
Abstract
Medical device (MD) developers currently use the Technology Readiness Levels (TRLs) to determine the status of technology. However, the broad definition of each level makes it difficult to ascertain readiness of MDs. The TRL is also indifferent to the strict regulatory requirements for MDs, requirements for human systems integration, and market competition. This paper proposes a technology readiness framework for communicating and planning the development of Class III MDs called the Medical Device Readiness Levels (MDRLs). Five exit criteria were staged in a meaningful sequence within the framework: safety, clinical effectiveness, usability, comfort, and affective response. It also incorporates the stakeholders’ perspectives, mindful of the users’ varying needs in manipulating the MD. The usefulness of the framework was affirmed by professionals involved in MD development.
Relevance to human factors/ergonomics theory
Biomedical devices (BMDs) are designed to address specific medical concerns and require intimate interaction with users especially the patient. Technology readiness assessment methods used in BMD development do not address the unique characteristics of BMDs and its regulatory process. This paper proposes several human factors considerations in technology readiness assessment and placed them in a hierarchy to help BMD developers prioritize goals at different stages of development.
1. Introduction
In the technology development process, readiness assessment is employed to determine the status of technology, assess risks, and make decisions concerning funding and transition of technology (U.S. Department of Defense Citation2011). The Technology Readiness Level (TRL) scale was developed to assess the maturity of technologies in the aerospace industry (Mankins Citation1995) and has been used in the assessment of medical devices (MD). Tapia-Siles, Coleman, and Cuschieri (Citation2016) evaluated several technologies for replacing flexible colonoscopy in screening asymptomatic colorectal cancer using the TRL levels to identify promising ones while Ryan et al. (Citation2015) assessed the readiness of electrospinning, additive manufacturing, and lithography implantable devices. Recent advancements in the field of Organs-on-a-Chip (OOAC) were also evaluated using TRLs (Allwardt et al. Citation2020). The TRL has been adopted as it provides standards of technology maturity that developers and managers can use to have a shared understanding and keep track of progress (Nolte Citation2008).
Unlike other technologies, users interact intimately with the MDs to address medical concerns towards achieving health and safety. MD development (MDD) technology managers face challenges applying the TRL, which was developed initially to assess space technologies’ readiness (Mankins Citation1995). The technology is administered to the patient or introduced into the human body in the healthcare setting. The development process requires stringent management from conception to market release (Wu et al. Citation2016). One of the challenges in MDD is keeping track of the regulatory requirements throughout its life cycle that vary across countries depending on the risk level of the device (De Ciurana Gay, Özel, and Serenó Citation2016). The USA, for example, requires rigorous testing of high-risk devices compared to the European Union (Kramer, Xu, and Kesselheim Citation2012). The US Food and Drug Administration (FDA) classified MDs into three categories: Class I – devices present minimal risk; Class II – devices have moderate risk; and Class III – devices carry the highest risk, represent a completely novel design, and support or sustain human life (Van Norman Citation2016). Class III devices that are entirely new in the market and do not have a predicate require premarket approval (PMA) from the FDA (Van Norman Citation2016). The PMA application must be supported by scientific evidence proving its safety and effectiveness through a series of clinical trials (Hwang et al. Citation2016; Kaplan et al. Citation2004; Van Norman Citation2016). These standards are unique to MDs and are not captured by the TRLs. Only one was successfully marketed out of five colonoscopy devices that achieved TRL 8 and 9 (Tapia-Siles, Coleman, and Cuschieri Citation2016). The Invendoscope (SC20), for example, reached TRL 9 but was abandoned for another design iteration (Sivananthan et al. Citation2020). OOAC developers and end-users cannot agree on a level of readiness using TRL. The technology developers did not even give a level of TRL 8–9 to their commercially available OOAC (Ryan et al. Citation2015).
We further evaluated the use of TRLs in MDD by interviewing professionals who have played key roles in MDD, have a working knowledge of TRL, have used this in the past, and are familiar with the technical and medical aspects of the MD. We found that the TRLs cannot adequately represent the technology readiness of MDs. The transition from TRL 5–6 in MDs requires a series of clinical trials to prove safety and effectiveness. These clinical trials are costly and adverse incidents can seriously affect the development process. Device manufacture is contingent on FDA approval via 510 (k) or PMA routes for Class II and III devices, respectively.
Having been developed in the aerospace industry, the TRLs did not consider the human dimension in assessing technology and its integration into the environment. MDs are distinct from other technologies because the core system is the human body receiving the treatment. Since the device is meant to support or sustain life, ensuring the user’s safety and optimum performance are critical considerations in the development process. The MD should be effective and efficacious to achieve the expected health outcome of the patient while maintaining a positive treatment journey. The interaction of the device and the users influence the patient experience, so the design should account for their perspectives. Human systems integration (HSI) strategies of including the user roles, structures, processes, and constraints in the design process must be used to achieve total systems performance (Phillips Citation2010).
The human dimension and system integration limitations of the TRL were addressed by the Human Readiness Levels (HRLs) (Acosta Citation2010; Endsley Citation2014; Phillips Citation2010; Salazar et al. Citation2020; See et al. Citation2017). The HRL was developed to ‘evaluate, track, and communicate the readiness of a technology for safe and effective human use throughout design’ (Human Factors and Ergonomics Society Citation2021). It considers human system integration (HSI) in assessing technology maturity. The scales of the HRL were aligned to the TRL levels so that human systems design and evaluation activities are integrated with the design and evaluation of the technological system (Human Factors and Ergonomics Society Citation2021). The human-centered approach of the HRL meets the need for assessing MDs to ensure the safety of patients and other users of the device. The lower HRLs examined the needs of potential users in the whole development process, including human systems integration (HSI) activities.
Although the HRLs recognize the needs of end-users in the development process, like the TRLs, the HRLs are indifferent to the regulations of MDD, making it difficult for the managers to compare technologies, and make decisions regarding plans for future investment. Large companies that develop MDs typically have several pipelines, making them lose focus on the regulatory requirements. A survey of UK MD companies showed that most of these companies lack understanding of regulatory requirements (Lee and Boyer Citation2019). MDs that do not comply with these requirements and are released prematurely harm the company’s reputation because of recalls due to safety and effectiveness issues (Hwang et al. Citation2016).
Despite its human-centered approach to readiness assessment, the HRL’s exit criteria did not specify key performance measures that indicate progress in attaining goals related to MDD. Proof of clinical effectiveness, for example, was not among the exit criteria, although this is part of the requirement for the PMA application of Class III devices. Predictors of market acceptance such as comfort and satisfaction were also not included in the HRLs or TRLs. MDs are meant to benefit a more significant number of patients, so there is a need to have relevant indicators of success. The Pillcam Colon made it to the market as a colonoscopy substitute because it did not just address clinical needs but also patients’ wants, such as the absence of pain and discomfort (Tapia-Siles, Coleman, and Cuschieri Citation2016). The fulfilment of these high-level needs discriminates between market acceptance and failure.
Readiness assessment using HRLs and TRLs does not go beyond the actual use of technology. In contrast, in MDD, post-marketing surveillance is part of the regulatory process for high-risk MDs. Device users, hospitals, and health professionals are required to inform regulatory bodies and device manufacturers of adverse events related to its use (Van Norman Citation2016). Post-marketing surveillance is necessary to ensure that the device is safe and works as intended.
Based on the preceding analysis, we propose assessing MD readiness featuring a hierarchy of needs reflected as exit criteria at specific readiness levels following FDA regulations because it is stricter than the EU (Marešová et al. Citation2020). The USA and EU are the two largest markets for MDs globally (Marešová et al. Citation2020). The needs are recognized from the interaction of the MD with various users such as the patient, healthcare worker (HCW), and support staff (SS). As used in this paper, MD refers to a Class III device with a higher risk level in terms of illness and injury and requires PMA (Van Norman Citation2016).
This paper is divided into four parts: the context of MD development, MD readiness dimension, development of the MD Readiness Levels (MDRL), and evaluation of MDRL in the MDD process.
2. MD development in a nutshell
Countries worldwide impose strict controls on MD at all stages of its lifecycle to ensure the safety and well-being of the people for whom these MDs will be used. The design and manufacture of MDs follow specific regulations and technical standards to ensure user safety and device effectiveness. Regulations differ among countries in terms of classification based on the risk level, quality system, clinical investigation, premarket approval, and post-market management (Chen et al. Citation2018). The control of the quality management system was harmonized with the recognition of the ISO 13485:2003(Wu et al. Citation2016).
The development of MDs generally goes through the following phases: ‘concept, prototype, preclinical, clinical analyses to test, improve, retest, validate, optimize, and finalize before beginning the manufacturing process’ (Özel et al. Citation2016). At the initial conceptualization phase, the primary consideration is addressing the clinical need, which is usually linked to attaining the desired health outcomes (HO) of the patient or improving existing ones. For example, due to the risks and patient discomfort brought about by conventional colonoscopy, several alternatives have been developed to make colorectal cancer screening more acceptable to patients (Tapia-Siles, Coleman, and Cuschieri Citation2016).
At the preclinical phase, a prototype is built to validate the concept in the laboratory. Once the MD is shown to work optimally in the laboratory, it is tested on animals. Stringent regulations are followed for animal testing. Negative and positive test results are the bases for redesigns and further retesting. Only when the animal studies pass the toxicity and biocompatibility test criteria can the MD be used for human testing at the clinical phase. The MD is usually tested first on a small group of healthy persons. The effect of the MD on these individuals is analyzed to determine the required adjustments.
The improved MD is then validated on a larger sample of persons who exhibit the disease condition it is designed to treat. The actual application of the MD on people reveals detailed effects on the targeted bodily functioning that may warrant further improvements on the MD until safety and effectiveness are assured. These clinical results would have to pass the regulatory requirements in the country where they will be used.
Each phase is an iterative process of retesting/validating to improve and optimize the MD until it meets patient requirements and needs while ensuring the safety of the patient and HCWs involved. For instance, the Pillcam COLON developed to investigate small bowel pathology is a technology that has undergone several improvements since its release in 2006 (Hong et al. Citation2018). Its early version showed low sensitivity in detecting colonic lesions compared to conventional methods (Van Gossum et al. Citation2009). The PillCam 2 was introduced after three years providing better sensitivity and specificity for colorectal polyps (Eliakim et al. Citation2009).
3. Medical device readiness dimensions
Given the limitations of the TRL and HRL in assessing MD readiness, we propose a framework that integrates regulatory requirements in technology and human readiness assessment. We considered five critical dimensions stipulated in various MD regulatory documents or deemed necessary for market adoption: safety, clinical effectiveness, usability, comfort, and affective response mapped to stakeholders based on the factors affecting their interaction with the MD. summarizes the unique characteristics of the patient, healthcare worker (HCW), and support staff (SS) in relation to the use of a MD. Factors 2–4 were adapted from socio-technical factors in complex treatment settings proposed by Henriksen, Joseph, and Zayas-Cabán (Citation2009).
Table 1. Factors affecting MD use.
The most critical consideration for MD development is the patient who is the recipient of the medical intervention. Medical procedures generally cause discomfort that is tolerated by the patient in view of a positive health outcome. The patient is usually a passive recipient of the MD while the HCW manipulates it according to established standards of operation. HCW refers to people who provide direct patient care and use the MD for the patient, such as doctors, nurses, or hospital technicians who position, activate, monitor, and troubleshoot the device during operation (Knisely et al. Citation2020). SS are clinical assistants and maintenance personnel that do not handle the MD directly while it is in operation but may assist the HCW in the upkeep and maintenance of the MD. SS knowledge of device operation is low since their primary concern is to prevent MD failure and prevent accidents. The interests of these people were tackled in various regulatory requirements on the manufacture and deployment of MDs. While the manufacture of MDs primarily aims to improve patient care, the successful deployment of a MD also depends on the role of HCWs and SS, specifically their participation in testing and evaluation (World Health Organization Citation2003).
Class III MDs are rarely manipulated or handled by patients on their own because they pose a high risk to users. The HCW assists or handles the device requiring specialized knowledge regarding its operation. The Global Harmonization Task Force, a group that harmonizes the implementation of MD regulations across the globe, specified the need to train intended users on the use of MDs so as not to compromise the clinical condition or safety of patients (World Health Organization Citation2003). SS are trained on the principles and methods of maintenance, calibration, initial repairs, and device management concepts (Bahreini, Doshmangir, and Imani Citation2019) but not necessarily on the operation of the device.
shows the relationship of the proposed dimensions to the various perspectives enumerated. Colored areas indicate the dimension’s relevance to a perspective.
Figure 1. Dimensions and perspectives in the MDRL.
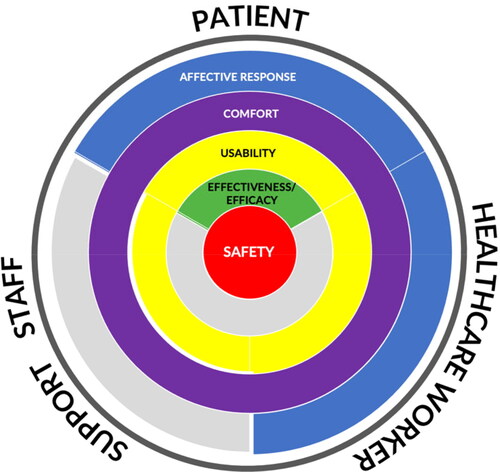
The first dimension in the MDRL is safety situated at the core of the circle. It is the essential requirement in MD development spanning the needs of all stakeholders. MD regulation underscores safety standards for patients and users in all aspects of use, including maintenance and handling (World Health Organization Citation2003). The clinical effectiveness of a MD, defined as producing the intended effect relative to the medical condition, is applicable only to the afflicted patient (World Health Organization Citation2003). Usability affects the safe use of a MD in terms of preventing errors and is relevant only to the patient and HCW. A MD may be judged safe to use and clinically effective but can endanger users because of improper operation due to usability problems related to components, accessories, displays, controls, packaging, product labels, etc. (Food and Drug Administration Citation2016).
Comfort is not part of MD regulatory requirements; however, the physical and functional definition of comfort is related to the physical dimension of usability when the MD comes in contact with the patient’s body or is used by the HCW and SS. Pain, pressure, and irritation are physical indicators of comfort, while handling, stability, and maintenance are functional indicators (Doutres et al. Citation2019). The SS assigned to clean the MD or replace parts, for instance, may not be comfortable due to ergonomics issues. This exit criterion was separated from usability due to differences in metrics.
Affective response is not mentioned in any MD regulation but is vital in the deployment stage where the device has been marketed, and user feedback is obtained. A MD that is pleasant to use and elicits positive affective responses such as enjoyment, fun, and surprise or minimizes distress is likely to be preferred by patients and HCWs. SS is not mapped to this dimension because they do not use the MD directly. The limited interaction is not likely to elicit affective responses comparable to the patient and HCW.
4. Medical device readiness levels (MDRL)
There are nine levels of MDRL proposed to leverage users’ familiarity with the structure of the TRL. provides the levels and definitions of the MDRL in relation to the TRL framework used by the US Department of Defense Technology Readiness Assessment (ASDRE Citation2011) and the HRL (Human Factors and Ergonomics Society Citation2021). shows the MDRLs apropos the TRLs and HRLs. Human systems integration and performance issues are identified as early as MDRL1 until the end of the development process. Levels 1–2 and 2–3 of both TRL and HRL were subsumed in MDRL 1 and 2, respectively. Levels 8–9 are unique only to MDRL. Higher levels of TRL and HRL focus on mission success and total system integration, respectively, which are different from the market acceptance goal of the MDRL. HRL was not mapped to MDRL at levels 3–4 due to the absence of human involvement at these levels.
Figure 2. Mapping of MD development progress using TRL and HRL.
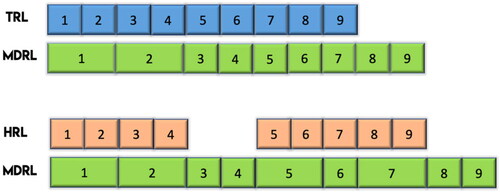
Table 2. Comparison of TRL, HRL and MDRL.
The MDRL was conceptualized for Class III devices because they have the most rigorous requirements. However, the MDRL can also be used to assess the readiness of Class I and II by foregoing irrelevant levels. For example, not all Class II devices require premarket notification. They may not require clinical trials (MDRL 4–7) if the manufacturer can provide data demonstrating substantial equivalence to an existing legally marketed device (Kaplan et al. Citation2004).
The MDRL aims to guide technology managers in the successful transition from research to application. Technology maturity assessment using the MDRL considers the regulations for manufacturing MDs at various stages, such as the need for three clinical trials for high-risk MDs, Food and Drug Administration (FDA) approval before deployment, and post-market surveillance to further monitor safety and clinical effectiveness. To address the issues of market adoption and sustained use, we proposed adding comfort and affective response dimensions as exit criteria in the MDRL. The exit criteria proposed serve as clear targets for technology managers to achieve an exit level and guide them in identifying the proper strategies and metrics to achieve their targets. At the point of exit, there is assurance that the requirement is fulfilled, and higher-level issues are addressed to move to another level.
shows the key tasks and exit criteria for each level and maps the relationship between the placement of the exit criteria against the TRL framework. In the same figure, we also indicated where premarket approval would be obtained.
Figure 3. MDRL key tasks and exit criteria.
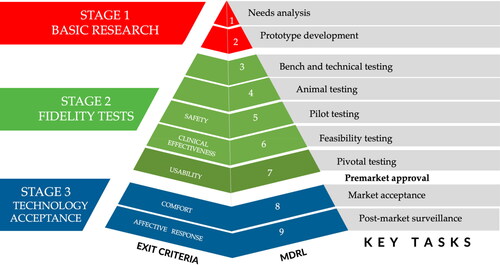
We propose staging technology readiness through the following exit criteria:
MDRL 5 – Safety: Parameters that must be validated to be safe for human use before the first clinical trial with human participants. (e.g. toxicity and biocompatibility test criteria)
MDRL 6 – Clinical effectiveness: Parameters related to the technology’s mission and objective, and existence, also known as cardinal requirements. (e.g. colonoscopy device’s ability to take clear and accurate images of the colon; prosthesis’ functionality mimicking comparably the body part it replaced)
MDRL 7 – Usability: Parameters that pose issues arising from interaction with the MD interface that can cause errors (e.g. unclear instructions, inconsistent screens, poor user control of a device, etc.).
MDRL 8 – Comfort: Parameters related to discomfort mitigation in using the MD such as contact stress, poor fit, acute and chronic pain (e.g. musculoskeletal symptom).
MDRL 9 – Affective Response: Parameters that trigger an emotional response when using the technology. (e.g. enjoyment, surprise, embarrassment, fear, trust)
Given the requirement of post-market surveillance after the FDA approval, the testing is scaled up to further validate the technology with larger samples through mass marketing. We further grouped the five dimensions into fundamental (MDRL 5–7) and advanced readiness (MDRL 8–9) to differentiate pre- and post-FDA approval readiness conditions. We included safety, clinical effectiveness, and usability as part of fundamental readiness where functionality and safety are met. These are basic requirements before releasing the MD to the market for the general population.
Advanced readiness includes the higher needs of stakeholders that promote sustained use. A technology that exits MDRL 8 would be projected to have higher market acceptance since users of the system are less likely to develop discomfort when using the technology. Similarly, technologies that exit MDRL 9 would be expected to gain greater adoption as they elicit a positive emotional response.
We provide the detailed description of our proposed exit criteria from MDRL 5–7 as follows:
4.1. Safety – MDRL 5
Larson et al. (Citation2017) defined safety architecture as ‘the documented organization of a device’s hardware and software elements that serve to detect, notify, mitigate and record device states that could result in patient harm’. The evidence required by FDA for safety depends on the MD risk classification, i.e. low-risk, moderate-risk, high-risk (Rathi and Gray Citation2019). High-risk MDs pose a severe threat to life and bodily functions, not primarily due to any faulty architectural design of the MD, but because of the nature of the MD, e.g. heart implants. For high-risk MDs, the rigor of testing is similar for medicines that go through laboratory tests, animal trials, and three clinical trials prior to market launch.
We took reference from the activities in Tapia-Siles et al.’s TRL categorization for evaluating micro-robots as substitutes for colonoscopy to examine the rigor needed for developing MD and concluded the need to achieve safe use at MDRL 5 before the MD interacts with the human body and its functioning (Tapia-Siles, Coleman, and Cuschieri Citation2016).
While we acknowledge that the safe use of the technology is a constant consideration throughout the development cycle, the technology risks for human use should be examined through the knowledge gained from MDRL 1 through MDRL 4 before entering clinical trials where healthy individuals are recruited as trial participants to ascertain the effectiveness of the technology.
4.2. Clinical effectiveness – MDRL 6
In a MD, clinical effectiveness is related to the health outcomes (HO) of the patient. For example, heart valve MDs are meant to make the heart pump blood effectively. MD effectiveness is measured through clinical and physiological indicators gathered from the patient. HO is a fundamental requirement that must be achieved at an early stage before the design is further enhanced to improve the experience in using and handling the technology. This naturally fits the exit criteria at MDRL 6 before the technology is tried in an operational setting.
We assessed the need for HO to be achieved entirely to exit MDRL 6, but it needs to be manifested as early as MDRL 4. Since the application of the MD on humans happens during the clinical stage at MDRL 6, surrogate measures for HO may be used in the preclinical stages when the MD is used in the laboratory during bench and technical testing and during animal testing. As early as the conceptualization stage, HO is considered as the primary endpoint in designing the MD. If HO is not eventually attained, the MD fails and loses its reason for being. Under such circumstances, the technology should not transit to MDRL 7 for trial in an operational setting.
4.3. Usability – MDRL 7
The lack of a usability study can result in technical glitches and patient mortality as in the Therac-25 accidents with patients suffering severe radiation burns (Leveson Citation1995; Leveson and Turner Citation1993). Some MDs are designed to be used alone by the patient at home or with the aid of a caregiver. A case in point is the Continuous Positive Airway Pressure (CPAP) machine used for sleep apnea, a potentially serious disorder. The correct setup of the device and proper fitting of the mask on the face is important to ensure that the MD will deliver the continuous positive pressure to the airways for the entire duration of sleep. A usability study conducted on this MD showed that patients find it difficult to get it ready for use, and caregivers find it hard to maintain (Fung et al. Citation2015).
Examining usability provides information on the needed training and instructional design to ensure a smooth and safe transition to operations for these users. They augment the safety analysis by addressing the lack of information on probabilities associated with user errors. Usability analysis is vital for discovering and pre-empting safety issues that arise in the complex interaction between subsystems. Hence, we propose conducting it at MDRL 7 to manage limited analytical resources.
We also positioned the technology as ready to obtain premarket approval upon the successful results obtained in the usability analysis. Unlike non-MDs, the validation of MD requires large samples of human participants at Stage 3, which is affirmed through a premarket approval. Tapia-Siles, Coleman, and Cuschieri (Citation2016) suggested having the FDA 510 (k) or equivalent approval at TRL 8. We take a similar position in our framework and placed premarket approval after meeting the usability exit criteria at MDRL 7.
4.4. Comfort – MDRL 8
Comfort, in our framework, is limited to the physical and functional dimensions of comfort. They are either (1) physically uncomfortable experiences that are temporary and acute or (2) physical strains that can lead to pain over prolonged use with or without chronic pain. Both have an impact on physical well-being but do not cause mortality. This dimension is closely related to the quality of user experience that can affect the sustained use of the MD. A technology that is functional and safe to use may not be preferred if the user experiences physical strain and discomfort.
For a patient, discomfort refers to pain, stress, irritation, annoyance, and physical intrusion experienced by the patient during and after the procedure, whereas discomfort for HCW and SS refers to work-related musculoskeletal discomfort (Keester and Sommerich Citation2017; Sommerich et al. Citation2020). Issues of comfort for HCW arise due to the need to exert force, maintain static posture or perform repetitive action. On the other hand, SS may be bothered by the weight and bulk of the equipment. Handling of these bulky MDs sometimes causes physical stress or musculoskeletal disorders.
Physical discomfort, although not fatal, can cause injuries that range from mild bruises to chronic pain. This dimension is perhaps more pertinent when examining the MD from the HCW perspective. Physical discomfort would result in workplace attrition and affects the sustained use of the technology for treating patients. We positioned comfort at MDRL 8 to indicate foreseeable sustained use of the MD in the market.
4.5. Affective response – MDRL 9
The highest level in our MDRL framework projects the market endurance of the MD due to the absence of negative affective response and the presence instead of a satisfying experience – a lofty target for a MD.
Affective design methods can be incorporated in technology development by empathizing with users and understanding their emotional needs. Affective products are more likely to be used and kept by consumers and increase product attachment (Bascos et al. Citation2020). Affective response refers to the patient’s prospective or actual psychological state while using the MD. It is an important aspect of treatment as it determines the uptake of technology and its perceived benefits (Kiviniemi, Jandorf, and Erwin Citation2014). This dimension is related to the psychological definition of comfort while using the MD, such as acceptability, pleasant, relaxing, happy to use, ‘got used to’ from the perspective of the patient (Doutres et al. Citation2019). Discomfort causes distress, and a bad episode can cause anxiety on the prospect of repeated use. A patient that experienced bruising after injection, for example, felt distressed and was unwilling to continue treatment (Cramer et al. Citation2006). Fear of treatment or procedure is derived from a previous negative experience and can only be overcome by successive positive treatment experiences (Carr, Lemanek, and Armstrong Citation1998).
Affective response is crucial for patients, especially for technologies that can cause intense emotional experiences. This dimension can also be relevant to HCW, especially for MDs to which they may form strong personal attachment (e.g. a physician’s stethoscope).
5. MDRL evaluation in the MDD process
We validated the usefulness of the MDRL in the MDD process by interviewing people who play a key role in MDD, have working knowledge on the use of the Technology Readiness Level or a related scale, have used the scale in a MDD project, and have a working knowledge of the technical and medical aspects of the device under development. The interview aimed to determine the value of the exit criteria in the MDD process and their sequence in the development of Class III MDs. Each level of the MDRL was evaluated in terms of clarity of the description of activities, appropriateness of the exit criteria sequence in the development process, and completeness of information in the description.
5.1. Participants
Two medical doctors and one engineer were interviewed. Doctor A is a surgeon that led three MDD projects. Doctor B is another surgeon who worked in the area of technology transfer and served as a clinician in a MDD project. Engineer A worked in a technology transfer office and was also involved in developing several MDs.
5.2. Procedure
Each participant was interviewed online via Zoom. The interview process started with introducing the MDRL’s purpose, process, levels, and exit criteria. The questionnaire used for the interview was adapted from Phillips (Citation2010).
Preliminary questions inquired about their experience in assessing readiness levels of medical devices, models/readiness scales used previously, challenges encountered with existing readiness scales, and suggestions for improvement.
Participants provided comments about the exit criteria by answering the following questions:
Are the exit criteria identified in the MDRL important in the development process?
How would the exit criteria help in the MD development process?
Are the exit criteria appropriately timed in the development process of Class III devices per FDA regulations (high-risk devices or class C-D in other countries)?
They also assessed each exit criteria and each level of the MDRL in terms of clarity, timing, or sequence within the medical device’s development life cycle and the information’s completeness.
5.3. Results
All the participants agreed that the MDRL would be useful in assessing MD readiness. Safety, clinical effectiveness, and usability are important exit criteria that are currently being assessed in the actual MDD process. Two of the participants strongly agreed on the inclusion of comfort and affective response as exit criteria, especially in ensuring the market success of the MD. Doctor B highlighted the need to measure affective response, not just market indicators of the MD, because of its importance in the eventual adoption of the MD. All the levels and exit criteria were judged to be appropriately timed and sequenced in the MDD process, respectively.
Descriptions of activities in 8 out of 9 MDRLs were judged to be clear. Engineer A expressed the need to consider ethical considerations in Level 1 because a treatment alternative can address a medical challenge but may need to be abandoned due to ethical issues.
The following suggestions were also obtained from the interviews:
Include an activity to validate the needs identified in Level 1 before moving to Level 2
Address ethical issues in the needs analysis
Indicate the activities related to comfort testing in the clinical trials
Consider comfort and affective response as non-mandatory exit criteria for Levels 8 and 9 because it might prolong the MDD process.
Integrate manufacturing readiness in the framework because it is a source of risk and can affect the MDD process.
5.4. Discussion
The usefulness of the MDRL was validated by professionals working in the field of MDD. The suggestions provided can be used to operationalize the MDRL since our current work was limited to the development of the levels, exit criteria, and description of activities. Future work can focus on identifying detailed activities of the exit criteria and documentary requirements to exit a level. Validation of the needs assessment and resolution of ethical issues, for instance, can be set as conditions for moving to Level 2, and measurement of comfort can be defined in clinical trials.
Comfort and affective response are not regulatory requirements and, therefore, not mandatory to obtain FDA approval. MD developers can set comfort and affective response metrics that are reasonable indicators of potential market adoption and the basis for further improving the device.
The suggestion to include manufacturing readiness lends additional thoughts to enhance the MDRL in addressing end-to-end development. To this end, the existing Manufacturing Readiness Level (OSD Manufacturing Technology Program Citation2018) can be reviewed to identify ways of factoring manufacturing concerns into the MDRL. Our current assessment is that manufacturing would likely follow a different readiness level scale that starts after MDRL 7. Future research can examine the benefits of combining manufacturing readiness with the MDRL.
There is general agreement that the MDRL proposed will provide an effective means for a MD working group to manage the development process. The framework contains almost all the necessary activities for the MDD program, and it would be helpful in guiding risk identification and market assessment.
The MDRL was validated using a limited sample size of two doctors and an engineer. The experiences of the participants may not be enough to identify the full advantages and assess the limitations of the proposed framework. A more comprehensive evaluation by actual users can be done in the future.
6. Conclusion
The development of high-risk MDs involves adherence to strict regulatory requirements. Existing readiness assessment scales such as TRL and HRL do not reflect these requirements making it difficult for MD developers to compare technologies and make future investment decisions. The TRL is popularly used in assessing MD readiness, but the broad definition of the levels makes it difficult for technology managers to agree on the level of readiness when used in the field. TRL also failed to consider human systems integration strategies in the development process, which was eventually addressed by the development of the HRL. At present, HRL is not being used in MD assessment, and despite HRLs inclusion of users’ needs, it is also indifferent to the regulatory requirements of MDs. The HRL’s exit criteria did not specify crucial metrics for attaining MD development goals.
We proposed the MDRL that built upon the TRL and HRL to provide a technology readiness framework for communicating and planning the development of Class III MDs. While other technologies view the human body as a system component, in MDs, the human body is the primary system that receives health benefits, hence the need for stringent testing.
The MDRL levels were found suitable for MD development, mirroring the TRL to take advantage of the popular nine levels used by technology managers. It also incorporated the human systems view of the HRL. Exit criteria were incorporated within the levels in five dimensions relevant to MD users and aligned with MD regulations. The five exit criteria were staged in a meaningful sequence that follows a hierarchy reflected in its timing in the development process. The most crucial dimensions for sustaining life are considered early in the development process, which we call fundamental readiness consisting of safety, clinical effectiveness, and usability. The other two dimensions: comfort and affective response, transcend the basic requirements and are called advanced readiness levels. The hierarchy parallels Maslow’s Hierarchy of Needs (MHN), where lower needs should be satisfied first before moving to the higher ones. Unlike the MHN, however, the basic requirement in healthcare is to preserve life. Higher needs are related to comfort and pleasure, which are ancillary to survival.
The MDRL also incorporates the perspectives of the stakeholders, mindful of the fact that the MD exists in a system of people. MD connects all the stakeholders in the system since each can contribute to the failure of use if their needs have not been incorporated in the development. The approach taken by the MDRL coincides with the Stakeholder Theory that acknowledges the moral obligation of the business to its stakeholders (Freeman Citation1984). This moral obligation was ensured in the MDRL by involving relevant stakeholders at each stage of the development process.
The MDRL went beyond the demands of current readiness levels by incorporating the FDA’s requirements and the last two levels: market acceptance and post-market surveillance. Without attaining Level 8, the MD may not be ready for widespread adoption. Level 9 ensures the positive evaluation of users in the general population in the long run, giving greater confidence that the technology would stay longer in the market.
The MDRL at this point is limited to the basic concept of readiness in MDD. Specific support documents have not been identified to establish the requirements for exiting levels 5–9. The inclusion of the user’s perspectives can extend development time and may not be attractive for companies. However, the extra work can serve the benefit of discovering unique issues that elude patient-exclusive testing.
The framework is yet to be used in the field and further reviewed by MD technology managers in the industry. Findings from the actual application of the MDRL are expected to enrich the scale and make it more usable. We further propose the extension of the MDRL to drug discovery and development.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Notes on contributors
Rosemary Ruiz Seva
Rosemary R. Seva, PhD is a full professor of Industrial Engineering at the John Gokongwei College of Engineering - De La Salle University, Philippines. She heads the IEA Technical Committee on Affective Design and is the president of the Human Factors and Ergonomics Society of the Philippines. Dr. Seva graduated from the Nanyang Technological University with a PhD in Affective Design.
Angela Li Sin Tan
Angela Li Sin Tan, C.ErgHF, PhD, is the programme director of the Human Performance Programme at the Defence Medical and Environmental Research Institute - DSO National Laboratories, Singapore. She is a chartered ergonomist and a past president of the Human Factors and Ergonomics Society of Singapore. Dr. Tan graduated from the Nanyang Technological University with a PhD in Cognitive Systems Engineering.
Lourdes Marie Sequerra Tejero
Lourdes Marie Tejero, RN, MA, MTM, PhD, is a full professor and director of Technology Transfer and Business Development Office at the University of the Philippines Manila (UPM). She served as dean of UPM College of Nursing, and director of the National Graduate Office of Health Sciences. Dr. Tejero graduated from University of California (UC) Berkeley and UC San Francisco with Master of Translational Medicine, a combination of bioengineering, medicine, and business. She completed her PhD in Nursing from UPM.
Maria Lourdes Dorothy S. Salvacion
Maria Lourdes Dorothy S. Salvacion, DDM, MHPEd, is a dentist and an assistant professor at the National Teacher Training Center for the Health Professions (NTTCHP) - University of the Philippines Manila (UPM). She serves as the college secretary and the chair of the Office of Curriculum and Instruction. Dr Salvacion graduated from NTTCHP with a Master in Health Professions Education. She completed her B.Sc in Dental Medicine from UPM.
References
- Acosta, H. 2010. “Human Readiness Levels: Implementing HSI – Connecting Some Dots.” [Panel Discussion] In 81st Annual Scientific Meeting of the Aerospace Medical Association, Phoenix, AZ.
- Allwardt, V., A. J. Ainscough, P. Viswanathan, S. D. Sherrod, J. A. McLean, M. Haddrick, and V. Pensabene. 2020. “Translational Roadmap for the Organs-on-a-Chip Industry toward Broad Adoption.” Bioengineering 7 (3): 112–127. doi:10.3390/bioengineering7030112.
- ASDRE. 2011. “Technology Readiness Assessment (TRA) Guidance.” In.: US Department of Defense.
- Bahreini, R., L. Doshmangir, and A. Imani. 2019. “Influential Factors on Medical Equipment Maintenance Management: In Search of a Framework.” Journal of Quality in Maintenance Engineering 25 (1): 128–143. doi:10.1108/JQME-11-2017-0082.
- Bascos, N., A. D. Aguilar, A. Lucena, and R. Seva. 2020. “Designing Ecobags for Product Attachment.” In Joint Conference of the Asian Council on Ergonomics and Design (ACED) and Southeast Asian Network of Ergonomics Societies (SEANES) 2020. Virtual. Manila, Philippines.
- Carr, T. D., K. L. Lemanek, and F. D. Armstrong. 1998. “Pain and Fear Ratings: Clinical Implications of Age and Gender Differences.” Journal of Pain and Symptom Management 15 (5): 305–313. doi:10.1016/S0885-3924(97)00370-9.
- Chen, Y. J., C. M. Chiou, Y. W. Huang, P. W. Tu, Y. C. Lee, and C. H. Chien. 2018. “A Comparative Study of Medical Device Regulations: US, Europe, Canada, and Taiwan.” Therapeutic Innovation & Regulatory Science 52 (1): 62–69. doi:10.1177/2168479017716712.
- Cramer, J. A., B. J. Cuffel, V. Divan, A. Al-Sabbagh, and M. Glassman. 2006. “Patient Satisfaction with an Injection Device for Multiple Sclerosis Treatment.” Acta Neurologica Scandinavica 113 (3): 156–162. doi:10.1111/j.1600-0404.2005.00568.x.
- De Ciurana Gay, J., T. Özel, and L. Serenó. 2016. “Overview.” Biomedical Devices, 1–22.
- Doutres, O., F. Sgard, J. Terroir, N. Perrin, C. Jolly, C. Gauvin, and A. Negrini. 2019. “A Critical Review of the Literature on Comfort of Hearing Protection Devices: Definition of Comfort and Identification of Its Main Attributes for Earplug Types.” International Journal of Audiology 58 (12): 824–833. doi:10.1080/14992027.2019.1646930.
- Eliakim, R., K. Yassin, Y. Niv, Y. Metzger, J. Lachter, E. Gal, B. Sapoznikov, et al. 2009. “Prospective Multicenter Performance Evaluation of the Second-Generation Colon Capsule Compared with Colonoscopy.” Endoscopy 41 (12): 1026–1031. doi:10.1055/s-0029-1215360.
- Endsley, M. 2014. “Human System Integration: Challenges and Opportunities.”
- Food and Drug Administration 2016. “Applying Human Factors and Usability Engineering to Medical Devices: Guidance for Industry and Food and Drug Administration Staff.” In Washington DC: U.S. Department of Health and Human Services.
- Freeman, E. 1984. Strategic Management: A Stakeholder Approach/R. Edward Freeman, Pitman Series in Business and Public Policy. Boston: Pitman.
- Fung, C. H., J. L. Martin, R. D. Hays, J. C. Rodriguez, U. Igodan, S. Jouldjian, J. M. Dzierzewski, B. J. Kramer, K. Josephson, and C. Alessi. 2015. “Development of the Usability of Sleep Apnea Equipment-Positive Airway Pressure (USE-PAP) Questionnaire.” Sleep Medicine 16 (5): 645–651. doi:10.1016/j.sleep.2015.01.019.
- Henriksen, K., A. Joseph, and T. Zayas-Cabán. 2009. “The Human Factors of Home Health Care: A Conceptual Model for Examining Safety and Quality Concerns.” Journal of Patient Safety 5 (4): 229–236. doi:10.1097/PTS.0b013e3181bd1c2a.
- Hong, S. N., S.-H. Kang, H. J. Jang, and M. B. Wallace. 2018. “Recent Advance in Colon Capsule Endoscopy: What’s New?” Clinical Endoscopy 51 (4): 334–343. doi:10.5946/ce.2018.121.
- Human Factors and Ergonomics Society. 2021. “ANSI/HFES 400: Human Readiness Level Scale in the System Development Process.” In. US: Human Factors and Ergonomics Society.
- Hwang, T J., E. Sokolov, J. M. Franklin, and A. S. Kesselheim. 2016. “Comparison of Rates of Safety Issues and Reporting of Trial Outcomes for Medical Devices Approved in the European Union and United States: Cohort Study.” BMJ 353: i3323. doi:10.1136/bmj.i3323.
- Kaplan, A. V., D. S. Baim, J. J. Smith, D. A. Feigal, M. Simons, D. Jefferys, T. J. Fogarty, R. E. Kuntz, and M. B. Leon. 2004. “Medical Device Development: From Prototype to Regulatory Approval.” Circulation 109 (25): 3068–3072. doi:10.1161/01.Cir.0000134695.65733.64.
- Keester, D. L., and C. M. Sommerich. 2017. “Investigation of Musculoskeletal Discomfort, Work Postures, and Muscle Activation among Practicing Tattoo Artists.” Applied Ergonomics 58: 137–143. doi:10.1016/j.apergo.2016.06.006.
- Kiviniemi, M. T., L. Jandorf, and D. O. Erwin. 2014. “Disgusted, Embarrassed, Annoyed: Affective Associations Relate to Uptake of Colonoscopy Screening.” Annals of Behavioral Medicine 48 (1): 112–119. doi:10.1007/s12160-013-9580-9.
- Knisely, B. M., C. Levine, K. C. Kharod, and M. Vaughn-Cooke. 2020. “An Analysis of FDA Adverse Event Reporting Data for Trends in Medical Device Use Error.” Proceedings of the International Symposium on Human Factors and Ergonomics in Health Care 9 (1): 130–134. doi:10.1177/2327857920091024.
- Kramer, D. B., S. Xu, and A. S. Kesselhedim. 2012. “Regulation of Medical Devices in the United States and European Union.” The New England Journal of Medicine 366 (9): 848–855. doi:10.1056/NEJMhle1113918.
- Larson, B. R., P. Jones, Y. Zhang, and J. Hatcliff. 2017. Principles and Benefits of Explicitly Designed Medical Device Safety Architecture. Biomed Instrum Technol 51 (5): 380–389. doi:10.2345/0899-8205-51.5.380.
- Lee, E. M., and S. Boyer. 2019. “The State of Medical Device Regulation (MDR) Readiness in UK SMEs.” In UK: ISO Life Sciences.
- Leveson, N. G. 1995. Safeware: System Safety and Computers. Appendix A: Medical Devices: The Therac-25. Boston: Addison-Wesley.
- Leveson, N. G., and C. S. Turner. 1993. “An Investigation of the Therac-25 Accidents.” Computer Magazine. 26 (7): 18–41. doi:10.1109/MC.1993.274940.
- Mankins, J. C. 1995. “Technology Readiness Levels, White Paper.” In Advanced Concepts Office, Office of Space Access and Technology: National Aeronautics and Space Administration.
- Marešová, P., B. Klímová, J. Honegr, K. Kuča, W. N. H. Ibrahim, and A. Selamat. 2020. “Medical Device Development Process, and Associated Risks and Legislative Aspects-Systematic Review.” Frontiers in Public Health 8: 308. doi:10.3389/fpubh.2020.00308.
- Nolte, W. L. 2008. Did I Ever Tell You about the Whale? Or Measuring Technology Maturity. Charlotte: Information Age Publishing, Inc.
- OSD Manufacturing Technology Program. 2018. “Manufacturing Readiness Level (MRL) Deskbook.”
- Özel, T., P. J. Bártolo, E. Ceretti, J. D. De Ciurana Gay, C. A. Rodríguez, and J. V. L. Da Silva. 2016. Biomedical Devices: Design, Prototyping, and Manufacturing. Hoboken, NJ: Wiley.
- Phillips, E. L. 2010. “The Development and Initial Evaluation of the Human Readiness Level Framework.” Naval Postgraduate School.
- Rathi, V. K., and S. T. Gray. 2019. “Device Safety.” Otolaryngologic Clinics of North America 52 (1): 103–114. doi:10.1016/j.otc.2018.08.013.
- Ryan, C. N., K. P. Fuller, A. Larrañaga, M. Biggs, Y. Bayon, J. R. Sarasua, A. Pandit, and D. I. Zeugolis. 2015. “An Academic, Clinical and Industrial Update on Electrospun, Additive Manufactured and Imprinted Medical Devices.” Expert Review of Medical Devices 12 (5): 601–612. doi:10.1586/17434440.2015.1062364.
- Salazar, G., J. See, H. Handley, and R. Craft. 2020. “Understanding Human Readiness Levels.” Paper presented at the Human Factors and Ergonomics Society 2020 Annual Meeting, Chicago, IL.
- See, J., R. Craft, J. Morris, and V. Newton. 2017. “Incorporating Human Readiness Levels at Sandia National Laboratories.” Paper presented at the 2017 Resilience Week (RWS), 18–22. Sept. 2017.
- Sivananthan, A., B. Glover, L. Ayaru, K. Patel, A. Darzi, and N. Patel. 2020. “The Evolution of Lower Gastrointestinal Endoscopy: Where Are We Now?” Therapeutic Advances in Gastrointestinal Endoscopy 13: 2631774520979591. doi:10.1177/2631774520979591.
- Sommerich, C. M., S. A. Lavender, K. D. Evans, E. B. N. Sanders, S. Joines, S. Lamar, R. Z. R. Umar, W.-T. Yen, and S. Park. 2020. “Collaborating with Radiographers to Address Their Work-Related Musculoskeletal Discomfort.” Applied Ergonomics 85: 103069. doi:10.1016/j.apergo.2020.103069.
- Tapia-Siles, S. C., S. Coleman, and A. Cuschieri. 2016. “Current State of Micro-Robots/Devices as Substitutes for Screening Colonoscopy: Assessment Based on Technology Readiness Levels.” Surgical Endoscopy 30 (2): 404–413. doi:10.1007/s00464-015-4263-1.
- U.S. Department of Defense. 2011. “Technology Readiness Assessment (TRA) Guidance.” https://www.afacpo.com/AQDocs/TRA2011.pdf.
- Van Gossum, A., M. Munoz-Navas, I. Fernandez-Urien, C. Carretero, G. Gay, M. Delvaux, M. G. Lapalus, et al. 2009. “Capsule Endoscopy versus Colonoscopy for the Detection of Polyps and Cancer.” The New England Journal of Medicine 361 (3): 264–270. doi:10.1056/NEJMoa0806347.
- Van Norman, G. A. 2016. “Drugs, Devices, and the FDA: Part 2: An Overview of Approval Processes: FDA Approval of Medical Devices.” JACC. Basic to Translational Science 1 (4): 277–287. doi:10.1016/j.jacbts.2016.03.009.
- World Health Organization. 2003. “Medical Device Regulations: Global Overview and Guiding Principles.” France: World Health Organization.
- Wu, Y. H., F. A. Li, Y. T. Fan, and P. W. Tu. 2016. “A Study of Medical Device Regulation Management Model in Asia.” Expert Review of Medical Devices 13 (6): 533–543. doi:10.1080/17434440.2016.1184970.
