
Abstract
The most representative kidney lesion of IgG4-related disease (IgG4-RD) is plasma cell-rich tubulointerstitial nephritis (TIN) with distinctive imaging findings including multiple low-density lesions on contrast-enhanced computed tomography. In addition, membranous glomerulonephritis is a representative glomerular lesion of this disease. Recent advances have clarified that inflammation with IgG4-positive plasma cell infiltrates is not restricted to the renal parenchyma, but can be seen in outside the renal capsule, around medium-sized arteries such as lobar arteries, around nerves, and in the renal pelvis and periureter. Hypocomplementemia is a very important feature of IgG4-TIN, and serum complement level might serve as a convenient biomarker to predict relapse. Although good responsiveness to glucocorticoid has been considered characteristic of IgG4-RD, delayed start of treatment is associated with partial scarring in the kidneys on imaging study. Therefore, steroid therapy should be immediately initiated as soon as the diagnosis of IgG4-TIN is made. Future analyses of pathogenesis will be needed to more precisely define the optimal therapeutic strategies for the various subsets of Ig4-RD patients.
1. Introduction
The kidney is one of the most frequently affected organs in IgG4-related disease (IgG4-RD) [Citation1]. The frequency of kidney involvement ranges from 7.0 to 24.6% of cases [Citation2–9]. The most common manifestation is plasma cell-rich tubulointerstitial nephritis (TIN), called IgG4-related TIN (IgG4-TIN) [Citation10–13]. Differing from drug induced TIN or TIN associated with Sjögren’s syndrome, the lesion is not diffuse but rather an aggregation of focal lesions that are surrounded by normal renal parenchyma [Citation10–13]. In some cases, with IgG4-TIN, despite the renal lesions being apparent on diagnostic imaging, only minor abnormalities are evident on renal biopsy because of this characteristic inhomogeneous distribution of kidney lesions. In addition to TIN, a variety of glomerular lesions have been reported in patients with IgG4-RD [Citation14–22]. Of these, membranous glomerulonephritis (MGN) is the most common [Citation23]. Renal pelvic lesion, named as IgG4-related pyelitis [Citation24], is characteristic of IgG4-RD and contributes to its diagnosis [Citation11]. Very rarely, the ureter can also be involved [Citation25,Citation26], and hydronephrosis is an urgent complication requiring immediate intervention. In this review, we provide an up-to-date overview of the kidney lesions of IgG4-RD including renal pelvic and periureteral lesions and hydronephrosis resulting from IgG4-related retroperitoneal fibrosis [Citation27].
2. Definition of IgG4-related kidney disease
IgG4-related kidney disease (IgG4-RKD) is the comprehensive term for kidney lesions encompassing parenchymal and pelvic lesions [Citation11]. There is some controversy as to whether periureteral and renal pelvic lesions are involved in IgG4-RKD or in IgG4-related retroperitoneal fibrosis.
3. IgG4-related tubulointerstitial nephritis
a. Clinical features
Decreased renal function without proteinuria or hematuria is the most typical clinical feature of this disease. However, mild proteinuria and/or hematuria have been reported in about half of patients [Citation11]. The amount of proteinuria is usually less than 500 mg/gCr except when membranous glomerulonephritis (MGN) or other glomerular lesions coexist. When MGN is accompanied by IgG4-RD, a nephrotic level of proteinuria may be detected. Most patients with IgG4-RD have very mild or no symptoms, and in some cases, kidney lesions are incidentally detected during the closer examination of other organ involvement using contrast-enhanced computed tomography in patients with already diagnosed IgG4-RD.
Serologically, the most important feature is elevated serum IgG4 concentrations. Although this is a common feature of IgG4-RD, normal serum IgG4 levels are sometimes encountered in patients with IgG4-related periaortitis/retroperitoneal fibrosis or type 1 autoimmune pancreatitis. In contrast, serum IgG4 is usually high when the kidney is involved. In our previous analysis, the average serum IgG4 level was 991 mg/dL (measured by turbidimetry using a commercially available kit from The Binding Site Limited) in patients with IgG4-related kidney disease [Citation11]. Thus, very high serum IgG4 is an outstanding characteristic of IgG4-TIN. Reflecting an allergic predisposition, many patients have serum IgE elevation and/or eosinophilia [Citation10–12]. Saeki et al. reported that about 50% of patients with IgG4-RKD had eosinophilia [Citation10]. However, recently, they showed that the frequency of elevated serum IgE levels or eosinophilia is similar in IgG4-RD patients both with and without clinically apparent allergies such as allergic rhinitis [Citation28]. Hypergammaglobulinemia is also a distinctive feature of IgG4-RD [Citation11]. Other subclasses of IgG in addition to IgG4 such as IgG1 or IgG3 may also be increased in this disease. In contrast, serum IgA or IgM levels are usually within the respective normal ranges [Citation29]. Although about 30% of patients have antinuclear antibodies, they usually do not have disease specific autoantibodies [Citation11].
Another very important serological feature is hypocomplementemia [Citation11]. Although hypocomplementemia is a feature of IgG4-RD, only about 16–34% of patients have low serum complement levels. In contrast, more than 50% of patients with active IgG4-TIN have low concentrations of complement [Citation11]. Very importantly, severe hypocomplementemia is a representative feature of IgG4-TIN. Yamada et al analyzed 334 patients with IgG4-RD and found that 30 of them (10.1%) had severe hypocomplementemia defined as C3 < 50 mg/dL and kidney involvement was strongly related to severely low C3 with odds ratio of 6.09 (95% CI 2.81–14.2) [Citation2]. With regard to the mechanism of hypocomplementemia, activation of the classical pathway has been assumed [Citation30]. On the other hand, involvement of the mannose-binding lectin (MBL) pathway is still controversial [Citation30,Citation31]. Since IgG4 is thought not to be able to activate complement, IgG subclasses other than IgG4 such as IgG1 or IgG3 are likely implicated in the formation of immune complexes resulting in hypocomplementemia. Indeed, our previous analysis demonstrated that serum C3 levels in patients with kidney lesions were significantly inversely correlated with serum levels of non-IgG4-IgG (calculated as the total IgG minus IgG4) but not significant with IgG4 [Citation2]. This finding suggests the significant role of IgG1 or IgG3 in the complement activation in IgG4-TIN. Interestingly, Shiokawa et al. very recently found that laminin 511-E8 is a target antigen of autoimmune pancreatitis (IgG4-related pancreatitis) and most of these antibodies belonged not to IgG4 but IgG1 subclass [Citation32]. Moreover, they showed that immunization of mice with human laminin 511-E8 induced a pancreatic lesion resembling human AIP. Their findings also suggest the significance of IgG1 in the pathogenesis of IgG4-RD. Very importantly, serum complement levels decrease again when IgG4-TIN recurs. Saeki et al. followed 14 patients with IgG4-TIN and hypocomplementemia and found that three patients had a redecrease of serum complement levels when they experienced recurrence [Citation33]. Thus, serum complement level might become a useful biomarker to monitor relapse in IgG4-TIN.
b. Imaging features
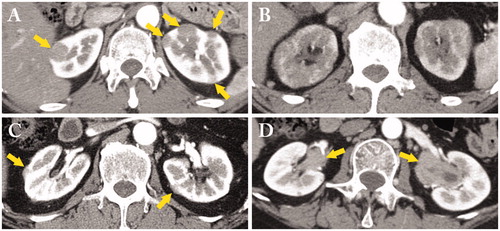
Contrast-enhanced (CE) computed tomography (CT) is the most important modality to detect and evaluate IgG4-TIN [Citation11,Citation34]. The distribution of lesions in each kidney is usually demonstrated as multiple low-density lesions (). When the lesions are small, they are called small cortical hypodense nodules. However, contrast-enhancement should be avoided in patients with decreased renal function because of the risk of contrast nephropathy. When contrast-enhancement cannot be used, bilateral diffuse kidney enlargement despite renal dysfunction is another feature. Since the border between affected and unaffected areas is very unclear with bilateral enlarged kidneys, diffuse patchy involvement is a more suitable name in such cases () [Citation15,Citation35]. Extrarenal lesions and those located on the renal capsule are another salient feature of this disease, and are called ‘rim-like lesion of the kidney’ () [Citation36–40]. This radiological finding corresponds to the capsule-like low-density rim of Type 1 autoimmune pancreatitis (AIP). Finally, a solitary mass lesion has also been reported [Citation41,Citation42]. This lesion is very difficult to differentiate from malignant tumor, and nephrectomy is sometimes required.
Figure 1. A variety of contrast-enhanced computed tomography (CT) findings in IgG4-related kidney disease. (A) Multiple low-density lesions in the bilateral kidneys (arrows); (B) Diffuse patchy involvement of the bilateral kidneys; (C) Rim-like lesion of the kidney (arrows); (D) Renal pelvis thickening with smooth intraluminal surface (arrows).
c. Histopathological features
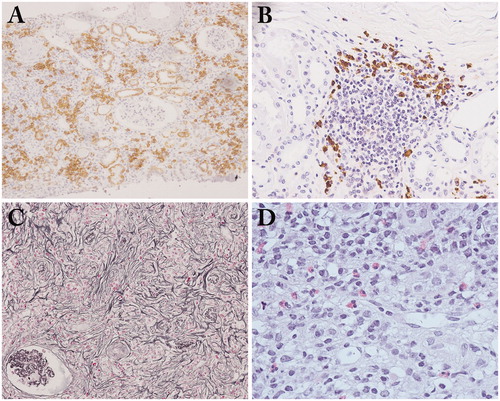
Plasma cell-rich TIN with fibrosis is a typical pathological finding () [Citation10–13]. Generally, IgG4/IgG-positive plasma cell ratio >40% and IgG4-positive plasma cells >10/high power field are sufficient for the diagnosis of IgG4-TIN when a needle biopsy is used () [Citation43]. The most specific histopathological feature is storiform fibrosis (). This type of fibrosis is rarely seen in other kinds of TIN such as drug induced TIN or TIN associated with Sjogren’s syndrome. Irregular fibers surrounding several inflammatory cells on periodic acid-methenamine silver staining (‘bird’s eye fibrosis’), are sometimes seen instead of typical storiform fibrosis in the kidney [Citation13,Citation44]. Obliterative phlebitis is a common pathological feature of IgG4-RD that is often seen in other involved organs, but rarely in the kidney. Eosinophil infiltration is another important feature of IgG4-TIN (). However, it is not specific for IgG4-TIN because drug-induced TIN also sometimes shows numerous tissue eosinophils in the interstitium. Germinal centers are also sometimes detected in IgG4-TIN ().
Figure 2. Light microscopy findings in IgG4-related tubulointerstitial nephritis. (A) Copious plasma cell infiltrates in the interstitium (CD138 immunostaining × 100); (B) Many IgG4-positive plasma cell infiltrates with germinal center in the subcapsular interstitium (IgG4 immunostaining ×200); (C) a representative example of storiform fibrosis (periodic acid-methenamine-silver staining ×100); (D) Many eosinophil infiltrates in the interstitium (hematoxylin and eosin staining ×400).
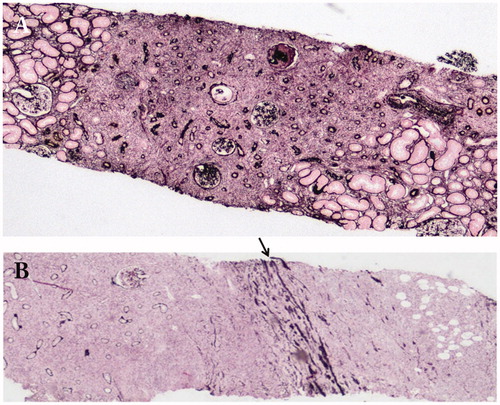
The specific distribution of renal parenchymal lesions is an important histopathological feature of this disease. In IgG4-TIN, the margin between affected and unaffected areas is very clearly demarcated () [Citation11,Citation38], with this finding thought to correspond to the imaging feature of multiple low-density lesions (). In addition, lymphoplasmacytic cell infiltration into and beyond the renal capsule is another unique distributional feature of this disease () [Citation37,Citation38], with lesions located outside the renal capsule thought to correspond to the imaging feature of a rim-like lesion of the kidney ().
Figure 3. The specific distribution of renal parenchymal lesions. (A) The margin between affected and unaffected areas is very clearly demarcated (periodic acid-methenamine-silver staining ×40); (B) Lymphoplasmacytic cells infiltrate into and beyond the renal capsule (arrow: renal capsule) (periodic acid-methenamine-silver staining ×40).
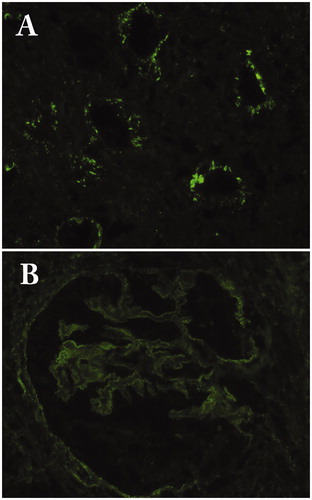
Immunoglobulin and complement deposition in the tubular basement membrane (TBM) is an important feature of IgG4-TIN [Citation12,Citation13]. Raissian et al. demonstrated that more than 80% of patients have IgG or C3 deposits in the TBM [Citation12]. Deposits are limited to the parenchyma with inflammation but are not noted in areas with only minor abnormalities. In contrast, IgG and complement deposits are seen in the TBM with or without tubulointerstitial inflammation in the interstitium in lupus patients. The major component of IgG is IgG4, but IgG1 or IgG3 is also deposited [Citation12,Citation13]. In addition, although less frequently, C1q deposition may also be detected (). In electron microscopy, electron dense deposits are found in the TBM in accordance with the immunfluorescent findings.
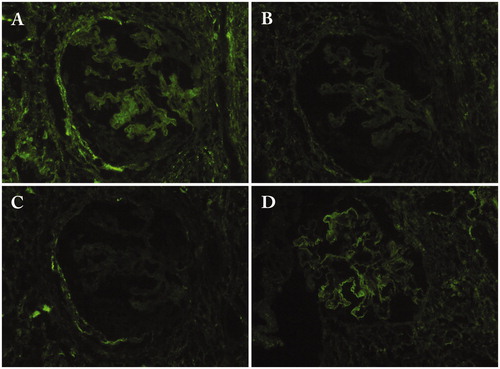
Figure 4. Immunofluorescence findings. (A) Granular deposits of C1q in the tubular basement membrane (×200); (B) Granular deposits of IgG4 in the glomerular basement membrane in IgG4-related membranous glomerulonephritis (×400).
4. Glomerular disease
MGN is the most common glomerular disease accompanied by IgG4-RD [Citation17,Citation23]. About 7% of patients with kidney lesions have MGN [Citation23]. Since almost all cases with IgG4-RD do not have anti-M-type phospholipase A2 receptor antibody, which is a marker of primary MGN, it seems to be a secondary MGN [Citation45]. While IgG4 is the most dominant subclass of deposited IgG (), other IgG subclasses are also deposited in the glomerular basement membrane () [Citation15]. C3 is also deposited followed by C1q in fewer cases [Citation15]. In most cases, MGN is accompanied by IgG4-TIN, but there is a subgroup of IgG4-RD that has MGN without TIN. Interestingly, the response to glucocorticoid therapy is different between IgG4-TIN and IgG4-MGN. Miyata et al. reported two cases with IgG4-TIN and IgG4-MGN in whom IgG4-TIN responded well to steroid while proteinuria persisted, with these patients needing additional immunosuppression [Citation46]. They reviewed 15 cases receiving such a combination and found that in 7 cases more than 1.5 g/day of proteinuria persisted while renal function was partially recovered with steroid therapy [Citation46].
Figure 5. Immunofluorescence findings of the glomerulus in a patient with IgG4-related membranous glomerulonephritis. (A) IgG1; (B) IgG2; (C) IgG3; (D) IgG4.
Since IgG4-RD is thought to be a T helper type 2 (Th2) dominant disease [Citation47], several Th2-related kidney diseases may coexist. These include IgA-vasculitis (Henoch-Schonlein purpura nephritis) [Citation19,Citation20] and minimal change disease [Citation21,Citation22]. In addition, a variety of other glomerular lesions have been reported, i.e. IgA nephropathy [Citation14], membranoproliferative glomerulonephritis [Citation16], and endocapillary proliferative glomerulonephritis [Citation18].
5. Periarterial lesions, arteritis, and perineural lesion
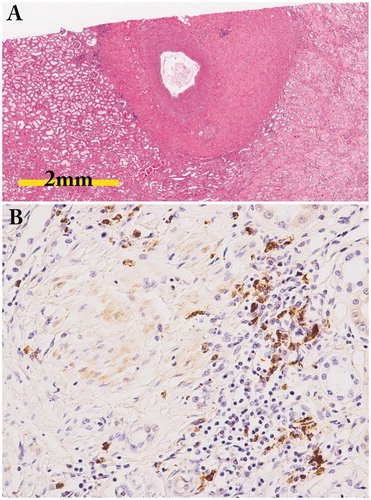
In analyses of autopsied cases, periarterial lesions around medium-sized arteries have been shown () [Citation48]. However, whether renal vasculitis is detectable or not in IgG4-RD is still controversial [Citation43,Citation49]. Perineural lesion is an important pathological feature of IgG4-RD. Notohara et al. analyzed 6 cases with IgG4-RD with gastric lesions (5 cases) or esophageal lesion (one case) histopathologically and found that all cases showed inflammatory cell infiltration around the myenteric nerve plexus [Citation50]. Fujii et al. experienced an autopsy case of IgG4-RD and found the characteristic distribution of inflammatory cells preferentially around the peripheral nerves [Citation51]. Thus, the perineural lesion is one of the characteristic histopathological features of this disease. Although perineural lesion has rarely been evaluated in IgG4-RKD, it can be detected in autopsy or nephrectomy cases ().
Figure 6. Periarterial lesion and perineural lesion. (A) Marked periarterial fibrosis of interlobar artery (periodic acid-Schiff staining); (B) IgG4-positive plasma cell infiltration in the perineural lesion in a patient with IgG4-ureteritis who underwent nephrectomy (IgG4 immunostaining ×400).
6. IgG4-related pyelitis and periureteral lesion
Renal pelvis thickening with smooth intraluminal surface is a frequently observed imaging finding characteristic to IgG4-RD () [Citation11,Citation52,Citation53]. This lesion is sometimes confused with malignant tumor including lymphoma, and may require nephrectomy for its differentiation. While the lesion is usually unilateral, synchronous or metachronous involvement of bilateral pelvises has also been reported [Citation52]. Histopathologically, it is an inflammatory nodular lesion resembling lymphoid organ with many lymphoid follicles [Citation52]. Lymphoplasmacytic infiltration with more than 40% IgG4+ PCs is a common and essential finding with other organs in IgG4-RD. In addition, these lesions have sometimes been accompanied by eosinophil infiltration or obliterative phlebitis. In some such cases, fibrosis with hyalinization or storiform fibrosis is found. Periureteral lesion is very rare, but when found is very difficult to differentiate from ureteral malignancy [Citation25,Citation26].
7. IgG4-related retroperitoneal fibrosis
Retroperitoneal fibrosis/periaortitis is another frequent manifestation of IgG4-RD. Approximately 9.6–27.0% of patients with IgG4-RD have this lesion [Citation2–9]. Abdominal, back, flank, or lumbar pain, lower extremity edema, decreased urinary excretion, low grade fever, appetite loss, and weight loss are representative clinical symptoms [Citation27]. The frequency of hydronephrosis has been noted to be 33–67% [Citation27]. In past reviews of case reports or case series of IgG4-related retroperitoneal fibrosis with hydronephrosis, 25% (7/28) of cases had bilateral hydronephrosis, and 75% had unilateral involvement (left: 15/28, 54%, right: 6/28, 21%) [Citation27]. This result is very similar to previously reported data about idiopathic retroperitoneal fibrosis (bilateral 27.6%; unilateral 72.4%).
8. Differential diagnosis
a. ANCA-associated vasculitis
Histopathologically, ANCA-associated vasculitis (AAV) can accompany plasma cell-rich TIN with many IgG4+ plasma cells [Citation12,Citation14,Citation54,Citation55]. In addition, patients with eosinophilic granulomatosis with polyangiitis (EGPA) [Citation54] or granulomatosis with polyangiitis (GPA) [Citation55] sometimes have elevated serum IgG4 levels. In particular, several reports have shown substantial serum IgG4 level elevation in some cases with EGPA. Moreover, IgG4-RD and EGPA share several clinical, serological, and imaging features including bronchial asthma, allergic rhinitis, chronic sinusitis, eosinophilia, pulmonary infiltrates, and tissue infiltrating eosinophils [Citation54]. These findings indicate that the differential diagnosis between IgG4-RD and EGPA is difficult in some cases with EGPA. However, the swift response to glucocorticoid therapy routinely noted in IgG4-RD may provide a useful clue to differentiate these two diseases.
Vasculitis-related histopathological findings include necrotizing vasculitis, granuloma, and moderate to severe neutrophil infiltration. Therefore, the existence of any of these lesions accompanied by the presence of ANCA indicates the diagnosis of AAV even in the presence of copious IgG4+ PC infiltration [Citation43].
Recently, several cases possibly manifesting some overlap of IgG4-RD and AAV have been reported. Della-Torre et al. experienced a patient with IgG4-RD and positive antiproteinase 3 (PR3) ANCA and concluded that he had concomitant GPA based on histological evidence of AAV in the lung [Citation56]. Danlos et al. analyzed 18 patients fulfilling both ACR and Chapel Hill criteria for AAV and IgG4-RD Comprehensive Diagnostic Criteria and found that two patients had typical features of IgG4-TIN concomitant with one of the pathological characteristic features of AAV in the kidney, neutrophilic infiltrate [Citation57]. One of these cases had AIP, which is not included within the spectrum of AAV, with this finding suggesting some possible overlap between these two diseases. However, whether truly overlapping cases, defined as cases in which all of the major clinical features cannot be explained by either IgG4-RD or AAV alone, exist or not will have to be determined by further investigations.
b. Multicentric Castleman disease
Idiopathic multicentric Castleman disease (MCD) usually affects lymph nodes, lungs, and skin [Citation58–62]. The kidney is also sometimes affected as an extra-nodular organ, and plasma cell-rich TIN with many IgG4+ PCs has been reported [Citation63]. Since serum IgG4 levels are also increased, differentiating this disease from IgG4-TIN is challenging. Although not all patients with MCD and plasma cell-rich TIN have many IgG4+ PC infiltrates, histopathological findings are very similar in the subgroup of patients with many IgG4+ PCs [Citation63]. Interestingly, fibrosis is usually absent or very mild, and accompanying storiform fibrosis has not been reported.
In contrast to the histopathological similarity, the clinical and serological features differ widely between these two diseases. Clinically, reflecting the continuous systemic inflammation, elevated C-reactive protein (CRP), hypoalbuminemia, anemia, and elevated serum IgA levels are distinctive features of MCD from IgG4-RD [Citation29]. The suboptimal response to glucocorticoid therapy is also an important feature of MCD [Citation63]. Generally, the addition of tocilizumab or other immunosuppressants is required to induce remission in patients with MCD.
c. Other disease (imaging, histopathological)
Several diseases show imaging features that are very similar to those of IgG4-TIN. These include malignant lymphoma, renal infarction, chronic pyelonephritis, GPA, polyarteritis nodosa, and urinary tract carcinomas [Citation11,Citation64]. However, the differential diagnosis is relatively easy in most cases based on careful clinicopathological correlation.
Plasma cell-rich TIN is an important extraglandular manifestation of Sjögren’s syndrome. However, IgG4+ PC infiltrates are absent or very few in Sjögren’s syndrome with TIN [Citation65]. Moreover, SSA/Ro is usually negative in IgG4-TIN. Therefore, these two diseases can be easily differentiated.
9. Treatment
Glucocorticoid is the first line drug for the treatment of IgG4-RD [Citation65]. The 30–40 mg/day or 0.6 mg/kg/day of prednisolone is usually employed as a starting dose. A good response to steroid is a common feature of IgG4-RD, and may be useful in supporting the diagnosis in some cases [Citation66–68]. Regarding the mechanism of the good response to steroid seen in IgG4-RD, Iguchi et al. analyzed glucocorticoid receptor in the affected organs and found that strong and abundant expression of glucocorticoid receptor was seen in fibro/myofibroblasts, CD4-positive T cells and IgG4-positive plasma cells in the kidney of IgG4-RD but rarely or only faintly in the kidney of Sjögren’s syndrome [Citation69]. Of even greater significance is a poor or negative response, which may raise doubts regarding the diagnosis of IgG4-RD. However, complete recovery of decreased renal function is not always expected. Saeki et al. conducted a long-term follow-up study with steroid therapy. They followed 34 patients with steroid treatment and found that patients with an eGFR before therapy of more than or equal to 60 showed continuously no changed average eGFR throughout the observation period [Citation33]. In contrast, in patients with decreased renal function before therapy (eGFR less than 60), eGFR immediately recovered one month after starting steroid therapy, but recovered renal function was maintained without further improvement until the final visit suggesting that only partial recovery of renal function is obtained in such patients even after swiftly starting steroid therapy [Citation33]. To minimize the adverse effects of steroid, the addition of an immunosuppressant such as azathioprine is recommended [Citation66].
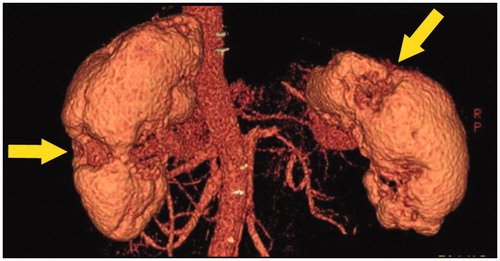
An imaging study revealed the reason why only partial recovery was obtained in patients with already decreased renal function before therapy. Mizushima et al. analyzed 23 patients with IgG4-RKD with glucocorticoid treatment for more than 24 months, and found that 11 of them developed renal cortical atrophy () 24 months after the start of steroid therapy [Citation70]. The risk factors for the development were pretreatment renal insufficiency and serum IgE levels. The imaging finding of partial renal atrophy seems to be the renal damage histopathologically corresponding to diffuse fibrosis. Therefore, the results of this study suggest that to prevent residual renal damage, steroid administration should be initiated immediately in patients with IgG4-TIN.
Figure 7. Three-dimensional CT findings. Multiple partially severely atrophied areas (arrows) were seen despite relatively preserved renal function.
In some cases, with IgG4-RD without renal involvement, renal parenchymal lesions newly developed as a recurrence of the disease during the maintenance steroid therapy [Citation71]. Since kidney is one of the most frequently affected organs of this diseases, annual imaging follow-up is necessary even if kidney is not involved at the first diagnosis.
To detect recurrence or newly developed lesions in the kidney, NAG, and β2 microglobulin (β2MG) have been thought to be promising biomarkers [Citation72]. However, previous reports showed that such urinary biomarkers are not necessarily elevated in IgG4-TIN patients [Citation71]. In addition, Mizushima et al. documented that urine NAG and β2MG concentrations fluctuate despite successful corticosteroid therapy [Citation67]. Thus, these urinary markers seem not to be useful for the follow-up of IgG4-RKD.
To avoid corticosteroid toxicity, rituximab is an alternative choice with promising accumulating data [66]. Carruthers et al. treated 30 IgG4-RD patients including 7 patients with renal lesion with two doses of rituximab (1000 mg each) prospectively and found that 97% of them responded well to rituximab [Citation73]. However, Wallace et al analyzed 60 patients with IgG4-RD treated with rituximab retrospectively and realized that 21 patients (37%) experienced relapse [Citation74]. In addition, to the best of our knowledge, only one case report has described the successful treatment of IgG4-TIN with rituximab [Citation75].
Therefore, maintenance therapy is very important for the treatment strategy for IgG4-RD in either standard corticosteroid-based therapy or rituximab-based one.
10. Conclusion
High frequency of recurrence is a very important feature of IgG4-RD. Since serum IgG4 is not an ideal biomarker to monitor the recurrence of IgG4-RD, further studies to identify more useful biomarkers are urgently needed to more rationally manage the treatment of IgG4-RD. Serum complement level is one candidate for IgG4-TIN, but is not sufficient by itself. Future analyses of pathogenesis will be needed to more precisely define the optimal therapeutic strategies for the various subsets of Ig4-RD patients.
Conflict of interest
None.
Acknowledgments
The authors thank Mr. John Gelblum for his critical reading of the manuscript.
Additional information
Funding
References
- Nakashima H, Kawano M, Saeki T, Ubara Y, Hisano S, Nagata M, et al. Estimation of the number of histological diagnosis for IgG4-related kidney disease referred to the data obtained from the Japan Renal Biopsy Registry (J-RBR) questionnaire and cases reported in the Japanese Society of Nephrology Meetings. Clin Exp Nephrol. 2017;21(1):97–103.
- Yamada K, Yamamoto M, Saeki T, Mizushima I, Matsui S, Fujisawa Y, et al. New clues to the nature of immunoglobulin G4-related disease: a retrospective Japanese multicenter study of baseline clinical features of 334 cases. Arthritis Res Ther. 2017;19:262.
- Inoue D, Yoshida K, Yoneda N, Ozaki K, Matsubara T, Nagai K, et al. IgG4-related disease: dataset of 235 consecutive patients. Medicine (Baltimore). 2015;94(15):e680.
- Chen Y, Zhao JZ, Feng RE, Shi JH, Li XM, Fei YY, et al. Types of organ involvement in patients with immunoglobulin G4-related disease. Chin Med J. 2016;129(13):1525–32.
- Sekiguchi H, Horie R, Kanai M, Suzuki R, Yi ES, Ryu JH. IgG4-related disease: retrospective analysis of one hundred sixty-six patients. Arthritis Rheumatol. 2016;68(9):2290–9.
- Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, Pillai S, Stone JH. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67(9):2466–75.
- Lin W, Lu S, Chen H, Wu Q, Fei Y, Li M, et al. Clinical characteristics of immunoglobulin G4-related disease: a prospective study of 118 Chinese patients. Rheumatology (Oxford). 2015;54(11):1982–90.
- Zen Y, Nakanuma Y. IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34(12):1812–9.
- Fernández-Codina A, Martínez-Valle F, Pinilla B, López C, DeTorres I, Solans-Laqué R, et al. IgG4-related disease: results from a multicenter Spanish Registry. Medicine (Baltimore). 2015;94(32):e1275.
- Saeki T, Nishi S, Imai N, Ito T, Yamazaki H, Kawano M, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010;78(10):1016–23.
- Kawano M, Saeki T, Nakashima H, Nishi S, Yamaguchi Y, Hisano S, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15(5):615–26.
- Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC, Takahashi N, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011;22(7):1343–52.
- Yamaguchi Y, Kanetsuna Y, Honda K, Yamanaka N, Kawano M, Nagata M, et al. Characteristic tubulointerstitial nephritis in IgG4-related disease. Hum Pathol. 2012;43(4):536–49.
- Kawano M, Mizushima I, Yamaguchi Y, Imai N, Nakashima H, Nishi S, et al. Immunohistochemical characteristics of IgG4-related tubulointerstitial nephritis: detailed analysis of 20 Japanese cases. Int J Rheumatol. 2012;2012:1.
- Kawano M, Saeki T. IgG4-related kidney disease-an update. Curr Opin Nephrol Hypertens. 2015;24(2):193–201.
- Morimoto J, Hasegawa Y, Fukushima H, Uesugi N, Hisano S, Saito T, et al. Membranoproliferative glomerulonephritis-like glomerular disease and concurrent tubulointerstitial nephritis complicating IgG4-related autoimmune pancreatitis. Intern Med. 2009;48(3):157–62.
- Saeki T, Imai N, Ito T, Yamazaki H, Nishi S. Membranous nephropathy associated with IgG4-related systemic disease and without autoimmune pancreatitis. Clin Nephrol. 2009;71(2):173–8.
- Katano K, Hayatsu Y, Matsuda T, Miyazaki R, Yamada K, Kawano M, et al. Endocapillary proliferative glomerulonephritis with crescent formation and concurrent tubulointerstitial nephritis complicating retroperitoneal fibrosis with a high serum level of IgG4. Clin Nephrol. 2007;68(11):308–14.
- Tamai R, Hasegawa Y, Hisano S, Miyake K, Nakashima H, Saito T. A case of IgG4-related tubulointerstitial nephritis concurrent with Henoch-Schönlein purpura nephritis. Allergy Asthma Clin Immunol. 2011;7(1):5.
- Ito K, Yamada K, Mizushima I, Aizu M, Fujii H, Mizutomi K, et al. Henoch-Schönlein purpura nephritis in a patient with IgG4-related disease: a possible association. Clin Nephrol. 2013;79(03):246–52. Mar
- Okuyama Y, Uchida HA, Tenta M, Nunoue T, Umebayashi R, Morinaga H, et al. Autoimmune pancreatitis and minimal change nephrotic syndrome: an unusual association?. Nephrology (Carlton). 2015;20(3):225–6.
- Yamada K, Zoshima T, Ito K, Mizushima I, Hara S, Horita S, et al. A case developing minimal change disease during the course of IgG4-related disease. Mod Rheumatol. 2017;27(4):712–5.
- Alexander MP, Larsen CP, Gibson IW, Nasr SH, Sethi S, Fidler ME, et al. Membranous glomerulonephritis is a manifestation of IgG4-related disease. Kidney Int. 2013;83(3):455–62.
- Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64(10):3061–7.
- Kim SA, Lee SR, Huh J, Shen SS, Ro JY. IgG4-associated inflammatory pseudotumor of ureter: clinicopathologic and immunohistochemical study of 3 cases. Hum Pathol. 2011;42(8):1178–84.
- Marando A, D'Ambrosio G, Catanzaro F, La Rosa S, Sessa F. IgG4-related disease of the ureter: report of two cases and review of the literature. Virchows Arch. 2013;462(6):673–8.
- Mizushima I, Inoue D, Kawano M. Retroperitoneal fibrosis/periaortitis and hydronephrosis. In: Saito T, Stone JH, Nakashima H, Saeki T, Kawano M, editors. IgG4-Related Kidney Disease. Tokyo: Springer; 2016. p. 159–71.
- Saeki T, Kobayashi D, Ito T, Tamura M, Yoshikawa S, Yamazaki H. Comparison of clinical and laboratory features of patients with and without allergic conditions in IgG4-related disease: a single-center experience in Japan. Mod Rheumatol. 2018;28(5):845–8.
- Sato Y, Kojima M, Takata K, Morito T, Mizobuchi K, Tanaka T, et al. Multicentric Castleman’s disease with abundant IgG4-positive cells: a clinical and pathological analysis of six cases. J Clin Pathol. 2010;63(12):1084–9.
- Muraki T, Hamano H, Ochi Y, Komatsu K, Komiyama Y, Arakura N, et al. Autoimmune pancreatitis and complement activation system. Pancreas 2006;32(1):16–21.
- Sugimoto M, Watanabe H, Asano T, Sato S, Takagi T, Kobayashi H, et al. Possible participation of IgG4 in the activation of complement in IgG4-related disease with hypocomplementemia. Mod Rheumatol. 2016;26(2):251–8.
- Shiokawa M, Kodama Y, Sekiguchi K, Kuwada T, Tomono T, Kuriyama K, et al. Laminin 511 is a target antigen in autoimmune pancreatitis. Sci Transl Med. 2018;10(453):pii–eaaq0997. Aug 8. doi: 10.1126/scitranslmed.aaq0997.
- Saeki T, Kawano M, Mizushima I, Yamamoto M, Wada Y, Nakashima H, et al. The clinical course of patients with IgG4-related kidney disease. Kidney Int. 2013;84(4):826–33.
- Takahashi N, Kawashima A, Fletcher JG, Chari ST. Renal involvement in patients with autoimmune pancreatitis: CT and MR imaging findings. Radiology 2007;242(3):791–801.
- Horita S, Fujii H, Mizushima I, Fujisawa Y, Hara S, Yamada K, et al. A case of IgG4-related tubulointerstitial nephritis and membranous glomerulonephritis during the clinical course of gastric cancer: imaging features of IgG4-related kidney disease. Mod Rheumatol. 2016;27:1–5. [Epub ahead of print]
- Inoue D, Kawano M, Yamada K. Kidney and urinary tract lesions. In: Umehara H, Okazaki K, Stone JH, Kawa S, Kawano M, editors. IgG4-Related Disease. Tokyo: Springer Japan; 2014. p. 99–105.
- Inoue K, Okubo T, Kato T, Shimamura K, Sugita T, Kubota M, et al. IgG4-related stomach muscle lesion with a renal pseudotumor and multiple renal rim-like lesions: a rare manifestation of IgG4-related disease. Mod Rheumatol. 2018;28(1):188–92.
- Kawano M, Yamada K. IgG4-related kidney disease and IgG4-related retroperitoneal fibrosis. Semin Liver Dis. 2016;36:283–90.
- Cho YJ, Jung WY, Lee SY, Song JS, Park HJ. Perirenal capsule and scrotal involvement in immunoglobulin G4-related kidney disease: case-based review. Rheumatol Int. 2018;38(10):1941–8.
- Bianchi D, Topazio L, Gaziev G, Iacovelli V, Bove P, Mauriello A, et al. IgG4-related kidney disease: report of a case presenting as a renal mass. Case Rep Surg. 2017;2017:9690218.
- Shoji S, Nakano M, Usui Y. IgG4-related inflammatory pseudotumor of the kidney. Int J Urol. 2010;17(4):389–90.
- Cai YI, Li HZ, Zhang YS. IgG4-related inflammatory pseudotumor of the kidney mimicking renal cell carcinoma: a case report. Oncol Lett. 2016;11(5):3438–40.
- Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181–92.
- Yoshita K, Kawano M, Mizushima I, Hara S, Ito Y, Imai N, et al. Light-microscopic characteristics of IgG4-related tubulointerstitial nephritis: distinction from non-IgG4-related tubulointerstitial nephritis. Nephrol Dial Transplant. 2012;27(7):2755–61.
- Khosroshahi A, Ayalon R, Beck LH Jr, Salant DJ, Bloch DB, Stone JH. IgG4-related disease is not associated with antibody to the phospholipase A2 receptor. Int J Rheumatol. 2012;2012:139409.
- Miyata KN, Kihira H, Haneda M, Nishio Y. IgG4-related tubulointerstitial nephritis associated with membranous nephropathy in two patients: remission after administering a combination of steroid and mizoribine. Case Rep Nephrol. 2014;2014:678538.
- Zen Y, Fujii T, Harada K, Kawano M, Yamada K, Takahira M, et al. Th2 and regulatory immune reactions are increased in immunoglobin G4-related sclerosing pancreatitis and cholangitis. Hepatology. 2007;45(6):1538–46.
- Hara S, Kawano M, Mizushima I, Harada K, Takata T, Saeki T, et al. Distribution and components of interstitial inflammation and fibrosis in IgG4-related kidney disease: analysis of autopsy specimens. Hum Pathol. 2016;55:164–73.
- Sharma SG, Vlase HL, D'Agati VD. IgG4-related tubulointerstitial nephritis with plasma cell-rich renal arteritis. Am J Kidney Dis. 2013;61(4):638–43.
- Notohara K, Kamisawa T, Uchida K, Zen Y, Kawano M, Kasashima S, et al. Gastrointestinal manifestation of immunoglobulin G4-related disease: clarification through a multicenter survey. J Gastroenterol. 2018;53(7):845–53.
- Fujii M, Sato Y, Ohara N, Hashimoto K, Kobashi H, Koyama Y, et al. Systemic IgG4-related disease with extensive peripheral nerve involvement that progressed from localized IgG4-related lymphadenopathy: an autopsy case. Diagn Pathol. 2014;9(1):41.
- Harada K, Ubara Y, IgG4-related kidney diseases and conditions: renal pelvic and ureteral diseases. In: Saito T, Stone JH, Nakashima H, Saeki T, Kawano M, editors. IgG4-Related Kidney Disease. Tokyo: Springer; 2016. p. 145–57.
- Inenaga J, Ueno T, Kawada M, Imafuku A, Mise K, Sumida K, et al. IgG4-related disease: a mass lesion in the intrarenal sinus near the renal pelvis. Intern Med. 2015;54(15):1897–900.
- Yamamoto M, Takahashi H, Suzuki C, Tabeya T, Ohara M, Naishiro Y, et al. Analysis of serum IgG subclasses in Churg-Strauss syndrome-the meaning of elevated serum levels of IgG4. Intern Med. 2010;49(14):1365–70.
- Chang SY, Keogh KA, Lewis JE, Ryu JH, Cornell LD, Garrity JA, et al. IgG4-positive plasma cells in granulomatosis with polyangiitis (Wegener’s): a clinicopathologic and immunohistochemical study on 43 granulomatosis with polyangiitis and 20 control cases. Hum Pathol. 2013;44(11):2432–7.
- Della-Torre E, Lanzillotta M, Campochiaro C, Bozzalla E, Bozzolo E, Bandiera A, et al. Antineutrophil cytoplasmic antibody positivity in IgG4-related disease: a case report and review of the literature. Medicine (Baltimore). 2016;95(34):e4633.
- Danlos FX, Rossi GM, Blockmans D, Emmi G, Kronbichler A, Durupt S, et al. Antineutrophil cytoplasmic antibody-associated vasculitides and IgG4-related disease: a new overlap syndrome. Autoimmun Rev. 2017;16(10):1036–43.
- van Rhee F, Voorhees P, Dispenzieri A, Fosså A, Srkalovic G, Ide M, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018;132(20):2115–24.
- Otani K, Inoue D, Fujikura K, Komori T, Abe-Suzuki S, Tajiri T, et al. Idiopathic multicentric Castleman’s disease: a clinicopathologic study in comparison with IgG4-related disease. Oncotarget 2018;9:6691–706.
- Xu D, Lv J, Dong Y, Wang S, Su T, Zhou F, et al. Renal involvement in a large cohort of Chinese patients with Castleman disease. Nephrol Dial Transplant. 2012;27(suppl_3):iii119–25.
- Takeuchi M, Sato Y, Takata K, Kobayashi K, Ohno K, Iwaki N, et al. Cutaneous multicentric Castleman’s disease mimicking IgG4-related disease. Pathol Res Pract. 2012;208(12):746–9.
- Ogoshi T, Kido T, Yatera K, Oda K, Kawanami T, Ishimoto H, et al. Assessment of pathologically diagnosed patients with Castleman’s disease associated with diffuse parenchymal lung involvement using the diagnostic criteria for IgG4-related disease. Lung 2013;191(6):575–83.
- Zoshima T, Yamada K, Hara S, Mizushima I, Yamagishi M, Harada K, et al. Multicentric castleman disease with tubulointerstitial nephritis mimicking IgG4-related disease: two case reports. Am J Surg Pathol. 2016;40(4):495–501.
- Ozaki K, Miyayama S, Ushiogi Y, Matsui O. Renal involvement of polyarteritis nodosa: CT and MR findings. Abdom Imaging. 2009;34(2):265–70.
- Saito T, Differential diagnosis (3): Sjogren’s syndrome. In: Saito T, Stone JH, Nakashima H, Saeki T, Kawano M, editors. IgG4-Related Kidney Disease. Tokyo: Springer; 2016. p. 271–277.
- Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015;67(7):1688–99.
- Mizushima I, Yamada K, Fujii H, Inoue D, Umehara H, Yamagishi M, et al. Clinical and histological changes associated with corticosteroid therapy in IgG4-related tubulointerstitial nephritis. Mod Rheumatol. 2012;22(6):859–70.
- Saeki T, Kawano M, Mizushima I, Yamamoto M, Wada Y, Ubara Y, et al. Recovery of renal function after glucocorticoid therapy for IgG4-related kidney disease with renal dysfunction. Clin Exp Nephrol. 2016;20(1):87–93.
- Iguchi T, Takaori K, Mii A, Sato Y, Suzuki Y, Yoshifuji H, et al. Glucocorticoid receptor expression in resident and hematopoietic cells in IgG4-related disease. Mod Pathol. 2018;31(6):890–9.
- Mizushima I, Yamamoto M, Inoue D, Nishi S, Taniguchi Y, Ubara Y, et al. Factors related to renal cortical atrophy development after glucocorticoid therapy in IgG4-related kidney disease: a retrospective multicenter study. Arthritis Res Ther. 2016;18:273.
- Takeji A, Yamada K, Inoue D, Mizushima I, Hara S, Ito K, et al. A case of IgG4-related kidney disease with predominantly unilateral renal atrophy. CEN Case Rep. 2018. doi:10.1007/s13730-018-0355-9. [Epub ahead of print]
- Nishi H, Shibagaki Y, Hirano K, Akahane M, Kido R, Nangaku M, et al. Laboratory and imaging features of kidney involvement in autoimmune pancreatitis: incidence, correlation, and steroid therapy response. Clin Nephrol. 2010;73(4):253–9.
- Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74(6):1171–7.
- Wallace ZS, Mattoo H, Mahajan VS, Kulikova M, Lu L, Deshpande V, et al. Predictors of disease relapse in IgG4-related disease following rituximab. Rheumatology (Oxford). 2016;55(6):1000–8.
- McMahon BA, Novick T, Scheel PJ, Bagnasco S, Atta MG. Rituximab for the treatment of IgG4-related tubulointerstitial nephritis: case report and review of the literature. Medicine (Baltimore). 2015;94(32):e1366.