
Abstract
IgG4-related disease (IgG4-RD) is a newly defined multi-organ disease proposed by Japanese physicians. IgG4-RD is characterized by elevated serum levels of IgG4 and massive infiltration of IgG4-expressing plasma cells in the affected organs. Recent studies have shown that abnormal adaptive immune responses mediated by T helper type 2 cells, regulatory T cells, follicular helper T cells, cytotoxic CD4+ T cells, and plasmablasts are involved in IgG4-RD immunopathogenesis. In addition to adaptive immune responses, innate immune responses play pathogenic roles in IgG4-RD. Plasmacytoid dendritic cells (pDCs), M2 macrophages, and basophils are activated to produce various kinds of cytokines in IgG4-RD. Recent studies highlight the importance of type I IFN and IL-33 produced by pDCs in IgG4-RD immunopathogenesis.
Introduction
IgG4-related disease (IgG4-RD) is a multi-organ disorder characterized by elevated serum levels of IgG4, increased infiltration of IgG4-expressing plasma cells in the affected organs, and storiform fibrosis [Citation1]. Various kinds of single-organ diseases such as autoimmune pancreatitis (AIP), retroperitoneal fibrosis, and sialoadenitis, are now considered organ-specific manifestations of IgG4-RD [Citation1,Citation2]. As gastroenterologists and rheumatologists increase the awareness and recognition for IgG4-RD, our knowledge regarding its clinicopathological features has been expanding rapidly, enabling establishment of diagnostic criteria for IgG4-RD [Citation3]. It is now widely accepted that IgG4-RD targets almost all organs including the pancreas, biliary tree, kidney, lung, salivary glands, prostate, aorta, thyroid, periorbital tissue, breast, meninges, and skin [Citation1].
Although IgG4-RD is attracting strong attention as a novel disease entity, the immunopathogenesis of this disorder is poorly defined. Earlier, elucidation of abnormal immune responses leading to excessive production of IgG4 antibody (Ab) was considered to provide mechanistic insights into IgG4-RD, as enhanced IgG4 Ab response is a hallmark of this autoimmune disorder. However, IgG4 has non-inflammatory, or rather anti-inflammatory properties, compared to other IgG subtypes [Citation4]. These non-inflammatory properties of IgG4 are mediated by its unique biological characteristics such as ability to exchange the Fab-arm and inability to activate complement factors. Therefore, it is possible that excessive IgG4 production is an epiphenomenon secondary to primary inflammatory responses. Shiokawa et al. [Citation5] reported that IgG1 rather than IgG4 plays a pathogenic role in IgG4-related AIP. They examined the pathogenicity of IgG subtypes in IgG4-related AIP by injecting IgG1 and IgG4 isolated from IgG4-related AIP patients into neonatal mice and found that subcutaneous injection of IgG isolated from patients, but not from healthy controls, resulted in pancreatic injury in mice. Although pancreatic injury was induced by injecting IgG1 or IgG4, the degree of pancreatic injury was much greater after IgG1 injection compared to that after IgG4 injection. Furthermore, pancreatic injury mediated by IgG1 was suppressed by co-injection with patient IgG4. In line with this elegant study, Hubers et al. [Citation6] showed that proinflammatory activities of IgG1 against Annexin A11, which is one of the autoantigen candidates in IgG4-related AIP, may be attenuated due to IgG4 binding with Annexin A11. Thus, these recent studies strongly suggest that IgG1 rather than IgG4 plays pathogenic roles in IgG4-RD, and that elucidation of abnormal immune responses leading to excessive IgG4 production is not sufficient to understand the immunopathogenesis of this disease.
Adaptive immune responses underlying the immunopathogenesis of IgG4-RD have been studied extensively. Accumulation of T helper type 2 (Th2) cells [Citation7,Citation8], regulatory T cells (Treg cells) [Citation7,Citation8], or follicular helper T (Tfh) cells [Citation9–11] into peripheral blood and disease-ridden organs is observed in patients suffering from IgG4-RD. In addition to Th2 cells, Treg cells, and Tfh cells a unique population of CD4+ cytotoxic T lymphocytes (CTLs) producing IFN-γ, IL-1β, and TGF-β1 is involved in the chronic fibro-inflammatory responses in IgG4-RD [Citation12,Citation13]. Moreover, oligoclonal expansion of plasmablasts bearing somatic mutations is one of the characteristic B cell responses in patients suffering from IgG4-RD [Citation14,Citation15]. Interestingly, the number of these adaptive immune cells correlates with the serum levels of IgG4 and the tissue expression levels of IgG4. Thus, it can be inferred that adaptive immune responses mediated by Th2 cells, Treg cells, Tfh cells, CD4+CTLs, and plasmablasts play pathogenic roles in IgG4-RD. However, the induction mechanisms of such abnormal adaptive immune responses have not been completely understood. In this regard, recent investigations have highlighted the importance of innate immune responses in preceding and/or augmenting the fibro-inflammatory responses of IgG4-RD [Citation16]. Here, we discuss the roles of cytokines produced by innate immune cells in IgG4-RD immunopathogenesis.
Possible triggers for innate immune responses
Microbe-associated molecular patterns (MAMPs) and damage-associated molecular patterns (DAMPs) are recognized by pattern recognition receptors (PRRs) expressed on cells mediating innate immunity, such as antigen-presenting cells (APCs) and epithelial cells (ECs) [Citation17–20]. Recent studies highlight the roles of PRRs activated by MAMPs in IgG4-RD. A significant population of patients suffering from IgG4-RD exhibits lesions in the gastrointestinal tract and an association of IgG4-RD with gastrointestinal involvement (IgG4-GID) was proposed by Notohara et al. [Citation21]. Considering that the gastrointestinal tract mucosa is always exposed to MAMPs derived from intestinal microflora, it is likely that innate immune responses driven by MAMPs underlie the IgG4-RD immunopathogenesis. Experimental models demonstrate the successful induction of AIP by repeated exposure to heat-killed Escherichia coli and polyinosinic-polycytidylic acid (poly (I:C), a mimic of MAMPs) in C57BL6 mice and MRL/MpJ mice, respectively [Citation22,Citation23]. Such studies led us to examine the involvement of innate immune responses in IgG4-RD development.
Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) are prototypical PRRs expressed in APCs. We previously reported a case of IgG4-RD with significant infiltration of IgG4-expressing plasma cells into the ileum and colon [Citation24]. Interestingly, peripheral blood mononuclear cells (PBMCs) isolated from this patient produced high levels of IgG4 in the presence of TLR2, TLR4, and TLR5 ligands, accompanied by downregulation of IFN-γ and upregulation of IL-10 responses [Citation24]. Thus, these data suggested the possible involvement of Th2 responses against intestinal microflora in the development of IgG4-RD. We then provided more evidence regarding the involvement TLR and NLR activation in enhanced IgG4 responses during IgG4-RD using co-culture systems comprising peripheral blood monocytes and B cells isolated from healthy controls and IgG4-RD patients [Citation25]. Surprisingly, IgG4 production by B cells obtained from healthy controls was significantly enhanced when co-cultured with monocytes from IgG4-RD patients, but not when co-cultured with monocytes from healthy controls, in the presence of TLR4 or NOD2 ligands. Collectively, these recent studies support the hypothesis that innate immune responses driven by TLR and NLR ligands may underlie the IgG4-RD immunopathogenesis.
Cytokines mediating innate immunity in IgG4-RD
Detection of PAMPs and DAMPs by PRRs leads to release of pro-inflammatory cytokines via activation of transcription factors such as nuclear factor-κB (NF-κB) and interferon regulatory factors (IRFs) [Citation17,Citation18]. Pro-inflammatory cytokines, mainly those produced by APCs, are considered to be involved in the fibro-inflammatory responses of IgG4-RD.
IL-6
IL-6 is one of the prototypical pro-inflammatory cytokines produced by a wide variety of hematopoietic and non-hematopoietic cells [Citation26]. IL-6 has pleiotropic functions in immune and non-immune cells, and excessive production of this cytokine is associated with autoimmune disorders such as rheumatoid arthritis and Castleman’s disease. In fact, tocilizumab, a humanized anti-IL-6 receptor Ab, is widely used for treating such disorders [Citation26]. Sato et al. compared the serum levels of IL-6 in IgG4-RD and in multicentric Castleman’s disease and found that IL-6 serum levels were markedly lower in patients suffering from IgG4-RD than in those suffering from Castleman’s disease [Citation27]. Another report also showed that serum IL-6 was detected only in some patients suffering from IgG4-RD with multiple organ involvement [Citation28]. These reports suggest that IL-6 does not play a major role in the inflammatory responses in IgG4-RD. In contrast, IL-6 expression was significantly higher in vascular lesions, as well as in the serum of patients suffering from IgG4-related aortic aneurysms than in patients harboring non-IgG4-related aneurysms [Citation29,Citation30]. In situ hybridization identified mesenchymal cells and macrophages as IL-6-producing cells in patients suffering from IgG4-related aortic aneurysms. Moreover, IL-6 serum levels were directly correlated with C-reactive protein levels and adventitial thickness [Citation29,Citation30]. Therefore, IL-6 produced by mesenchymal cells and macrophages appears to play a pathogenic role in IgG4-related vascular disease.
IL-10
Macrophages are classified as M1 and M2 macrophages [Citation31]. Differentiation of M1 and M2 macrophages is promoted in the presence of Th1 and Th2 cytokines, respectively [Citation31]. As IgG4-RD is characterized by Th2 rather than Th1 cytokines [Citation7,Citation8], differentiation and activation of M2 macrophages is more likely to be induced in IgG4-RD. In fact, the number and frequency of M2 macrophages expressing CD163 is significantly higher in the salivary glands of patients suffering from IgG4-RD compared to that in patients suffering from primary Sjogren syndrome (pSS) [Citation32]. The frequency of M2 macrophages is significantly correlated with the degree of fibrosis [Citation32]. Interestingly, most of such M2 macrophages, accumulate in the salivary glands, and express the pro-fibrogenic factors IL-10 and IL-13 [32]. Thus, M2 macrophages contribute to tissue fibrosis via IL-10 and IL-13 production in IgG4-RD.
BAFF and APRIL
The B cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL) are critical survival factors for B cells [Citation33]. APCs, such as macrophages and dendritic cells (DCs), serve as sources of these cytokines [Citation33]. Elevated serum levels of BAFF were observed in various kinds of autoimmune disorders, such as systemic lupus erythematosus (SLE) and pSS [Citation33]. In vitro studies revealed that IgG4 class-switch DNA recombination is enhanced in the presence of IL-4 and BAFF or APRIL [Citation34], which strongly suggests that BAFF and APRIL are involved in B cell survival as well as in IgG4 production. In addition, two Japanese groups, including ours, have recently reported that serum levels of BAFF and APRIL were markedly elevated in patients suffering from IgG4-RD compared to those in healthy controls and patients suffering from chronic pancreatitis (CP) [Citation35,Citation36]. Interestingly, serum BAFF levels were decreased and APRIL levels were increased following glucocorticoid (GC) therapy in patients suffering from IgG4-RD [Citation35]. Therefore, enhanced expression of BAFF and APRIL is one of the most prominent features of serum cytokine profiles in patients suffering from IgG4-RD.
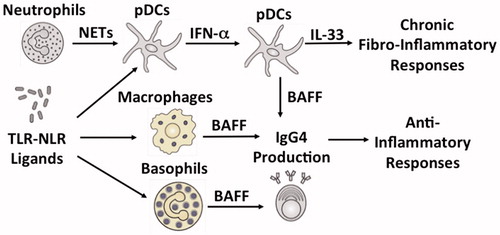
Activation of TLRs and NLRs expressed on APCs plays a critical role in BAFF production in patients suffering from IgG4-RD (). We tried to identify PRRs with the ability to enhance IgG4 production by B cells [Citation25]. PBMCs isolated from healthy controls preferentially produced IgG4 but not IgG1, upon stimulation with muramyl dipeptide (MDP), a NOD2 ligand. Stimulation of PBMCs with lipopolysaccharide (LPS), a TLR4 ligand, or peptidoglycan, a TLR2 ligand, induced both IgG1 and IgG4 responses. Cell-depletion and purification studies, in which CD3+ T cells, CD14+ monocytes, and CD19+ B cells were co-cultured in various combinations in the presence of MDP, revealed that MDP-mediated activation of NOD2 in monocytes, induced IgG4 production by B cells in a T cell-independent manner [Citation25]. Interestingly, such T cell-independent IgG4 production requires BAFF expression by monocytes [Citation25]. Thus, these data suggested that NOD2-mediated BAFF expression in monocytes induced IgG4 production by B cells in healthy controls. PBMCs isolated from patients suffering from IgG4-RD produced a large amount of both IgG1 and IgG4 upon stimulation with NOD2 and TLR ligands compared to the PBMCs isolated from healthy controls [Citation25]. Moreover, peripheral blood monocytes isolated from patients suffering from IgG4-RD induced IgG4 production in B cells obtained from healthy controls in the presence of NOD2 and TLR4 ligands [Citation25]. Taken together, our in vitro studies support the hypothesis that excessive production of monocyte-derived BAFF via activation of TLRs and NLRs is involved in the enhanced IgG4 responses in patients suffering from IgG4-RD. However, enhanced BAFF expression is not limited to APCs such as macrophages and DCs. Peripheral blood basophils isolated from patients suffering from IgG4-RD also enhanced IgG4 production by healthy control B cells in the presence of TLR2 or TLR4 ligands in a BAFF-dependent and T cell-independent manner [Citation37]. Moreover, Yanagawa et al. [Citation38] recently reported that basophils expressing TLR2 and/or TLR4 are localized in the pancreatic lesions of patients suffering from IgG4-RD. Therefore, it seems likely that sensing of MAMPs through TLRs and NLRs expressed in monocytes and basophils plays a pathological role in IgG4-RD immunopathogenesis via activation of BAFF-mediated signaling pathways.
Figure 1. Innate immunity cytokines in IgG4-related disease. Plasmacytoid dendritic cells (pDCs) produce type I IFN (IFN-α) in response to neutrophil extracellular traps (NETs) and Toll-like receptor (TLR)-nucleotide binding oligomerization domain-like receptor (NLR) ligands. pDCs produce B cell activating factor (BAFF) and IL-33 in an IFN-α-dependent manner. The IFN-α-IL-33 axis in pDCs mediates chronic fibro-inflammatory responses during IgG4-related disease (IgG4-RD). BAFF is also produced by macrophages and basophils in response to TLR-NLR ligands. BAFF induces IgG4 production by B cells. IgG4 antibody plays anti-inflammatory roles rather than pro-inflammatory roles in IgG4-RD.
Type I IFN
Production of type I IFN (IFN-I) is induced after interaction of MAMPs with PRRs. Type I IFNs play a critical role in host defenses against microbial infection [Citation39]. Plasmacytoid dendritic cells (pDCs) have a unique ability to produce a large amount of IFN-I upon sensing viral nucleic acids with TLR7 and TLR9, and thus, pDCs are regarded as a major cellular source of IFN-I [Citation40]. Although eradication of viruses and some bacterial pathogens depends upon IFN-I production by innate immune cells, such as pDCs and ECs, IFN-I overproduction plays a pathogenic role in various kinds of autoimmune disorders. SLE is one such prototypical autoimmune disorder mediated by IFN-I (IFN-α) overproduction by pDCs [Citation40]. Indeed, pDCs-mediated IFN-I responses are significantly enhanced in SLE and inhibition of IFN-I responses using a neutralizing Ab against IFN-α [Citation41] or IFN-α receptor Ab [Citation42] resulted in remarkable therapeutic success in patients with SLE. As in the case with SLE, IFN-I overproduction underlies the immunopathogenesis of IgG4-RD.
The earliest evidence showing that the pDCs-mediated IFN-I response is a prominent feature in patients suffering from IgG4-RD, was provided by our group () [Citation36]. MRL/MpJ mice treated with repeated intraperitoneal injections of poly (I:C) exhibited pathological changes akin to human AIP including immune cell infiltration, destruction of pancreatic architecture, and fibrosis [Citation23]. Arai et al. [Citation36] tried to determine the type of immune cells that play substantial roles in the development of such experimental AIP and found that pancreatic lesions were characterized by massive accumulation of pDC antigen-1 (PDCA-1)+B220dim pDCs. Accumulation of pDCs in the pancreas led to enhanced expression of IFN-I (IFN-α and IFN-β), BAFF, and IFN-I-related chemokines such as C-X-C motif chemokine ligand (CXCL) 9, CXCL10, and CXCL11 [36]. Importantly, depletion of pDCs using 120G8 Ab or inhibition of IFN-I responses using anti-IFN-I receptor Ab protected MRL/MpJ mice from AIP development. Thus, Arai et al. provided strong evidence that the development of experimental AIP depends on IFN-I responses mediated by pDCs. Neutrophil extracellular traps (NETs), which are web-like structures composed of self-DNA and neutrophil-derived proteins, function as a strong inducer of pDCs-mediated IFN-I production [Citation43,Citation44]. Generation of NETs, defined by colocalization of double-stranded DNA and neutrophil-derived myeloperoxidase, was observed in the pancreas of MRL/MpJ mice treated with poly (I:C), but not in control mice [Citation36]. Generation of NETs was inhibited by reducing the number of pDCs or inhibition of IFN-I responses, suggesting the presence of a positive feedback loop between NETs induction and IFN-I responses.
Elevated serum levels of IFN-α were observed in patients suffering from IgG4-RD, but not in those suffering from CP or in healthy controls [Citation28,Citation36]. Peripheral blood pDCs isolated from patients suffering from IgG4-RD exhibited enhanced IgG4 production by healthy control B cells in the presence of NETs [Citation36]. pDCs isolated from patients suffering from IgG4-RD exhibited enhanced IgG4 production in a T cell-independent and IFN-I-dependent manner. Furthermore, pancreatic lesions of patients suffering from IgG4-RD are characterized by infiltration of blood dendritic cell antigen 2 (BDCA2)+ pDCs expressing IFN-α and BAFF [Citation36]. Infiltration of CD123+ pDCs was also observed in the salivary glands of patients suffering from IgG4-RD [Citation45]. Collectively, these data strongly suggest that chronic fibro-inflammatory responses of IgG4-RD are caused by pDCs-mediated IFN-I responses and that IgG4-RD is one of the prototypical autoimmune disorders mediated by IFN-I responses as in the case of SLE. However, the molecular mechanisms accounting for IFN-I production in pDCs remain to be elucidated.
IL-33
IL-33 is produced by both APCs and ECs in response to PAMPs and DAMPs exposure [Citation46]. IL-33 produced by innate immune cells activates both Th2 and fibrogenic responses [Citation46]. To address the role of IL-33 in AIP and IgG4-RD, we evaluated IL-33 expression in MRL/MpJ mice treated with multiple injections of poly (I:C). Expression levels of IL-33 and its downstream pro-fibrogenic mediators, such as IL-13 and TGF-β1, were markedly enhanced in the pancreas of MRL/MpJ mice suffering from AIP [Citation47]. Depletion of pDCs by 120G8 Ab or inhibition of IFN-I responses using anti-IFN-I receptor Ab prevented the development of pancreatic fibrosis, which was also associated with diminished IL-33 expression [Citation47]. Moreover, intraperitoneal injection of a neutralizing Ab against ST2, an IL-33 receptor, prevented the development of AIP in MRL/MpJ mice [Citation47]. IL-33 inhibition in poly (I:C)-treated MRL/MpJ mice reduced the pancreatic accumulation of pDCs and inhibited the expression of IL-13 and TGF-β1, thereby suppressing the fibro-inflammatory responses in experimental AIP. Thus, these animal studies strongly suggest that IL-33 contributes to the generation of chronic fibro-inflammatory responses unique to AIP ().
Pancreatic mononuclear cells (PMNCs) isolated from MRL/MpJ mice suffering from AIP, produced high levels of IFN-α and IL-33 upon stimulation with TLR ligands [Citation47]. IL-33 production by PMNCs depends upon IFN-I responses because inhibition of IFN-I responses by anti-IFN-I receptor Ab markedly suppressed IL-33 production [Citation47]. Depletion of pDCs from PMNCs led to marked reduction of IL-33 induced by TLR ligands, whereas pDCs purified from PMNCs produced high levels of IL-33 [Citation47]. Thus, these in vitro studies suggest that pDCs are a cellular source of both IFN-I and IL-33 in experimental AIP. Pancreatic localization of BDCA2+ pDCs expressing both IFN-α and IL-33 is observed in patients suffering from IgG4-RD, but not in patients suffering from CP. Taken together, these studies support the idea that the IFN-I-IL-33 axis in pDCs plays a pathogenic role in generating chronic fibro-inflammatory responses in patients suffering from IgG4-RD.
Enhanced IL-33 expression was also observed in the salivary glands of patients suffering from IgG4-RD [Citation45]. Moreover, serum levels of IL-33 declined in patients suffering from IgG4-RD after GC treatment [Citation45]. Tissue staining studies revealed infiltration of CD163+ M2 macrophages expressing IL-33 in the salivary glands of patients suffering from IgG4-RD. These data reported by Furukawa et al. suggest that IL-33 produced by M2 macrophages is involved in the tissue fibrosis in patients suffering from IgG4-RD. Although IL-33 is being established as one of the pathogenic cytokines in patients suffering from IgG4-RD, cellular sources of its downstream cytokines, such as IL-13 and TGF-β1, have not been elucidated. IL-13 is one of the prototypical pro-fibrogenic factors produced by a wide variety of cells including basophils, innate lymphoid cells, mast cells, and Th2 cells, and a strong correlation between IL-33 and IL-13 expression was observed in the salivary glands of patients suffering from IgG4-RD [Citation45]. In this regard, mast cells and M2 macrophages producing IL-13 are increased in the salivary glands of patients suffering from IgG4-RD [Citation32,Citation48]. In addition, peripheral blood basophils isolated from patients suffering from IgG4-RD produce high levels of IL-13 [Citation37]. Thus, mast cells, M2 macrophages, and basophils might contribute to tissue fibrosis via IL-13 production in response to IL-33.
Type I IFN-IL-33 axis in IgG4-RD
Activation of the IFN-I-IL-33 axis is one of the most prominent features in patients suffering from IgG4-RD, as well as in patients suffering from CP. In parallel to AIP, CP is another type of chronic fibro-inflammatory disorder of the pancreas and is caused by a wide variety of factors including alcohol consumption, smoking, and susceptive gene abnormalities [Citation19]. We recently established a novel model of experimental CP induced by repeated injections of a NOD1 ligand (FK156 or FK565) and cerulein (a cholecystokinin receptor (CCKR) agonist) [Citation19,Citation49–51]. In this model, synergistic actions of CCKR and NOD1 induce IFN-β (IFN-I) expression by pancreatic acinar cells. In response to IFN-I and TNF-α derived from acinar cells and macrophages, respectively, acinar cells produce high levels of IL-33 [Citation19,Citation49–51]. CP development is dependent on the signaling pathways mediated by IFN-I and IL-33 because mice deficient in the IFN-I receptor or those treated with anti-ST2 Ab are protected from CP development [Citation19,Citation49–51]. Moreover, expression of IL-33 was observed in acinar cells of the pancreas in patients suffering from CP [Citation47]. Thus, activation of the IFN-I-IL-33 axis in acinar cells plays a major role in CP progression. Although IgG4-related AIP and CP share common innate immune pathways such as enhanced expression of IFN-I and IL-33, the type of cells producing these cytokines is completely different in both disorders. Activation of the IFN-I-IL-33 axis in pDCs and acinar cells mediates fibro-inflammatory responses in IgG4-related AIP and CP, respectively. Future studies are thus required to elucidate the molecular mechanisms by which two distinct disorders of the pancreas are associated with overproduction of the same innate immunity cytokines derived from two different cellular sources. However, it should be noted that inhibition of the IFN-I-IL-33 axis might be beneficial for both IgG4-related AIP and CP.
The role played by pDCs-mediated IFN-I responses is well established in systemic autoimmune diseases such as SLE and pSS [Citation52]. The findings that pDCs-mediated IFN-I responses are involved in experimental AIP development and that enhanced expression of IFN-I via the pDCs activation is seen in patients suffering from IgG4-RD led us to define IgG4-RD as an IFN-I-related autoimmune disorder. Although SLE and IgG4-RD are linked by a common innate immune response, i.e., overproduction of IFN-I through pDCs activation, there are major differences between SLE and IgG4-RD in terms of clinical manifestations and pathological findings. First, enhanced IgG4 responses can be diagnosed only in patients suffering from IgG4-RD. Second, IgG4-RD affects almost all organs of the body, whereas SLE preferentially affects the skin, kidney, joints, lung, and central nervous system. Third, a unique form of fibrosis called storiform fibrosis is found only in patients suffering from IgG4-RD. Fourth, a decrease in levels of complements reflects the disease activity of SLE, whereas serum levels of C3, C4, and CH50 are normal in most cases of IgG4-RD. Thus, the clinical manifestations and pathological findings of IgG4-RD are completely different from those of SLE, despite the fact that both are characterized by pDCs-mediated IFN-I responses. Such differences between IgG4-RD and SLE might be partially explained by the activation of IL-33-mediated signaling pathways. As described above, activation of the IFN-I-IL-33 axis in pDCs is one of the prominent features of experimental AIP and IgG4-RD. In contrast, serum levels of IL-33 in patients suffering from SLE are comparable to those of healthy controls [Citation53]. Considering that IL-33 acts as a core mediator of inflammation and fibrosis, it is possible that activation of the IFN-I-IL-33 axis plays a major role in clinical manifestations and the development of pathology specific to IgG4-RD. Clinical trials targeting IFN-I and IL-33 in patients suffering from IgG4-RD are necessary to confirm this hypothesis.
Conflict of interest
None
Additional information
Funding
References
- Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–51.
- Kamisawa T, Chari ST, Lerch MM, Kim MH, Gress TM, Shimosegawa T. Recent advances in autoimmune pancreatitis: type 1 and type 2. Gut. 2013;62(9):1373–80.
- Kamisawa T, Okazaki K. Diagnosis and treatment of IgG4-related disease. Curr Top Microbiol Immunol. 2017;401:19–33.
- Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy. 2009;39(4):469–77.
- Shiokawa M, Kodama Y, Kuriyama K, Yoshimura K, Tomono T, Morita T, et al. Pathogenicity of IgG in patients with IgG4-related disease. Gut. 2016;65(8):1322–32.
- Hubers LM, Vos H, Schuurman AR, Erken R, Oude Elferink RP, Burgering B, et al. Annexin A11 is targeted by IgG4 and IgG1 autoantibodies in IgG4-related disease. Gut. 2018;67(4):728–35.
- Zen Y, Fujii T, Harada K, Kawano M, Yamada K, Takahira M, et al. Th2 and regulatory immune reactions are increased in immunoglobin G4-related sclerosing pancreatitis and cholangitis. Hepatology. 2007;45(6):1538–46.
- Tanaka A, Moriyama M, Nakashima H, Miyake K, Hayashida JN, Maehara T, et al. Th2 and regulatory immune reactions contribute to IgG4 production and the initiation of Mikulicz disease. Arthritis Rheum. 2012;64(1):254–63.
- Kubo S, Nakayamada S, Zhao J, Yoshikawa M, Miyazaki Y, Nawata A, et al. Correlation of T follicular helper cells and plasmablasts with the development of organ involvement in patients with IgG4-related disease. Rheumatology (Oxford). 2018;57(3):514–24.
- Akiyama M, Suzuki K, Yamaoka K, Yasuoka H, Takeshita M, Kaneko Y, et al. Number of circulating follicular helper 2 T cells correlates with IgG4 and interleukin-4 levels and plasmablast numbers in IgG4-related disease. Arthritis Rheumatol. 2015;67(9):2476–81.
- Kamekura R, Takano K, Yamamoto M, Kawata K, Shigehara K, Jitsukawa S, et al. Cutting edge: a critical role of lesional T follicular helper cells in the pathogenesis of IgG4-related disease. J Immunol. 2017;199(8):2624–9.
- Mattoo H, Mahajan VS, Maehara T, Deshpande V, Della-Torre E, Wallace ZS, et al. Clonal expansion of CD4(+) cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol. 2016;138(3):825–38.
- Maehara T, Mattoo H, Ohta M, Mahajan VS, Moriyama M, Yamauchi M, et al. Lesional CD4+ IFN-γ+ cytotoxic T lymphocytes in IgG4-related dacryoadenitis and sialoadenitis. Ann Rheum Dis. 2017;76(2):377–85.
- Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014;134(3):679–87.
- Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74(1):190–5.
- Watanabe T, Yamashita K, Kudo M. IgG4-related disease and innate immunity. Curr Top Microbiol Immunol. 2017;401:115–28.
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511.
- Strober W, Watanabe T. NOD2, an intracellular innate immune sensor involved in host defense and Crohn's disease. Mucosal Immunol. 2011;4(5):484–95.
- Watanabe T, Kudo M, Strober W. Immunopathogenesis of pancreatitis. Mucosal Immunol. 2017;10(2):283–98.
- Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8(4):279–89. Epub 2008/03/15.
- Notohara K, Kamisawa T, Uchida K, Zen Y, Kawano M, Kasashima S, et al. Gastrointestinal manifestation of immunoglobulin G4-related disease: clarification through a multicenter survey. J Gastroenterol. 2018;53(7):845–53.
- Haruta I, Yanagisawa N, Kawamura S, Furukawa T, Shimizu K, Kato H, et al. A mouse model of autoimmune pancreatitis with salivary gland involvement triggered by innate immunity via persistent exposure to avirulent bacteria. Lab Invest. 2010;90(12):1757–69.
- Qu WM, Miyazaki T, Terada M, Okada K, Mori S, Kanno H, et al. A novel autoimmune pancreatitis model in MRL mice treated with polyinosinic:polycytidylic acid. Clin Exp Immunol. 2002;129(1):27–34.
- Akitake R, Watanabe T, Zaima C, Uza N, Ida H, Tada S, et al. Possible involvement of T helper type 2 responses to Toll-like receptor ligands in IgG4-related sclerosing disease. Gut. 2010;59(4):542–5. Epub 2010/03/25.
- Watanabe T, Yamashita K, Fujikawa S, Sakurai T, Kudo M, Shiokawa M, et al. Activation of toll-like receptors and NOD-like receptors is involved in enhanced IgG4 responses in autoimmune pancreatitis. Arthrit Rheumat. 2012;64(3):914–24.
- Kang S, Tanaka T, Kishimoto T. Therapeutic uses of anti-interleukin-6 receptor antibody. Int Immunol. 2015;27(1):21–9.
- Sato Y, Kojima M, Takata K, Morito T, Asaoku H, Takeuchi T, et al. Systemic IgG4-related lymphadenopathy: a clinical and pathologic comparison to multicentric Castleman's disease. Mod Pathol. 2009;22(4):589–99.
- Yamamoto M, Takano K, Kamekura R, Suzuki C, Ichimiya S, Himi T, et al. Stage classification of IgG4-related dacryoadenitis and sialadenitis by the serum cytokine environment. Mod Rheumatol. 2018;28(6):1004–8.
- Kasashima S, Kawashima A, Zen Y, Ozaki S, Kasashima F, Endo M, et al. Upregulated interleukins (IL-6, IL-10, and IL-13) in immunoglobulin G4-related aortic aneurysm patients. J Vasc Surg. 2018;67(4):1248–62.
- Kasashima S, Kawashima A, Kasashima F, Endo M, Matsumoto Y, Kawakami K. Inflammatory features, including symptoms, increased serum interleukin-6, and C-reactive protein, in IgG4-related vascular diseases. Heart Vessels. In press.
- Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118(11):3522–30.
- Furukawa S, Moriyama M, Tanaka A, Maehara T, Tsuboi H, Iizuka M, et al. Preferential M2 macrophages contribute to fibrosis in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz's disease. Clin Immunol. 2015;156(1):9–18.
- Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. 2009;9(7):491–502.
- Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3(9):822–9.
- Kiyama K, Kawabata D, Hosono Y, Kitagori K, Yukawa N, Yoshifuji H, et al. Serum BAFF and APRIL levels in patients with IgG4-related disease and their clinical significance. Arthritis Res Ther. 2012;14(2):R86.
- Arai Y, Yamashita K, Kuriyama K, Shiokawa M, Kodama Y, Sakurai T, et al. Plasmacytoid dendritic cell activation and IFN-alpha production are prominent features of murine autoimmune pancreatitis and human IgG4-related autoimmune pancreatitis. J Immunol. 2015;195(7):3033–44.
- Watanabe T, Yamashita K, Sakurai T, Kudo M, Shiokawa M, Uza N, et al. Toll-like receptor activation in basophils contributes to the development of IgG4-related disease. J Gastroenterol. 2013;48(2):247–53.
- Yanagawa M, Uchida K, Ando Y, Tomiyama T, Yamaguchi T, Ikeura T, et al. Basophils activated via TLR signaling may contribute to pathophysiology of type 1 autoimmune pancreatitis. J Gastroenterol. 2018;53(3):449–60.
- Honda K, Takaoka A, Taniguchi T. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25(3):349–60.
- Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8(8):594–606.
- Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, et al. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75(11):1909–16.
- Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti-interferon-alpha receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. 2017;69(2):376–86.
- Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Translat Med. 2011;3(73):73ra19.
- Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Translat Med. 2011;3(73):73ra20.
- Furukawa S, Moriyama M, Miyake K, Nakashima H, Tanaka A, Maehara T, et al. Interleukin-33 produced by M2 macrophages and other immune cells contributes to Th2 immune reaction of IgG4-related disease. Sci Rep. 2017;7:42413.
- Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. 2014;31:31–7.
- Watanabe T, Yamashita K, Arai Y, Minaga K, Kamata K, Nagai T, et al. Chronic fibro-inflammatory responses in autoimmune pancreatitis depend on IFN-α and IL-33 produced by plasmacytoid dendritic cells. J Immunol. 2017;198(10):3886–96.
- Takeuchi M, Ohno K, Takata K, Gion Y, Tachibana T, Orita Y, et al. Interleukin 13-positive mast cells are increased in immunoglobulin G4-related sialadenitis. Sci Rep. 2015;5:7696.
- Watanabe T, Asano N, Kudo M, Strober W. Nucleotide-binding oligomerization domain 1 and gastrointestinal disorders. Proc Jpn Acad Ser B: Phys Biol Sci. 2017;93(8):578–99.
- Watanabe T, Sadakane Y, Yagama N, Sakurai T, Ezoe H, Kudo M, et al. Nucleotide-binding oligomerization domain 1 acts in concert with the cholecystokinin receptor agonist, cerulein, to induce IL-33-dependent chronic pancreatitis. Mucosal Immunol. 2016;9(5):1234–49.
- Tsuji Y, Watanabe T, Kudo M, Arai H, Strober W, Chiba T. Sensing of commensal organisms by the intracellular sensor NOD1 mediates experimental pancreatitis. Immunity. 2012;37(2):326–38.
- Ganguly D. Do type I interferons link systemic autoimmunities and metabolic syndrome in a pathogenetic continuum? Trends Immunol. 2018;39(1):28–43.
- Awada A, Nicaise C, Ena S, Schandene L, Rasschaert J, Popescu I, et al. Potential involvement of the IL-33-ST2 axis in the pathogenesis of primary Sjogren's syndrome. Ann Rheum Dis. 2014;73(6):1259–63.