
Abstract
Objectives: To assess the clinicopathologic features of Multicentric Castleman disease (MCD) patients in Japan.
Methods: We assessed baseline data for 342 Japanese MCD patients with a biopsy-proven diagnosis, enrolled in a prospective, observational study for tocilizumab treatment.
Results: Of 342 patients, 86.0% had plasma-cell type. None had a family history of MCD. Median disease duration of MCD was 3.7 years. Mean body weight and body mass index tended to be lower than those in the general Japanese population. The most common clinical presentations besides lymphadenopathy included fatigue (61.7%), pulmonary involvement (42.7%), and splenomegaly (41.8%). Secondary amyloidosis was reported in 34 patients (9.9%). Laboratory abnormalities included decreased hemoglobin and albumin, and increased acute-phase proteins, serum immunoglobulins, and interleukin-6 (IL-6). IL-6 levels among the MCD patients tested in this study were correlated with levels of albumin, hemoglobin, triglyceride, total cholesterol, C-reactive protein, fibrinogen and immunoglobulin G (Spearman’s correlation coefficient, |r| = 0.28–0.59).
Conclusion: The clinical features and laboratory abnormalities are similar to those previously reported in other countries, besides higher rates of pulmonary involvement, secondary amyloidosis, and ECG abnormalities. Our results imply that IL-6 is involved in MCD pathogenesis. These findings would be informative for diagnosis and appropriate treatment for MCD.
Introduction
Castleman disease (CD) is a heterogeneous group of rare lymphoproliferative disorders [Citation1–3]. Histologic examinations of affected lymph nodes in CD reveal three subtypes: hyaline-vascular type, plasma-cell type, and mixed type [Citation2,Citation3]. CD is clinically classified as unicentric CD (UCD) or multicentric CD (MCD). UCD is confined to a single lymph node chain or area, and is often curable by surgical excision of the mass [Citation1]. In contrast, MCD is characterized by a lymphadenopathy in multiple lymph nodes or multiple chains of lymph nodes [Citation3,Citation4]. MCD is more aggressive and frequently found in patients with human immunodeficiency virus (HIV) infection, in whom MCD has been associated with human herpesvirus 8 (HHV-8) infection and Kaposi’s sarcoma in the United States and Europe [Citation5–8]. MCD with HHV-8 infection is classified as HHV-8-related MCD, while MCD without HHV-8 infection is classified as idiopathic MCD.
Patients with MCD usually have systemic inflammatory manifestations. Symptoms associated with MCD include pyrexia, weight loss, night sweats, anorexia, skin changes, renal failure, hepatosplenomegaly, and pulmonary disease [Citation9–15]. Abnormal laboratory findings associated with MCD include anemia, thrombocytosis (or thrombocytopenia), hypoalbuminemia, hypocholesterolemia, hypergammaglobulinemia, proteinuria, increased acute-phase proteins, and increased interleukin-6 (IL-6) levels [Citation4,Citation11–16]. MCD is often associated with secondary amyloidosis and malignant lymphoma and has a poor prognosis if not treated [Citation4,Citation10–15].
Clinical abnormalities of MCD, as mentioned above, resemble those seen in transgenic mice carrying the human IL-6 genomic gene, for example, splenomegaly, lymph node enlargement, and high concentrations of IL-6 and immunoglobulin (Ig) G [Citation17–19]. Moreover, in clinical studies, a high amount of IL-6 was found in the serum of patients with both MCD and plasma-cell type UCD [Citation20,Citation21]. Clinical improvement and decrease in serum IL-6 were observed in a patient with plasma-cell type UCD following surgical removal of the affected lymph node, but not in a patient with MCD after the surgical removal of one of the abdominal large hyperplastic lymph nodes. Furthermore, a high level of IL-6 was found in the culture supernatant of the resected lymph nodes. These results indicate that IL-6 secreted by the affected lymph nodes is involved in the clinical abnormalities both in MCD and plasma-cell type UCD. In the United States, Canada, and most of Europe, the IL-6 inhibitor siltuximab is approved for the treatment of MCD in patients who are HIV- and HHV-8-negative [Citation22–24]. The humanized, monoclonal, anti-IL-6-receptor antibody tocilizumab is the only therapeutic drug, apart from corticosteroids, that is approved in Japan for the treatment of patients with CD with CD-associated symptoms and laboratory findings, who do not qualify for therapeutic lymphadenectomy. This is usually because they have lymphadenopathy in multiple lymph nodes or multiple chains of lymph nodes. Thus, patients with CD treated by tocilizumab in Japan generally have MCD. Since MCD is a rare disease, the number of patients enrolled in the tocilizumab clinical trial was relatively small [Citation11]. Therefore, patients with MCD who needed tocilizumab treatment were mandatorily enrolled in the tocilizumab postmarketing surveillance (PMS), supervised by the Ministry of Health, Labour and Welfare Japan, to further investigate the safety and effectiveness in the real world. Herein, we report the findings of a Japanese epidemiologic study of MCD using baseline data from the tocilizumab PMS study.
Methods
Among 384 Japanese patients enrolled in the PMS study (UMIN-CTR number, UMIN000023071) from June 2005 to July 2011, 24 patients who previously received tocilizumab in the clinical trial or as compassionate use were excluded from the present study. Patients with misdiagnosed MCD (9 patients; 5 with malignant lymphoma, 2 with another malignancy, 1 with UCD, and 1 with autoimmune disease) and patients without histopathology (10 patients) were also excluded. As a result of these exclusions, we assessed baseline data from 342 patients with MCD with a biopsy-proven diagnosis (). Characteristics of the patients were investigated using a case report form including age, sex, body weight, date of MCD onset, duration of MCD, family history of MCD, pathologic classification, medical history, medication in the 3 months before enrollment including chemotherapy and corticosteroid use, systemic symptoms, electrocardiograph (ECG) abnormalities, pulmonary involvement, hepatosplenomegaly, rash, secondary amyloidosis, and neurologic disorder. In addition to these symptoms, laboratory data were assessed. We also evaluated the number of severe MCD in this study using symptoms and laboratory data because the recently published consensus guideline for the treatment of idiopathic MCD defines severity criteria of idiopathic MCD [Citation25].
Figure 1. Enrollment, disposition, and pathologic classification in patients with multicentric Castleman disease. Twenty-four patients previously received tocilizumab in the clinical trial or compassionate use for tocilizumab. Nine patients with misdiagnosed MCD (5 with malignant lymphoma, 2 with another malignancy, 1 with UCD, and 1 with autoimmune disease). *One patient was excluded for two reasons: one of the five patients with malignant lymphoma had also previously received tocilizumab.
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (www.clinicalstudydatarequest.com).
For further details on Chugai's Data Sharing Policy and how to request access to related clinical study documents, see here (www.chugai-pharm.co.jp/english/profile/rd/ctds_request.html).
Results
Patient disposition
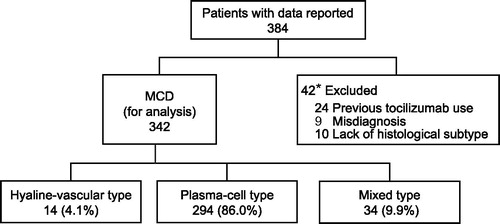
Of 342 patients, 59.1% (202/342) were male. Median age in available data (n = 342) was 48.0 years, and none had a family history of CD (). Median age at onset of MCD (n = 284) was 43.0 years (range, 2.0–82.0 years) (), with peak onset in patients in their 30s and 40s (). Median disease duration in patients with MCD (n = 288) was 3.7 years, and disease duration in 41 of 172 male patients (23.8%) and 17 of 116 female patients (14.7%) was over 10 years (; ). The mean height of the patients with MCD was similar to that of the general Japanese population (). The mean body weight (n = 319) and the mean body mass index (BMI) of patients (n = 277) in this study were 58.1 ± 12.1 kg and 21.8 ± 3.5 kg/m2, respectively (). The mean body weight and the mean BMI of the patients with MCD tended to be lower than those in the general Japanese population, except for men in their 20s and women in their 70s (). Combination chemotherapy was used off-label to treat MCD by 7.9% of patients (27/342) in the 3 months before enrollment, and 73.1% (250/342) were taking corticosteroids at a median dose of 15.0 mg/day (0.3 mg/kg/day) in the same period ().
Figure 2. Age at disease onset and duration of disease in patients with multicentric Castleman disease. The distributions of age at disease onset (A) and of disease duration (B) in male (A: n = 170, B: n = 172) and female (A: n = 114, B: n = 116) patients with MCD in available data. Dark gray bars indicate male patients and light gray bars indicate female patients.
Figure 3. Mean body weight, height, and body mass index of patients with multicentric Castleman disease compared with the general Japanese population. Differences in the mean body height (A, B), weight (C, D) and body mass index (E, F) between this study population and the general Japanese population [Citation46]. The bar indicates standard deviation. Statistical analysis has not been performed because of a large difference in the sample size between this study population and the general Japanese population. Dark gray bars indicate the study population and light gray bars indicate the general Japanese population.
![Figure 3. Mean body weight, height, and body mass index of patients with multicentric Castleman disease compared with the general Japanese population. Differences in the mean body height (A, B), weight (C, D) and body mass index (E, F) between this study population and the general Japanese population [Citation46]. The bar indicates standard deviation. Statistical analysis has not been performed because of a large difference in the sample size between this study population and the general Japanese population. Dark gray bars indicate the study population and light gray bars indicate the general Japanese population.](/cms/asset/cdfaf1b0-dd2d-4a06-8fbb-c98bd9edad64/imor_a_1704983_f0003_b.jpg)
Table 1. Characteristics of the patients.
Disease characteristics of MCD
Histopathologic features in this study revealed that 4.1% of patients (14/342) had hyaline-vascular type, 86.0% (294/342) had plasma-cell type, and 9.9% (34/342) had mixed type (). Regardless of their histopathologic features, all 342 patients had at least one symptom or one laboratory abnormality related to MCD. According to severity criteria published in the treatment guideline, MCD was severe in 108 of 342 patients (31.6%) () [Citation25].
Lymphadenopathy was observed in all the patients in this study (). In total, 98.2% of patients (336/342) had at least one clinical feature besides lymphadenopathy (). Clinical features observed in more than 2% of patients in this study are shown in . Clinical features were divided into six categories: liver disorder (108/342; 31.6%), kidney and urological disorder (113/342; 33.0%), heart disease (63/342; 18.4%), respiratory disorder (168/342; 49.1%), infection (34/342; 9.9%), and others (326/342; 95.3%). Across all six categories, commonly reported clinical features in order of descending frequency were fatigue (defined according to Eastern Cooperative Oncology Group performance status ≥1 [211/342; 61.7%] and ≥2 [70/342; 21.0%]), pulmonary involvement (146/342; 42.7%), splenomegaly (143/342; 41.8%), hepatomegaly (91/342; 26.6%), rash (80/342; 23.4%), hypertension (73/342; 21.3%), pyrexia (59/342; 17.3%), ECG abnormalities (53/342; 15.5%), diabetes mellitus (53/342; 15.5%), decreased appetite (52/342; 15.2%), night sweats (43/342; 12.6%), and hyperuricemia (37/342; 10.8%). Ascites (7/342; 2.0%), pleural effusion (9/342; 2.6%), and edema (5/342; 1.5%, data not shown in ) were observed as fluid accumulation. Secondary amyloidosis was reported in 34 patients (9.9%). Regarding infection, HIV testing in 160 patients revealed that two patients had concurrent HIV infection. HHV-8 tests in 83 patients including the two HIV-positive patients were all negative.
Table 2. Clinical features (≥2.0%).
Median (range) laboratory test values with available data were as follows: albumin, 2.5 g/dL (0.5–4.3); hemoglobin, 9.0 g/dL (4.5–15.6); triglycerides, 61.0 mg/dL (22.0–293.0); total cholesterol, 117.0 mg/dL (51.0–320.0); C-reactive protein, 67.8 mg/L (0.0–375.5); fibrinogen, 575.5 mg/dL (1.5–1181.0); IgG, 4572.0 mg/dL (523.0–10070.0), IgA, 531.0 mg/dL (25.0–1741.0), and IgM, 241.5 mg/dL (32.0–2621.0); platelets, 324.0 × 103/µL (5.0 × 103–1117.0 × 103); estimated glomerular filtration rate-normalized body surface area (eGFR-NOR), 76.0 mL/min/1.73 m2 (4.3–823.3); IL-6, 20.1 pg/mL (2.8–326.0) (). IL-6 levels among the patients with MCD tested in this study were correlated with levels of albumin (r = −0.59), hemoglobin (r = −0.59), triglyceride (r = −0.38), total cholesterol (r = −0.29), C-reactive protein (r = 0.58), fibrinogen (r = 0.28), and IgG (r = 0.29) (; ). On the other hand, the correlation coefficient of IgA, IgM, platelets and eGFR-NOR with IL-6 were 0.17, −0.20, 0.03, and −0.06, respectively (; ).
Figure 4. Results of laboratory tests in patients with multicentric Castleman disease: correlation with IL-6. Laboratory tests included albumin (Alb; A), hemoglobin (Hb; B), triglycerides (TG; C), total cholesterol (T-cho; D), C-reactive protein (CRP; E), fibrinogen (Fib; F), immunoglobulin G (IgG; G), immunoglobulin A (IgA; H); immunoglobulin M (IgM; I), platelets (PLT; J), estimated glomerular filtration rate-normalized body surface area (eGFR-NOR; K). r indicates the Spearman’s product-moment correlation coefficient.
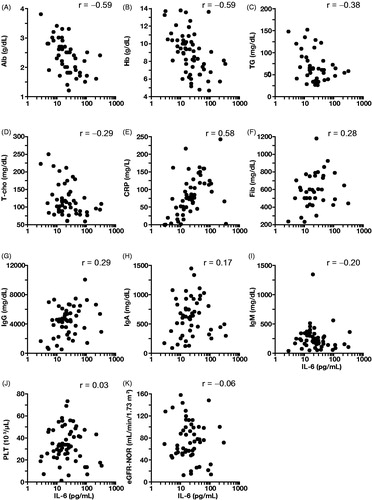
Table 3. Laboratory tests associated with multicentric Castleman disease.
Table 4. Laboratory tests: Spearman’s correlation coefficient with IL-6.
Discussion
Although international, evidence-based consensus diagnostic criteria for HHV-8 negative/idiopathic MCD were published in 2017 [Citation14], more investigations are necessary for criteria for MCD diagnosis due to lack of large epidemiologic data. This study assessed the clinicopathologic characteristics of 342 patients with MCD in Japan.
MCD affects people of all ages, with peak frequency during adulthood. The median age of onset in this study was 43.0 years (), which is also consistent with the median age of 38 (n = 28) and 46 years (n = 21) in previous studies in Japan [Citation11,Citation12]. A study in the United States has also reported the median age as 55 years (n = 59) [Citation8]. There were no patients with a family history of MCD in the present study. A genetic predisposition to MCD has not been reported so far, which is consistent with this result. Median duration of MCD in this study was 3.7 years, and 59.0%, 40.6%, and 20.1% of patients survived for more than 3, 5, and 10 years, respectively. Although the 5-year overall survival rate for MCD was 65% in the previous study and MCD has a relatively poor prognosis, some patients might be able to survive longer [Citation26]. In this study, 31.6% of MCD cases with available data were defined as severe () [Citation25]. The survival of these severe MCD cases needs to be followed up.
Our data were similar to the data previously reported in some parts but different in other parts. Firstly, in the limited number of patients that were tested for HIV and HHV-8 infection in the present study, only 2 of 160 were HIV-positive, and all 83 tested patients were negative for HHV-8, including two HIV-positive patients. On the other hand, it has been noted that MCD frequently occurs in patients with HIV in the United States and Europe, in whom MCD is associated with HHV-8 infection and Kaposi’s sarcoma, and these patients have a poor prognosis [Citation5–8]. The small proportion of HIV-positive patients with MCD in this study may be due to there being fewer patients with HIV infection in Japan than in other countries [Citation12,Citation27]. This suggests that there is a distinct regional difference in the causes of MCD in Japanese patients compared with patients from other countries, with most Japanese patients having idiopathic MCD. Second, pulmonary involvement was present in 42.7% of patients while there are few reports of pulmonary involvement in patients with MCD in other countries. Pulmonary lesions in patients with MCD are mainly lymphoproliferative [Citation11,Citation28,Citation29]. Common computed tomography findings are as bilateral hilar and mediastinal lymphadenopathy, ground-glass attenuation, air-space consolidation, and indistinct nodules and cysts [Citation28]. Furthermore, Johkoh et al. reported that substantial thickening of the bronchovascular bundles and interlobular septa is often found in MCD [Citation28]. In the past, lymphocytic interstitial pneumonia (LIP) was regarded as a histologic variant of diffuse pulmonary lymphoid hyperplasia but was subsequently moved to a category of rare idiopathic interstitial pneumonias [Citation30–33]. Therefore, the use of the term LIP has been very limited in the literature. Even though LIP has been rarely reported in Western countries, this study confirmed that pulmonary involvement is high in the Japanese MCD population, having also been observed in previous studies in 18 of 28 patients [Citation11], 7 of 21 patients [Citation12] and 5 of 7 patients [Citation34]. Recently complications of lung involvement were also reported in 13 of 27 idiopathic MCD patients (48%) in France [Citation35], which indicates that pulmonary involvement may also be seen outside of Japan if tested at diagnosis of MCD. Third, 9.9% of patients had secondary amyloidosis, most commonly renal in this study (). Secondary amyloidosis occurs in chronic inflammatory diseases such as vasculitis syndrome, [Citation36] in which the overexpression of IL-6 leads to overproduction of serum amyloid A by the liver. Secondary amyloidosis is not frequently reported in patients with MCD in countries other than Japan, but our results indicate that secondary amyloidosis may be one of the signature symptoms of MCD caused by overproduction of IL-6, although there is no significant relationship between serum IL-6 levels and amyloidosis. (Supplemental Figure). This is because (1) the number of samples for IL-6 measurement in this study was limited; (2) the severity of organ involvement was not evaluated in this study; (3) the serum IL-6 levels varied widely among patients and did not display a normal distribution (Gaussian distribution) [Citation37]; and (4) the serum samples were collected at only a single time point, although serum IL-6 levels vary in each individual. The fact that incidence of both pulmonary involvement and secondary amyloidosis is higher in Japan than in other countries may indicate that Japanese patients are more susceptible to pulmonary involvement and secondary amyloidosis, which could point to genetic factors [Citation38–41]. This result could imply a regional difference, but pulmonary involvement and secondary amyloidosis should also be carefully considered outside of Japan. Fourth, ECG abnormalities observed in 15.5% of patients in our study could be attributed to an underlying heart condition (); however concomitant heart conditions have been rarely reported [Citation42]. There are reports of patients who developed CD-associated cardiac dysfunction as a result of heart amyloidosis and pulmonary arterial hypertension [Citation43,Citation44]. Kawabata and colleagues observed that heart failure was observed in 14% of Japanese patients with MCD [Citation12]. Therefore, performing ECG should be considered at diagnosis of MCD regardless of whether clinical symptoms are present. With the exception of pulmonary involvement, secondary amyloidosis, and ECG abnormalities, the clinical features of the 342 patients with MCD in this study were consistent with MCD clinical features and laboratory abnormalities in previous reports from countries other than Japan [Citation13,Citation45].
Most of the clinical features and laboratory test abnormalities might be caused by overproduction of IL-6, a multifunctional cytokine. Overproduction of IL-6 may be responsible for the severe nutritional status observed in patients with MCD and may be involved in wasting syndrome and chronic inflammatory diseases [Citation11]. In this study, the body weight and BMI of patients with MCD of both sexes tended to be lower than the general population of Japan (), which is consistent with previous reports of patients with MCD [Citation11,Citation12,Citation14]. The mean and median serum IL-6 levels were 39.0 ± 61.4 pg/mL and 20.1 pg/mL, respectively (range, 2.8–326.0 pg/mL) (), although IL-6 data were not always available for all patients in this study. Serum IL-6 levels in this study were correlated with the levels of albumin, hemoglobin, C-reactive protein, triglyceride, total cholesterol, fibrinogen, and IgG (; ). The mean serum IL-6 level was consistent with that of 28 patients with MCD before administration of tocilizumab (24.5 ± 6.5 pg/mL) in the previous study [Citation37]. These values were beyond the normal range (the normal upper limit of serum IL-6 is 4.0 pg/mL) but were not very high even compared with that of patients with rheumatoid arthritis (58.4 ± 13.8 pg/mL) [Citation37]. We reported, however, that serum IL-6 in 28 patients with MCD from the previous study showed a greater increase at day 14 of tocilizumab treatment by blocking IL-6 receptor-mediated consumption of IL-6 (24.5 ± 6.5 pg/mL at baseline, 564.8 ± 127.6 pg/mL at day 14). This increased serum IL-6 level after the introduction of tocilizumab represents the actual endogenous IL-6 production and true disease activity of patients with MCD [Citation37]; therefore, the correlation between the laboratory abnormal values and the increased serum IL-6 levels by tocilizumab treatment would be much stronger than those of the present study. Measurement of IL-6 for differential diagnosis of MCD remains underutilized because it is not listed among the current diagnostic criteria, including those by the National Cancer Institute and Fajgenbaum et al. [Citation14]. However, in this respect, it should be noted that high IL-6 levels that cannot be otherwise explained may be one of the potential diagnostic criteria for MCD.
Although our study population is a good representation of patients with MCD, some limitations should be noted. First, data were obtained at the start of tocilizumab treatment rather than at diagnosis of MCD. Therefore, this study enrolled both untreated patients (27%) and patients whose symptoms were not controlled by other treatments such as corticosteroid therapy (73%). Second, pathologic diagnosis for MCD was not conducted at a central facility because this was an observational study. Third, although the proportion may be low, the MCD population in this study may include patients with HHV8-related MCD because not all patients were tested for HIV or HHV-8 infection.
In conclusion, the majority of clinical features and laboratory test abnormalities in patients with MCD are considered to be related to IL-6 overexpression, and basic information for differential diagnostic criteria of MCD might be identified from the clinicopathologic characteristics of patients in this study. Most of our findings for clinical features in patients with MCD were consistent with known disease characteristics, but differences in symptoms such as the presence of pulmonary involvement, secondary amyloidosis, ECG abnormalities and rates of HIV and HHV-8 infection, were revealed between Japanese patients and those of other countries. Further investigation is needed to confirm these differences. The results of this study provide empirical evidence on the presentation of MCD. Comparing the results of our study with other case reports or small-scale studies can provide a better understanding of the pathophysiology of MCD, enabling faster, more efficient diagnosis and effective treatment.
Authorship
All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. NN contributed study concept. TJ, SH, SO, M Mizuki, SN and NN contributed study design and protocol. TJ, SH, SO, M Mizuki, SN and NN contributed data collection and enrollment. M Murakami, TJ, SH, SO, M Mizuki, SN, MT, KK, AN and NN analyzed or interpreted data. M Murakami, MT, KK, AN and NN drafted the manuscript; All authors revised the manuscript.
Conflict of interest
M. Murakami has received personal fees from Chugai Pharmaceutical, grants from Chugai Pharmaceutical, AYUMI Pharmaceutical, and Sekisui medical, grants and personal fees from Eisai, grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan. TJ has nothing to disclose. SH has received personal fees from Chugai pharmaceutical. SO has received speakers bureau fees, or consulting fees from Chugai, Bristol-Myers Squibb, AbbVie, Astellas, Takeda, Tanabe-Mitsubishi, Daiichi-Sankyo and Janssen. M Mizuki has received personal fees from Chugai Pharmaceutical, and Takeda. SN has nothing to disclose. MT, KK, and AN are employees of Chugai Pharmaceutical. NN has received grants and personal fees from Chugai Pharmaceutical, Eisai, Sekisui medical and AYUMI Pharmaceutical; personal fees from Mitsubishi Tanabe, Pfizer, AbbVie, UCB Japan, Novartis, and grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Supplemental Material
Download MS Word (13.1 KB)Supplemental Material
Download PDF (100.6 KB)Acknowledgments
The authors thank all our patients, their families, and the physicians who participated in this study. The authors thank the late Dr. Junichi Azuma for his influential leadership in the Safety Evaluation Committee of tocilizumab for Castleman disease. The authors thank Dr. Misato Hashizume for drafting the manuscript. The academic authors vouch for the completeness and accuracy of the data and data analyses, and for the fidelity of the study to the protocol.
References
- Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer. 1956;9(4):822–30.
- Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972;29(3):670–83.
- Gaba AR, Stein RS, Sweet DL, Variakojis D. Multicentric giant lymph node hyperplasia. Am J Clin Pathol. 1978;69(1):86–90.
- Peterson BA, Frizzera G. Multicentric Castleman's disease. Semin Oncol. 1993;20(6):636–47.
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood. 1995;86(4):1276–80.
- Oksenhendler E. HIV-associated multicentric Castleman disease. Curr Opin HIV AIDS. 2009;4(1):16–21.
- Reddy D, Mitsuyasu R. HIV-associated multicentric Castleman disease. Curr Opin Oncol. 2011;23(5):475–81.
- Robinson D Jr, Reynolds M, Casper C, Dispenzieri A, Vermeulen J, Payne K, et al. Clinical epidemiology and treatment patterns of patients with multicentric Castleman disease: results from two US treatment centres. Br J Haematol. 2014;165(1):39–48.
- Frizzera G, Banks PM, Massarelli G, Rosai J. A systemic lymphoproliferative disorder with morphologic features of Castleman's disease. Pathological findings in 15 patients. Am J Surg Pathol. 1983;7(3):211–31.
- Frizzera G, Peterson BA, Bayrd ED, Goldman A. A systemic lymphoproliferative disorder with morphologic features of Castleman's disease: clinical findings and clinicopathologic correlations in 15 patients. J Clin Oncol. 1985;3(9):1202–16.
- Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627–32.
- Kawabata H, Kadowaki N, Nishikori M, Kitawaki T, Kondo T, Ishikawa T, et al. Clinical features and treatment of multicentric castleman's disease: a retrospective study of 21 Japanese patients at a single institute. J Clin Exp Hematopathol. 2013;53(1):69–77.
- Liu AY, Nabel CS, Finkelman BS, Ruth JR, Kurzrock R, van Rhee F, et al. Idiopathic multicentric Castleman's disease: a systematic literature review. Lancet Haematol. 2016;3(4):e163–75.
- Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646–57.
- Fujimoto S, Koga T, Kawakami A, Kawabata H, Okamoto S, Mizuki M, et al. Tentative diagnostic criteria and disease severity classification for Castleman disease: a report of the research group on Castleman disease in Japan. Modern Rheumatol. 2018;28(1):161–7.
- Soumerai JD, Sohani AR, Abramson JS. Diagnosis and management of Castleman disease. Cancer Control. 2014;21(4):266–78.
- Suematsu S, Matsuda T, Aozasa K, Akira S, Nakano N, Ohno S, et al. IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc Natl Acad Sci U S A. 1989;86(19):7547–51.
- Nishimoto N, Kishimoto T, Yoshizaki K. Anti-interleukin 6 receptor antibody treatment in rheumatic disease. Ann Rheumatic Dis. 2000;59(90001): i21–7.
- Katsume A, Saito H, Yamada Y, Yorozu K, Ueda O, Akamatsu K, et al. Anti-interleukin 6 (IL-6) receptor antibody suppresses Castleman's disease like symptoms emerged in IL-6 transgenic mice. Cytokine. 2002;20(6):304–11.
- Yoshizaki K, Matsuda T, Nishimoto N, Kuritani T, Taeho L, Aozasa K, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman's disease. Blood. 1989;74(4):1360–7.
- Beck JT, Hsu SM, Wijdenes J, Bataille R, Klein B, Vesole D, et al. Brief report: alleviation of systemic manifestations of Castleman's disease by monoclonal anti-interleukin-6 antibody. N Engl J Med. 1994;330(9):602–5.
- US Food and Drug Administration. FDA approves Sylvant for rare Castleman's disease. 2014. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125496Orig1s000TOC.cfm.
- Product monograph for SYLVANT. Health Canada; 2014. Available from: https://hpr-rps.hres.ca/reg-content/summary-basis-decision-detailTwo.php?linkID=SBD00193.
- European Commission. Community register of medicinal products for human use: SYLVANT authorisation. 2014. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/sylvant.
- van Rhee F, Voorhees P, Dispenzieri A, Fossa A, Srkalovic G, Ide M, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018;132(20):2115–24.
- Dispenzieri A, Armitage JO, Loe MJ, Geyer SM, Allred J, Camoriano JK, et al. The clinical spectrum of Castleman's disease. Am J Hematol. 2012;87(11):997–1002.
- Masaki Y, Nakajima A, Iwao H, Kurose N, Sato T, Nakamura T, et al. Japanese variant of multicentric castleman's disease associated with serositis and thrombocytopenia–a report of two cases: is TAFRO syndrome (Castleman- Kojima disease) a distinct clinicopathological entity? J Clin Exp Hematopathol. 2013;53(1):79–85.
- Johkoh T, Muller NL, Ichikado K, Nishimoto N, Yoshizaki K, Honda O, et al. Intrathoracic multicentric Castleman disease: CT findings in 12 patients. Radiology. 1998;209(2):477–81.
- Higuchi T, Nakanishi T, Takada K, Matsumoto M, Okada M, Horikoshi H, et al. A case of multicentric Castleman's disease having lung lesion successfully treated with humanized anti-interleukin-6 receptor antibody, tocilizumab. J Korean Med Sci. 2010;25(9):1364–7.
- American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161(2 Pt 1):646–64.
- American Thoracic S, European Respiratory S. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165(2):277–304.
- Travis WD, Costabel U, Hansell DM, King TE, Jr., Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
- Johkoh T, Fukuoka J, Tanaka T. Rare idiopathic intestinal pneumonias (IIPs) and histologic patterns in new ATS/ERS multidisciplinary classification of the IIPs. Eur J Radiol. 2015;84(3):542–6.
- Nishimoto N, Sasai M, Shima Y, Nakagawa M, Matsumoto T, Shirai T, et al. Improvement in Castleman's disease by humanized anti-interleukin-6 receptor antibody therapy. Blood. 2000;95(1):56–61.
- Oksenhendler E, Boutboul D, Fajgenbaum D, Mirouse A, Fieschi C, Malphettes M, et al. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br J Haematol. 2018;180(2):206–16.
- Scarpioni R, Ricardi M, Albertazzi V. Secondary amyloidosis in autoinflammatory diseases and the role of inflammation in renal damage. World J Nephrol. 2016;5(1):66–75.
- Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood. 2008;112(10):3959–64.
- Natsuizaka M, Chiba H, Kuronuma K, Otsuka M, Kudo K, Mori M, et al. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med. 2014;190(7):773–9.
- Furukawa H, Oka S, Shimada K, Tsuchiya N, Tohma S. Genetics of interstitial lung disease: Vol de Nuit (Night Flight). Clin Med Insights Circ Respir Pulm Med. 2015;9(Suppl 1):1–7.
- Baba S, Masago SA, Takahashi T, Kasama T, Sugimura H, Tsugane S, et al. A novel allelic variant of serum amyloid A, SAA1 gamma: genomic evidence, evolution, frequency, and implication as a risk factor for reactive systemic AA-amyloidosis. Hum Mol Genet. 1995;4(6):1083–7.
- Moriguchi M, Terai C, Koseki Y, Uesato M, Nakajima A, Inada S, et al. Influence of genotypes at SAA1 and SAA2 loci on the development and the length of latent period of secondary AA-amyloidosis in patients with rheumatoid arthritis. Hum Genet. 1999;105(4):360–6.
- Man L, Goudar RK. Reversal of cardiomyopathy with tocilizumab in a case of HIV-negative Castleman's disease. Eur J Haematol. 2013;91(3):273–6.
- Kanda J, Kawabata H, Yamaji Y, Ichinohe T, Ishikawa T, Tamura T, et al. Reversible cardiomyopathy associated with Multicentric Castleman disease: successful treatment with tocilizumab, an anti-interleukin 6 receptor antibody. Int J Hematol. 2007;85(3):207–11.
- Arita Y, Sakata Y, Sudo T, Maeda T, Matsuoka K, Tamai K, et al. The efficacy of tocilizumab in a patient with pulmonary arterial hypertension associated with Castleman's disease. Heart Vessels. 2010;25(5):444–7.
- Talat N, Schulte KM. Castleman's disease: systematic analysis of 416 patients from the literature. Oncologist. 2011;16(9):1316–24.
- Ministry of Health, Labour and Welfare. Japanese Government Statistics. Trends in mean height, body weight and body mass index in 2012.