizumab monotherapy in methotrexate inadequate responders: 24-week symptomatic and structural results of a 2-year randomised controlled strategy trial in rheumatoid arthritis (ACT-RAY). Ann Rheum Dis. 2013;72(1):43–50.
- Smolen JS, Aletaha D. Interleukin-6 receptor inhibition with tocilizumab and attainment of disease remission in rheumatoid arthritis: the role of acute-phase reactants. Arthritis Rheum. 2011;63(1):43–52.
- Moller HJ. Effectiveness studies: advantages and disadvantages. Dialogues Clin Neurosci. 2011;13(2):199–207.