
ABSTRACT
Background: We examined the efficacy and safety of saxagliptin as an add-on to insulin in Japanese patients with type 2 diabetes mellitus.
Research design and methods: We randomized 240 patients with type 2 diabetes mellitus on insulin monotherapy to 5-mg saxagliptin or placebo as add-on therapy for a 16-week, double-blind period. All patients received 5-mg saxagliptin and insulin for an additional 36 weeks (open-label extension). Change in hemoglobin A1c (HbA1c) at Week 16 was the main endpoint.
Results: At Week 16, the adjusted change in HbA1c from baseline increased by 0.51% with placebo and decreased by 0.40% with saxagliptin (difference −0.92% [95% confidence interval −1.07%, −0.76%; p < 0.001]). In patients receiving saxagliptin, reductions in HbA1c at Week 16 were maintained to Week 52, while switching from placebo to saxagliptin resulted in a similar reduction in HbA1c. The incidence of hypoglycemia was not markedly increased with saxagliptin versus placebo in the double-blind period and did not increase substantially during the open-label extension period. The efficacy and safety of saxagliptin was similar between the elderly and non-elderly patient groups.
Conclusions: Adding saxagliptin to ongoing insulin therapy improved glycemic control and was well tolerated in Japanese patients with type 2 diabetes.
1. Introduction
The Japan Diabetes Society (JDS) guidelines propose a target hemoglobin A1c (HbA1c) of <6.0% for normoglycemia and <7.0% to prevent complications in patients with diabetes [Citation1]. The guidelines also suggest a target of <8.0% if intensification of therapy is considered difficult. However, treatment objectives should be tailored to individual patients. For example, the JDS/Japan Geriatrics Society (JGS) Joint Committee on Improving Care for Elderly Patients with Diabetes recommends that the target for glycemic control be determined for each elderly patient according to background characteristics and health status (including age, cognitive function, and physical function), comorbidities, risk for severe hypoglycemia, and life expectancy [Citation2].
The JDS guidelines recommend initial diet and exercise therapy and other lifestyle improvements as treatment for patients with type 2 diabetes mellitus (T2DM), while initial pharmacotherapy may involve an oral antidiabetic drug (OAD), insulin, or a glucagon-like peptide-1 receptor agonist (GLP-1RA). If these treatments prove insufficient, the OAD dose should be increased or another OAD added. Alternatively, the patient may be switched to insulin or to an OAD combined with insulin, or switched to a GLP-1RA alone or in combination with an OAD or insulin.
Many Japanese T2DM patients share a common pathological feature of reduced insulin secretion [Citation3], where OAD therapy alone is often insufficient to achieve adequate glycemic control, and therefore, insulin therapy is needed. However, despite the opportunity to titrate the insulin dose and select an insulin type (e.g., long-acting, intermediate-acting, or premixed insulin) that suits the patient’s lifestyle and medical needs, insulin therapy alone does not necessarily provide sufficient or durable improvements in glycemic control. Thus, some patients might be candidates for insulin in combination with other drugs. In particular, the risk of hypoglycemia may limit the extent to which the insulin dose can be increased, while weight gain may occur with long-term insulin treatment. Accordingly, many patients in Japan are prescribed insulin therapy in combination with another OAD to achieve the recommended treatment targets.
Dipeptidyl peptidase (DPP)-4 inhibitors are widely used OADs [Citation3,Citation4]. The enzyme DPP-4 cleaves incretins, which stimulate insulin secretion and suppress glucagon secretion. Inhibition of DPP-4 with DPP-4 inhibitors promotes the action of incretins, which in turn attenuates hyperglycemia [Citation5]. In addition, DPP-4 inhibitors can be used safely even in elderly patients with T2DM [Citation6–Citation8], and are often used in combination with insulin in Japanese diabetic patients. Several clinical studies have examined the efficacy of DPP-4 [Citation9–Citation16] inhibitors as an add-on to insulin therapy. However, thus far, no studies have compared the safety and efficacy of combination therapy with DPP-4 inhibitors and insulin between Japanese elderly and non-elderly diabetic patients in a double-blind, placebo-controlled fashion that includes the meal tolerance test and self-monitoring of blood glucose.
Saxagliptin is a DPP-4 inhibitor approved in Japan for the treatment of T2DM, based in part on results from four studies of Japanese patients that examined the efficacy and safety of saxagliptin as monotherapy or add-on therapy to other OADs [Citation17–Citation20]. Until now, however, no studies have examined the efficacy or safety of saxagliptin in combination with insulin in Japan. We designed this study to test the hypothesis that saxagliptin and insulin combination therapy in Japanese T2DM patients would result in a significant decrease in HbA1c compared with insulin monotherapy, and that this level of adequate glycemic control would be maintained in the long term. We performed a 52-week clinical trial including a 16-week, double-blind period with patients on insulin monotherapy randomized to either 5-mg saxagliptin or placebo. This was followed by a 36-week extension when all patients received open-label saxagliptin with insulin. We further assessed the efficacy and safety of saxagliptin added to insulin monotherapy in elderly and non-elderly T2DM patient groups.
2. Methods
2.1. Ethics
This postmarketing clinical trial was performed in accordance with the Declaration of Helsinki, the Japanese Pharmaceutical Affairs Law, Good Clinical Practice and Good Post-marketing Study Practice. This trial was registered on www.clinicaltrials.jp (registration number: JapicCTI-132348).
2.2. Patients
All patients gave written informed consent to participate. Patients were eligible for this trial if they met the following inclusion criteria: age ≥20 years; diagnosis of T2DM; stable dietary and exercise therapy; fasting C-peptide ≥0.5 ng/mL at screening or at 2 weeks before starting the allocated treatment (Week −2); administration of a stable dose of insulin (daily dose of ≥8 units to ≤40 units) for ≥12 weeks before starting the allocated treatment, with a difference in the total daily insulin dose of ≤20% during the run-in period (patients could use long-acting, intermediate-acting, or premixed insulin [≤50% of insulin as short-acting or rapid-acting insulin as one component] formulations with up to three injections per day); fasting plasma glucose (FPG) at screening and at Week −2 of ≤220 mg/dL; and HbA1c of ≥7.5% to <10.0% at Week −2, and a difference in HbA1c between screening and Week −2 of ≤1.0%. Patients taking insulin monotherapy for ≥14 weeks at Week −6 were eligible, providing their HbA1c at screening was ≥7.5% to <10.0%. Patients taking one OAD for ≥8 weeks at Week −12, who had HbA1c ≥7.0% to ≤9.0% at Week −12 and who could tolerate a washout of the OAD, entered a 12-week washout period prior to randomization. Only patients who recorded ≥80% of the daily insulin doses in the entire run-in period using diaries were eligible for randomization. The major exclusion criteria were similar to those of other clinical trials of saxagliptin in Japan and included patients with: severe ketosis, diabetic coma or precoma, or type 1 diabetes mellitus; other conditions requiring glycemic control with insulin injection (severe infections, serious injuries, or perioperative patients); elevated liver enzymes at Week −2 (aspartate aminotransferase or alanine aminotransferase ≥2.5 times the upper limit of normal); kidney dysfunction (serum creatinine >1.4 mg/dL for men and >1.2 mg/dL for women, or creatinine clearance <50 mL/min at Week −2); difficulty with completing a 52-week trial (e.g., patients with progressive renal disease); low hemoglobin at Week −2 (<11.0 g/dL for men and <10.0 g/dL for women), or history of hemoglobinopathies; hypoglycemic symptoms at least twice a week ≤12 weeks before starting the allocated treatment; poorly controlled hypertension (mean systolic pressure of >160 mmHg or mean diastolic pressure of >100 mmHg calculated as the mean of values at screening and at Week −2); New York Heart Association stage III or IV heart failure; medical history of acute coronary syndrome, percutaneous coronary intervention or coronary artery bypass graft, cerebral stroke or transient ischemic attack, arteriosclerosis obliterans of the lower extremity accompanied by pain at rest, ischemic ulcer, or necrosis (classified as grade III or IV according to the Fontaine classification) ≤6 months before screening; and patients who used any other antihyperglycemic medication during the run-in period.
2.3. Trial design
This trial was performed at 62 centers in Japan. The trial design is illustrated in and comprised a run-in period, a 16-week double-blind randomized period, and a 36-week open-label extension period.
Figure 1. Study design.
OAD: oral antidiabetic drug
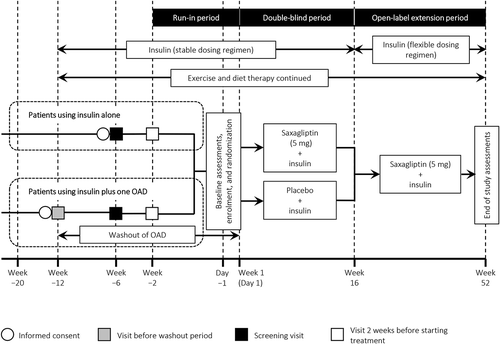
All patients were on insulin monotherapy for 12 weeks before randomization. The following measures were taken to ensure that patients were not exposed to abnormally high blood glucose concentrations. Patients with HbA1c ≥10.0% at Week −2 or FPG >220 mg/dL at Weeks −6 or −2 were ineligible for the study, while those who showed abnormally high blood glucose concentrations after washout were discontinued from the study at that time-point. Before randomization, the daily dose of insulin could be increased or decreased by 20% compared with the prescribed insulin at Week −12. After starting the study treatment, criteria for up-titration of insulin dose (rescue dose) were available. Patients with HbA1c ≥10.4% or FPG ≥270 mg/dL at two consecutive blood samples were discontinued from the study.
Patients were randomized to receive either saxagliptin or placebo on Day −1 using a dynamic allocation method in which patients were stratified according to the following factors: insulin type (long-acting, intermediate-acting, or premixed insulin) as the first factor and HbA1c (<8.5% or ≥8.5%) at Week −2 as the second factor. The randomization allocation sequence, enrollment, and assigning of patients to their interventions were all performed by Bell Medical Solutions (Tokyo, Japan).
Patients could be withdrawn from the trial at any time for the usual reasons in clinical trials (e.g., patient request, investigator decision, inclusion/exclusion criteria contravened, inability to undergo required assessments, adverse events, and pregnancy). In addition, patients were to be discontinued for the following reasons: HbA1c ≥10.4% or FPG ≥270 mg/dL, as measured by the central laboratory in two consecutive blood samples, or altered renal function (serum creatinine > 1.4 mg/dL for men or >1.2 mg/dL for women, or creatinine clearance <50 mL/min).
2.4. Treatments
Patients received either placebo or 5-mg saxagliptin once daily during the double-blind period (both drugs were identical in appearance and packaging), and all patients received 5-mg saxagliptin during the open-label extension period (placebo→saxagliptin and saxagliptin→saxagliptin).
Insulin was to be continued throughout the double-blind period at the same stable dose, provided the criteria for up-titration (rescue dose) or down-titration were not met. During the open-label extension period, the insulin dose could be varied at the physician’s discretion by reference to the criteria for up-titration or down-titration. No changes in insulin product were allowed at any stage of the trial unless they reached the criteria for up-titration as follows. Down-titration of the prescribed insulin dose by 1–4 units per day was permitted according to the investigator’s decision if the patient was deemed to be at increased risk of hypoglycemia. Although the number of down-titrations was not limited, the maximum cumulative reduction in insulin dose was 4 units (relative to the dose used in the run-in period). Down-titration was permitted in the following circumstances: the patient experienced hypoglycemic symptoms with self-monitored blood glucose (SMBG) <70 mg/dL in the absence of acute changes in daily life or physical activity, or two or more consecutive SMBG measurements of <80 mg/dL with a high risk of hypoglycemia as determined by the physician. Up-titration of the insulin dose was permitted if the centrally measured FPG was >240 mg/dL at Week ≤23 or ≥200 mg/dL at Week ≥24 in the absence of acute changes in daily life or physical activity. Patients who did not meet the criteria for up-titration or down-titration but required an increase or decrease in the insulin dose during the double-blind period based on the investigator’s decision were handled as protocol deviations but were not withdrawn from the study.
Patients were prohibited from using drugs likely to interfere with glucose levels (e.g., other OADs, GLP-1RAs, and steroids [except for local/nasal administration]), strong CYP3A4/5 inhibitors (except for external administration), erythropoiesis-stimulating drugs, other investigational drugs, and red blood cell transfusion.
2.5. Assessments and endpoints
All clinical and laboratory results were analyzed centrally by LSI Medience Corporation (Tokyo, Japan), except for SMBG, which was measured by patients at home. HbA1c and FPG were measured at each visit. 1,5-anhydroglucitrol, fasting C-peptide, and fasting glucagon were measured at baseline, and at Weeks 16, 32, and 52 (or at discontinuation). Patients were instructed to perform 7-point SMBG measurements (taken before and 2 h after each main meal, and at bedtime) at baseline and Weeks 16 and 52. Meal tolerance tests were performed at baseline and Week 16, in which patients consumed a standard meal comprising 8.8 g (19.8%) of fat, 15.2 g (15.2%) of protein, and 65.0 g (65.0%) of carbohydrate (total 400 kcal). Plasma glucose, C-peptide, and glucagon levels were measured before the meal, and at 30 min, 1 h, and 2 h after the meal. C-peptide and glucagon in either fasting state or during the meal tolerance test were measured by chemiluminescent immunoassay and radioimmunoassay, respectively.
The primary efficacy endpoint was the change in HbA1c from baseline to Week 16. Secondary efficacy endpoints included the changes in 2-h postprandial plasma glucose (PPG) and area under the plasma glucose concentration-time curve from 0 to 2 h (AUC0–120min) in the meal tolerance test, and the change in FPG from baseline to Week 16. Exploratory efficacy endpoints included the changes in HbA1c at each time-point and the proportion of patients with HbA1c <7.0% at each time-point during the double-blind and open-label extension periods.
Safety-related variables were recorded throughout the trial, and included treatment-emergent adverse events (TEAEs), clinical laboratory tests, and vital signs. Investigators assessed the AEs in terms of severity, duration, and whether they were drug-related. Hypoglycemia was confirmed by investigators based on hypoglycemic symptoms recorded in patient diaries. Low blood glucose without hypoglycemic symptoms (SMBG value <50 mg/dL) was classified as ‘blood glucose decreased.’ Twelve-lead electrocardiography was performed at baseline, Week 16, and Week 52 (or at discontinuation).
2.6. Sample size
The difference in the change from baseline in HbA1c at Week 16 between the saxagliptin + insulin group and the placebo + insulin group was assumed to be −0.4% with an SD of 1.0% based on the saxagliptin trials that were conducted outside Japan [Citation21–Citation25]. The sample size of 100 patients per group provided 90% power to detect the difference between saxagliptin and placebo using a t test at a two-sided significance level of 5%. Considering unevaluable patients (5%), the planned sample size was 105 patients per group.
2.7. Statistical analyses
The primary efficacy endpoint (change from baseline in HbA1c at Week 16) was analyzed using the analysis of covariance (ANCOVA) model with terms for treatment group, prior treatment with OADs, and type of insulin as fixed effects and baseline HbA1c as a covariate. Based on the ANCOVA model, the adjusted change in HbA1c from baseline and its 95% confidence intervals were calculated for each group. To demonstrate the superiority of saxagliptin, a t test based on the adjusted change from baseline was conducted at a two-sided significance level of 5%. The 95% confidence interval of the difference in the adjusted change from baseline between saxagliptin and placebo was also calculated. The changes in FPG and meal tolerance test-related variables were compared between the two groups in the same way as HbA1c. The last observation carried forward (LOCF) method was used to handle missing data at Week 16 for HbA1c and FPG. Values of HbA1c and FPG observed after the insulin dose increased were handled as missing values. The M value was calculated using SMBG values [Citation26].
Safety endpoints were analyzed descriptively in terms of the mean ± SD or number (percent) of patients. Data analyses were performed using SAS version 9.2 (SAS Institute, Cary, NC, USA). There were no changes to the preplanned statistical analysis after enrollment began.
We also performed post hoc analyses for further interpretation of the data obtained in this study after conducting the prespecified analyses. We calculated the C-peptide index (CPI; C-peptide divided by postprandial glucose in meal tolerance test and multiplied by 100) and insulin secretion rate (ISR) [Citation27] to evaluate beta-cell function. Subgroup analyses of the efficacy (except for change from baseline in HbA1c at Week 16, prespecified in the statistical analysis plan) and safety endpoints by age (e.g., <65 vs. ≥65 years) and by insulin type (premixed/intermediate-acting vs. long-acting) were performed. Regarding the exploratory and post hoc analyses, summary statistics were used to evaluate the profiles over time, and no formal statistical tests were conducted.
3. Results
3.1. Patients
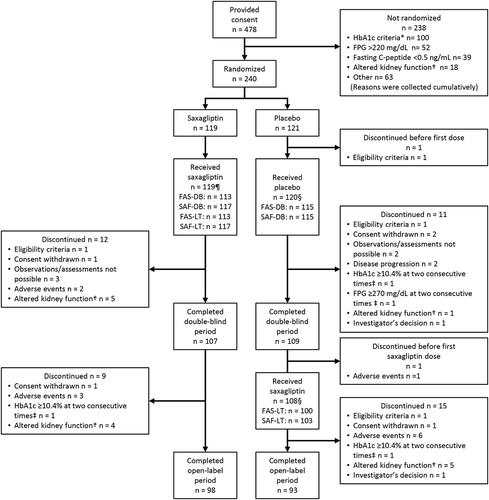
This trial was performed between November 2013 and December 2015. Of 478 patients who provided informed consent, 240 were randomized (119 to saxagliptin and 121 to placebo). The remaining 238 patients were not randomized, mainly due to not meeting HbA1c, FPG, C-peptide, or kidney dysfunction criteria (). One patient allocated to placebo discontinued before the first dose. A total of 107 and 109 patients in the saxagliptin and placebo groups, respectively, completed the double-blind period while 98 and 93, respectively, completed the open-label extension period (). The main reasons for discontinuing the trial included altered kidney function and inability to conduct observation or assessment in the double-blind period, and altered kidney function and TEAEs in the open-label extension period (). Seven patients whose data were not sufficiently reliable were excluded from all analysis sets. The numbers of patients included in the full and safety analysis sets in each treatment period are presented in . The baseline characteristics of patients are shown in and Supplemental Table 1 with no marked differences between the groups. Two-thirds of patients were using premixed insulin and the remainder was using long-acting insulin; only one patient used intermediate-acting insulin.
Table 1. Demographic and other baseline characteristics in the full analysis set from the double-blind treatment period.
Figure 2. Patient disposition.
Regarding the reasons why some patients were not randomized, these were collected cumulatively. Note that some patients had more than one reason for not being randomized. For this reason, the total number of patients does not add up to 238. *A hundred patients were not randomized because they did not meet HbA1c inclusion criteria at Weeks −12, −6, or −2. †Serum creatinine >1.4 mg/dL (males) or >1.2 mg/dL (females) or creatinine clearance <50 mL/min. ‡Centrally measured. Reasons for non-inclusion in the analysis sets following first dose of study drug (in the double-blind period) or open-label saxagliptin (in the open-label extension period) are as follows: ¶Saxagliptin. FAS-DB: Four patients without any evaluable HbA1c data obtained after starting the allocated treatment, two patients whose data were not sufficiently reliable. SAF-DB: Two patients whose data were not sufficiently reliable. FAS-LT: Four patients without any evaluable HbA1c data obtained after starting the allocated treatment, two patients whose data were not sufficiently reliable. SAF-LT: Two patients whose data were not sufficiently reliable. §Placebo. FAS-DB: Five patients whose data were not sufficiently reliable. SAF-DB: Five patients whose data were not sufficiently reliable. FAS-LT: Three patients without any evaluable HbA1c data obtained after starting open-label saxagliptin, five patients whose data were not sufficiently reliable. SAF-LT: Five patients whose data were not sufficiently reliable. FAS-DB, full analysis set during the double-blind period; FAS-LT, full analysis set during the long-term period; SAF-DB, safety analysis set during the double-blind period; SAF-LT, safety analysis set during the long-term period.
3.2. Insulin doses
The mean total daily insulin doses (95% confidence interval) (in units) at baseline, Week 16, and Week 52 for patients in the full analysis set (double-blind period) were 22.91 (21.24, 24.59) (n = 113), 22.67 (20.96, 24.37) (n = 106), and 23.06 (21.15, 24.97) (n = 96), respectively, in the saxagliptin→saxagliptin group. The corresponding values in the placebo→saxagliptin group were 23.73 (22.09, 25.36) (n = 115), 23.40 (21.71, 25.10) (n = 106), and 24.10 (22.13, 26.08) (n = 90). During the double-blind period, the insulin dose of one patient in the saxagliptin group (0.9%) was increased because of high HbA1c levels based on their own judgment, while one in the placebo group (0.9%) required dose increases and decreases applicable to a sliding-scale regimen during hospitalization because of an AE. No patients met the criteria for up-titration in either group. Six patients in the saxagliptin group (5.3%) and one in the placebo group (0.9%) met the criteria for down-titration. However, for one patient in the saxagliptin group (0.9%) and one in the placebo group (0.9%), the treating physicians decided to decrease the dose because these patients were considered at an increased risk of hypoglycemia.
3.3. Efficacy of saxagliptin
3.3.1. HbA1c
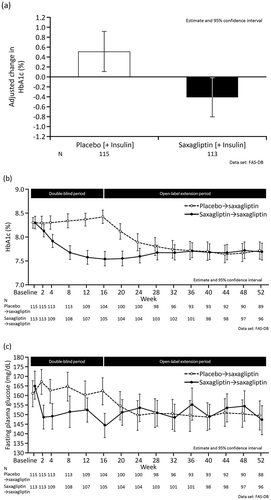
shows the adjusted changes in HbA1c from baseline in both groups. HbA1c increased by 0.51% in the placebo group and decreased by 0.40% in the saxagliptin group between baseline and Week 16 (LOCF), corresponding to a difference of −0.92% (95% confidence interval −1.07%, −0.76%; p < 0.001). As illustrated in , HbA1c was continuously lower in the saxagliptin group than in the placebo group during the double-blind period. In patients who used saxagliptin in both treatment periods, the reduction in HbA1c at Week 16 was maintained through to Week 52 ().
Switching from placebo to saxagliptin at Week 16 was associated with a reduction in HbA1c that was maintained through to the end of the open-label extension period (). This reduction was similar to that in patients who received saxagliptin in both periods.
Figure 3. (a) Adjusted change in HbA1c (%) from baseline to Week 16 (with last observation carried forward). (b) Changes in HbA1c over time in the double-blind period and open-label extension period. (c) Changes in fasting plasma glucose over time in the double-blind period and open-label extension period. HbA1c: hemoglobin A1c; FAS-DB: full analysis set during the double-blind period.
Results of the subgroup analysis of the changes in HbA1c at the end of the double-blind period are shown in . As indicated in this table, saxagliptin was associated with reductions in HbA1c in all subgroups of patients relative to placebo. Changes in HbA1c in patients taking saxagliptin in the double-blind period were largely maintained through the end of the open-label extension period; furthermore, patients administered placebo in the double-blind period and saxagliptin in the open-label extension period also had reductions in HbA1c by Week 52 (Supplemental Table 2).
Table 2. Subgroup analysis of the changes in HbA1c (%) from baseline to the end of the double-blind treatment period.
Overall, 22/105 patients (21.0%) in the saxagliptin group and 1/104 (1.0%) patients in the placebo group achieved HbA1c <7.0% at Week 16. For the long-term period, 16/96 patients (16.7%) in the saxagliptin→saxagliptin group (Weeks 0–52) and 14/89 (15.7%) in the placebo→saxagliptin group (Weeks 16–52 of treatment only) had achieved HbA1c < 7.0% at Week 52. The proportion of patients with therapeutic glycemic response by age (7.0% in those aged <65 or ≥65 years, 7.5% in those aged ≥65 and <75 years, and 8.0% in those aged ≥75 years) are shown in Supplemental Table 3.
3.3.2. Meal tolerance test
and Supplemental Table 4 show the changes in meal tolerance test-related variables at baseline and Week 16. As indicated, 2-h PPG and AUC0–120min for plasma glucose decreased in the saxagliptin group, but not in the placebo group. There was no difference between the two groups in the change in C-peptide from baseline. The reduction in concentrations and AUC0–120min for glucagon was numerically greater in the saxagliptin group than in the placebo group. The CPI at Week 16 in the saxagliptin group increased relative to baseline, while the CPI did not change in the placebo group. Increases in ISR at each plasma glucose concentration from baseline to Week 16 in the saxagliptin group were numerically greater than in the placebo group. A similar trend was observed in the subgroup analysis by age (≥65 years, <65 years) and by insulin type (premixed/intermediate-acting, long-acting) (Supplemental Tables 5 and 6, and Supplemental Figures 1 and 2).
Figure 4. Mean values during the meal tolerance test at baseline and at the end of the double-blind period in the full analysis set. (a) Plasma glucose (mg/dL) in the saxagliptin group at baseline and Week 16. (b) Plasma glucose (mg/dL) in the placebo group at baseline and Week 16. (c) Mean change in plasma glucose (mg/dL) from baseline at Week 16. (d) C-peptide (ng/mL) in the saxagliptin group at baseline and Week 16. (e) C-peptide (ng/mL) in the placebo group at baseline and Week 16. (f) Mean change in C-peptide (ng/mL) from baseline at Week 16. (g) Glucagon (pg/mL) in the saxagliptin group at baseline and Week 16. (h) Glucagon (pg/mL) in the placebo group at baseline and Week 16. (i) Mean change in glucagon (pg/mL) from baseline at Week 16. (j) C-peptide Index in the saxagliptin group at baseline and Week 16. (k) C-peptide Index in the placebo group at baseline and Week 16. (l) Insulin secretion rate (pmol/kg/min) at each plasma glucose concentration (mg/dL) in the saxagliptin group at baseline and Week 16. (m) Insulin secretion rate (pmol/kg/min) at each plasma glucose concentration (mg/dL) in the placebo group at baseline and Week 16. (l) and (m) are regression lines.
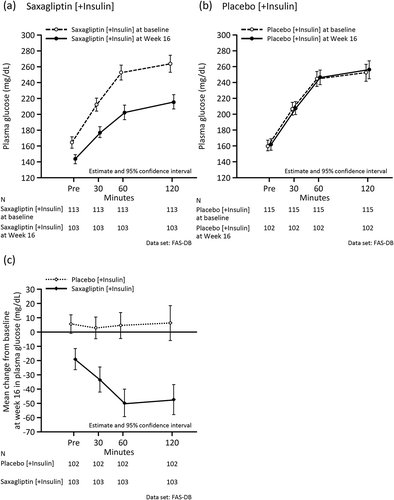
Figure 4. (continued).
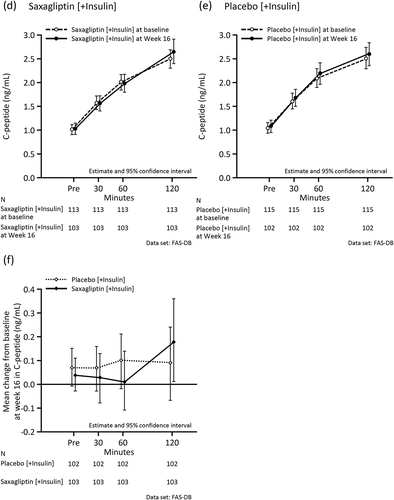
Figure 4. (continued).
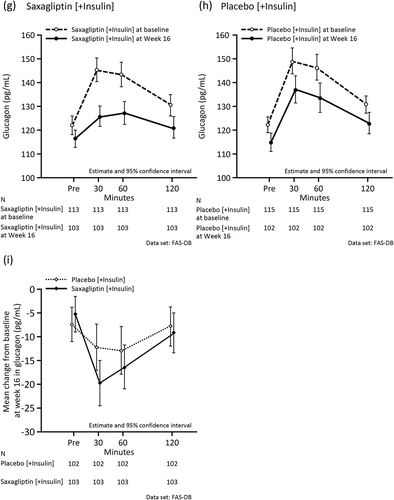
Figure 4. (continued).

3.3.3. FPG and SMBG
Consistent with the changes in HbA1c, we also observed a greater reduction in FPG from baseline to the end of the double-blind period () in the saxagliptin group. The adjusted change in FPG from baseline to Week 16 (LOCF) was 30.3 mg/dL (95% confidence interval 9.2, 51.5) in the placebo group and 11.2 mg/dL (95% confidence interval −9.5, 31.8) in the saxagliptin group, corresponding to a difference of −19.2 mg/dL (95% confidence interval −27.2, −11.1). FPG was also continuously lower in the saxagliptin group during the double-blind period and the reduction in FPG was maintained through to Week 52 ().
SMBG was measured at baseline, Week 16, and Week 52, and the results are shown in . SMBG values at each time of day decreased in the saxagliptin group but not in the placebo group in the double-blind period (, ). The reductions in SMBG observed in the saxagliptin group were maintained through Week 52 (). A similar trend was observed in the subgroup analysis by age. In Supplemental Figure 3, we show the SMBG values from baseline to Week 16 and through Week 52 in patients aged <65 years (Supplemental Figure 3a–c) and those aged ≥65 years (Supplemental Figure 3d–f).
Figure 5. Self-monitored blood glucose values in the double-blind (a, b) and the double-blind period and open-label extension periods (c) according to the treatment received.
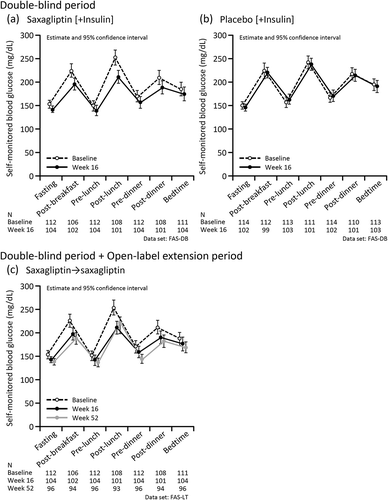
The mean M values (95% confidence interval) at baseline and Week 16 in the double-blind period were 46.16 (39.45, 52.88) (n = 105) and 33.75 (27.56, 39.93) (n = 99), respectively, in the saxagliptin group. The corresponding values in the placebo group were 45.44 (39.52, 51.36) (n = 108) and 41.38 (36.17, 46.59) (n = 99). In the long-term period, the mean M values (95% confidence interval) at baseline, Week 16, and Week 52 were 46.16 (39.45, 52.88) (n = 105), 33.75 (27.56, 39.93) (n = 99), and 29.29 (24.99, 33.59) (n = 92), respectively, in the saxagliptin→saxagliptin group. A decrease in the M value was also observed in the placebo→saxagliptin group: 41.21 (35.87, 46.56) (n = 95) at Week 16 and 33.09 (28.42, 37.75) (n = 82) at Week 52.
3.3.4. Fasting C-peptide, fasting glucagon, and 1,5-anhydroglucitol
In terms of other efficacy variables, saxagliptin was associated with no changes in fasting C-peptide or fasting glucagon, and an increase in 1,5-anhydroglucitol during the double-blind period, and these results were maintained through to Week 52 (). A similar trend was observed in the subgroup analysis by age (≥65 years, <65 years) (Supplemental Table 7).
Table 3. Clinical efficacy variables during the double-blind and open-label extension periods.
3.4. Safety
3.4.1. TEAEs
In the double-blind period, TEAEs and drug-related TEAEs occurred in 62.4% and 23.1% of patients in the saxagliptin group versus 53.0% and 21.7% of patients in the placebo group (). Using data from both treatment periods, the incidences of TEAEs and drug-related TEAEs were 84.6% and 40.2% for the saxagliptin→saxagliptin group (Weeks 0–52) and 71.8% and 26.2% for the placebo→saxagliptin group (Weeks 16–52 of treatment only).
Table 4. TEAEs in ≥5% of patients in either treatment group, serious TEAEs, and TEAEs of special interest.
TEAEs in ≥5% of patients in either group are summarized in , the most common being hypoglycemia and nasopharyngitis in both groups and in both treatment periods. Hypoglycemia was also the most common drug-related TEAE in both the double-blind period and the long-term period.
3.4.2. Hypoglycemia
In the double-blind period, hypoglycemia occurred as a TEAE and as a drug-related TEAE in 20.5% and 15.4% of patients in the saxagliptin group. The corresponding values in the placebo group were 14.8% and 13.0%. ‘Blood glucose decreased’ was reported as a TEAE and drug-related TEAE in 1.7% and 0.9% patients in the saxagliptin group, respectively, but was not observed in the placebo group. The incidence of hypoglycemia was not markedly increased with saxagliptin relative to placebo in the double-blind period. In the long-term period, the incidence of hypoglycemia and ‘blood glucose decreased’ was not markedly increased in the saxagliptin→saxagliptin group relative to the saxagliptin group in the double-blind period. Additionally, the incidence of hypoglycemia in the placebo→saxagliptin group in the open-label extension period was similar to that in the placebo group in the double-blind period (). The incidences of hypoglycemia and ‘blood glucose decreased’ by age and by insulin type in both treatment periods are shown in and Supplemental Table 8.
Table 5. Subgroup analysis of the incidence of hypoglycemia, blood glucose decreased, and serious TEAEs by age.
The severity of hypoglycemia and ‘blood glucose decreased’ was also assessed. In the double-blind period, one episode of hypoglycemia in the saxagliptin group was rated as severe and two episodes in the placebo group were rated as moderate. There were no moderate or severe cases of hypoglycemia or ‘blood glucose decreased’ in the open-label extension period. There were no withdrawals because of hypoglycemia or ‘blood glucose decreased’ in either treatment period.
The patient who developed severe hypoglycemia was a 42-year-old woman treated with twice-daily premixed insulin, which contains 50% of rapid-acting insulin. The episode occurred shortly after the administration of insulin with dinner 14 days after starting the allocated study treatment. At the time of hypoglycemia, the patient presented blurred vision and unconsciousness. She was treated with intravenous glucose infusion and recovered in 1 h and 40 min after the onset of symptoms. From the following day onward, she continued administration of the study drug with a decreased dose of insulin.
3.4.3. Laboratory variables and vital signs
Body weight remained unchanged during the trial (Supplemental Table 9). Among patients treated with saxagliptin in both treatment periods (saxagliptin→saxagliptin), the mean (±SD) body weight was 64.28 ± 10.87 kg (n = 117) at baseline, 65.09 ± 11.15 kg (n = 105) at Week 16, and 65.02 ± 10.99 kg (n = 96) at Week 52. The corresponding values in the placebo→saxagliptin group were 66.61 ± 15.27 kg (n = 115), 66.31 ± 15.17 kg (n = 106), and 67.08 ± 16.32 kg (n = 89), respectively. There were no clinically relevant changes in laboratory variables, other vital signs, or 12-lead electrocardiogram in either period.
4. Discussion
Administration of saxagliptin in combination with insulin was associated with improvements in glycemic control and meal tolerance test parameters during the 16-week, randomized, placebo-controlled double-blind period that were maintained through the end of the 36-week open-label extension period. Switching from placebo to open-label saxagliptin was also associated with improvements in both HbA1c and FPG during the open-label extension period that were similar to those observed in the saxagliptin group in the double-blind period. Furthermore, the glycemic improvement in this study is consistent with the results of studies of saxagliptin monotherapy [Citation19,Citation20] and combination therapy with OADs [Citation20].
Consistent improvements in HbA1c were observed in subgroups of patients divided by baseline characteristics, including age, insulin type, FPG, fasting C-peptide, and daily insulin dose, suggesting that the efficacy of saxagliptin in combination with insulin is largely independent of patient characteristics.
In the meal tolerance test, a decrease of postprandial glucose and suppression of glucagon secretion were observed in the saxagliptin group, although the mean changes of postprandial C-peptide from baseline to Week 16 were similar between both groups. However, from baseline to Week 16, the increases of CPI and ISR at each plasma glucose concentration in the saxagliptin group were greater compared with the placebo group. This suggests that saxagliptin may contribute to the improvement of beta-cell function. In the SAVOR-TIMI 53 study, Leibowitz et al. showed that saxagliptin improved glycaemia and prevented the reduction in HOMA2-beta values, which may result in less decline of beta-cell function in patients treated with saxagliptin than in those treated with placebo. However, their patients were not undergoing insulin treatment [Citation28]. In our study, we assessed beta-cell function in patients who were treated with insulin. Our findings suggest that treatment with insulin for patients with more advanced diabetes may result in improved beta-cell function. The blood glucose-lowering effect and increases of CPI and ISR were observed in the meal tolerance test regardless of age or insulin type. These results suggest that saxagliptin may improve beta-cell function in various clinical settings.
In this study, we focused on elderly patients and performed subgroup analyses of the efficacy endpoints. The results of these analyses showed that the efficacy of saxagliptin was similar between elderly patients (≥65 years) and non-elderly patients (<65 years).
This combination also proved to be well tolerated in terms of the low rates of TEAEs and drug-related TEAEs. The most common TEAE/drug-related TEAE was hypoglycemia in the double-blind period. During this period, the incidence was not substantially increased in the saxagliptin group compared with the placebo group, suggesting that many episodes of hypoglycemia were related to insulin rather than saxagliptin. Moreover, switching from placebo to saxagliptin at the start of the open-label extension period did not markedly increase the incidence of hypoglycemia. Additionally, only one episode of severe hypoglycemia was reported in the saxagliptin group in the double-blind period and two episodes of moderate hypoglycemia in the placebo group; all of the other episodes were classified as mild. These results suggest that saxagliptin does not substantially increase the incidence or severity of hypoglycemia in insulin-treated patients. Furthermore, the episodes of hypoglycemia were well tolerated regardless of patient age. The patient who developed severe hypoglycemia was a 42-year-old woman, indicating that the severity of hypoglycemia is not age-related.
In the present study, there was very little change in the insulin dose in both study arms. The reason for this is that no patients met the criteria for up-titration in either group in the double-blind period, which was as short as 16 weeks. In the open-label period, all subjects used saxagliptin and insulin in combination. Although the dose of insulin was changed in some patients, the mean dose of insulin was stable during the open-label period.
Several other studies have examined the efficacy and safety of DPP-4 inhibitors in combination with insulin in Japanese patients [Citation9–Citation16]. There appear to be similar trends between the present study results and the results from combination therapy studies using other DPP-4 inhibitors and insulin.
Some aspects of our trial warrant mention. In particular, insulin may be used in combination with other OADs in Japan, especially in patients with poorly controlled diabetes, but our trial evaluated only the combination of insulin and saxagliptin. By contrast, in trials of vildagliptin and anagliptin, patients could continue another OAD and insulin at the time of starting the DPP-4 inhibitor [Citation9,Citation13], and this may have influenced the changes in glycemic control during the trials. In clinical practice, it is feasible that some patients will be prescribed a DPP-4 inhibitor, insulin, and another OAD as intensive therapy.
It should be noted that the Japanese diabetes clinical practice guideline recommends not only metformin but also other OADs, insulin, and GLP-1RAs as the first-line agents for initial drug therapy because Japanese diabetes patients have a different pathology and lifestyle compared with diabetes patients in Western countries. Therefore, combination therapy with a DPP-4 inhibitor and insulin without metformin is the usual practice in Japan; no subjects took metformin during the present study. The mean BMI of patients in the present study was relatively low compared with the BMI of T2DM in Western countries. Our findings highlight the efficacy of saxagliptin in patients with low BMI and/or reduced insulin secretion, which are common features shared by East Asian T2DM patients [Citation3].
In our trial, the saxagliptin dose was fixed at 5 mg, whereas a sitagliptin trial permitted an increase in the dose to 100 mg in patients meeting the following criteria: FPG ≥140 mg/dL from Week 20 through to Week 32 or HbA1c ≥7.4% from Week 28 through to Week 32 [Citation10]. The sitagliptin dose was increased in 113/125 patients in the sitagliptin→sitagliptin group and in 113/129 patients in the placebo→sitagliptin group. This led to a further improvement in HbA1c, such that 20/172 patients with HbA1c ≥7.4% before the dose increase achieved HbA1c <7.4% at 16 weeks after the dose increase.
Fifteen patients were withdrawn from this study owing to changes in markers of renal dysfunction. Although non-Japanese trials have documented the efficacy and safety of saxagliptin in patients with renal impairment [Citation29,Citation30], a lower dose of saxagliptin (2.5 mg/day) is recommended in patients with moderate or severe renal impairment or end-stage renal disease (ESRD) [Citation31]. This lower dose is also permitted in Japanese patients with moderate or severe renal dysfunction or ESRD. In clinical practice, patients who receive 2.5 mg of saxagliptin are able to continue combination therapy with insulin. In this trial, to examine the efficacy and safety of a stable dose of saxagliptin as add-on therapy to insulin, the saxagliptin dose was fixed and patients were withdrawn from the study once their serum creatinine or creatinine clearance rate met the criteria for discontinuation. In clinical practice, it is possible to change the dose of saxagliptin, and kidney function could be assessed by several serum creatinine measurements and other clinical laboratory values. In this regard, our study does not reflect the actual clinical situation.
Another aspect of our trial that warrants mention is that this study was not planned to adjudicate major adverse cardiovascular events including hospitalization for heart failure; therefore, we cannot compare the safety results of the present study with those of the SAVOR-TIMI 53 study.
Our results highlight areas for future research. In our study, we assessed patients by age with a cut-off of 65 years. In the future, we hope to evaluate the safety and efficacy of saxagliptin in elderly patients with more advanced ages (e.g., ≥75 years). In our increasingly aging society, treatments more specifically tailored to the condition of each patient are required. The results from the wealth of studies on DPP-4 inhibitors also warrant further investigation to ascertain the relative suitability of different DPP-4 inhibitors for different populations and to assess the value of different combinations of OADs with or without insulin.
5. Conclusions
In conclusion, this trial revealed that adding saxagliptin to ongoing insulin therapy was associated with clinically meaningful improvements in glycemic control during the 16-week randomized double-blind period and was well tolerated in Japanese patients with T2DM, with a low rate of hypoglycemia, particularly severe hypoglycemia.
Of note, the improvements observed at 16 weeks were maintained through the end of the 36-week open-label extension period and the incidence of TEAEs and drug-related TEAEs, such as hypoglycemia, did not increase substantially during this period.
The efficacy of saxagliptin was also apparent in subgroups of patients, suggesting the reduction in HbA1c is largely independent of patient characteristics, such as age, insulin type, baseline FPG, fasting C-peptide, and insulin dose. The efficacy and safety of saxagliptin was similar between the elderly and the non-elderly patients.
Declaration of interest
T Kadowaki has received personal fees from Kyowa Hakko Kirin Co., Ltd. during the conduct of the study; grants and personal fees from Kyowa Hakko Kirin Co., Ltd.; grants, personal fees other support from Takeda Pharmaceuticals Co., Ltd.; grants and personal fees from MSD Co., Ltd.; personal fees from Boehringer Ingelheim Co., Ltd.; personal fees and other support from Novo Nordisk; grants, personal fees and other support from Mitsubishi Tanabe Pharma Corporation; grants, personal fees and other support from Daiichi Sankyo Co., Ltd.; personal fees and other support from Taisho Pharmaceuticals Co., Ltd.; grants and personal fees from Astellas Pharma Inc.; grants and personal fees from Sanofi K.K.; grants from Chugai Pharmaceuticals Co., Ltd.; grants and personal fees from Ono Pharmaceuticals Co., Ltd.; personal fees from Kowa Pharmaceuticals Co., Ltd.; grants and personal fees from Sumitomo Dainippon Pharma Co., Ltd.; personal fees from Sanwa Kagaku Kenkyusho Co., Ltd.; personal fees from Eli Lilly Japan Co., Ltd.; personal fees from Novartis Pharma Co., Ltd.; grants and personal fees from Taisho Toyama Pharmaceuticals Co., Ltd.; personal fees from Kissei Pharmaceuticals Co., Ltd.; personal fees from Kaken Pharmaceuticals Co., Ltd.; personal fees from Teijin Pharma Ltd.; and grants and personal fees from AstraZeneca K.K., outside the submitted work. S Muto, Y Ouchi and R Shimazaki are employees of Kyowa Hakko Kirin Co., Ltd. Y Seino reports personal fees from Kyowa Hakko Kirin Co., Ltd. during the conduct of the study; personal fees from Kao Corporation; personal fees from Taisho Pharmaceuticals Co., Ltd.; personal fees from Becton Dickinson and Company; grants and personal fees from Novo Nordisk Pharma Ltd.; personal fees from MSD Co., Ltd.; personal fees from Intarcia Therapeutics, Inc.; personal fees from Johnson & Johnson K.K.; personal fees from GlaxoSmithKline K.K.; personal fees from Takeda Pharmaceuticals Co., Ltd.; personal fees from Sanofi K.K.; grants and personal fees from Taisho Toyama Pharmaceutical Co., Ltd.; personal fees from Eli Lilly and Company; personal fees from Mitsubishi Tanabe Pharma Corporation; grants and personal fees from Ono Pharmaceutical Co., Ltd.; grants and personal fees from Kowa Pharmaceutical Co., Ltd.; personal fees from Astellas Pharma Inc.; personal fees from Boehringer Ingelheim Co., Ltd.; personal fees from Kyowa Hakko Kirin Co., Ltd.; personal fees from AstraZeneca K.K.; grants and personal fees from Sumitomo Dainippon Pharma Co., Ltd.; personal fees from Daiichi Sankyo Co., Ltd.; grants and personal fees from Terumo Corporation; personal fees from Arkray Inc.; grants from Arkray Marketing Inc.; and grants from Hayashibara Co., Ltd. outside the submitted work. Medical writing support, provided by Nicholas D Smith (Edanz Medical Writing) and Marion Barnett (Edanz Medical Writing) was utilized in the production of this manuscript and funded by Kyowa Hakko Kirin Co., Ltd. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Supplementary_material.zip
Download Zip (2.6 MB)Acknowledgments
The authors thank all the patients, investigators, and trial teams who were involved in this trial. The authors thank Dr. Roman Hovorka for developing and distributing the ISEC program.
Supplemental data
Supplemental data for this article can be accessed here.
Additional information
Funding
Notes on contributors
Takashi Kadowaki
All authors contributed to trial design, data interpretation, and drafting of the manuscript. Takashi Kadowaki was the coordinating investigator and Yutaka Seino was the medical advisor for this trial. Satsuki Muto, Yoshiumi Ouchi, and Ryutaro Shimazaki are employees of the sponsor. The sponsor was responsible for trial design, supply of trial products, monitoring, data collection, statistical analyses, and preparation of the clinical trial report.
References
- Araki E, Haneda M, Kasuga M, et al. New glycemic targets for patients with diabetes from the Japan diabetes society. Diabetol Int. 2016;7(4):327–330.
- Japan Diabetes Society (JDS)/Japan Geriatrics Society (JGS) Joint Committee on Improving Care for Elderly Patients with Diabetes.. Committee report: glycemic targets for elderly patients with diabetes. Diabetol Int. 2016;7(4):331–333.
- Seino Y, Kuwata H, Yabe D. Incretin-based drugs for type 2 diabetes: focus on East Asian perspectives. J Diabetes Invest. 2016;7(Suppl1):102–109.
- Deacon CF, Holst JJ. Dipeptidyl peptidase-4 inhibitors for the treatment of type 2 diabetes: comparison, efficacy and safety. Expert Opin Pharmacother. 2013;14(15):2047–2058.
- Wani JH, John-Kalarickal J, Fonseca VA. Dipeptidyl peptidase-4 as a new target of action for type 2 diabetes mellitus: a systematic review. Cardiol Clin. 2008;26(4):639–648.
- Strain WD, Lukashevich V, Kothny W, et al. Individualised treatment targets for elderly patients with type 2 diabetes using vildagliptin add-on or lone therapy (INTERVAL): a 24 week, randomised, double-blind, placebo-controlled study. Lancet. 2013;382(9890):409–416.
- Barnett AH, Huisman H, Jones R, et al. Linagliptin for patients aged 70 years or older with type 2 diabetes inadequately controlled with common antidiabetes treatments: a randomised, double-blind, placebo-controlled trial. Lancet. 2013;382(9902):1413–1423.
- Umezawa S, Kubota A, Maeda H, et al. Two-year assessment of the efficacy and safety of sitagliptin in elderly patients with type 2 diabetes: post hoc analysis of the ASSET-K study. BMC Endocr Disord. 2015;15:34.
- Hirose T, Suzuki M, Tsumiyama I. Efficacy and safety of vildagliptin as an add-on to insulin with or without metformin in Japanese patients with type 2 diabetes mellitus: a 12-week, double-blind, randomized study. Diabetes Ther. 2015;6:559–571.
- Kadowaki T, Tajima N, Odawara M, et al. Efficacy and safety of sitagliptin add-on therapy in Japanese patients with type 2 diabetes on insulin monotherapy. Diabetol Int. 2013;4(3):160–172.
- Kaku K, Mori M, Kanoo T, et al. Efficacy and safety of alogliptin added to insulin in Japanese patients with type 2 diabetes: a randomized, double-blind, 12-week, placebo-controlled trial followed by an open-label, long-term extension phase. Expert Opin Pharmacother. 2014;15(15):2121–2130.
- Katsuno T, Ikeda H, Namba M. Medium-term effect of add-on therapy with the DPP-4 inhibitor, sitagliptin, in insulin-treated Japanese patients with type 2 diabetes mellitus. Diabetes Ther. 2016;7:309–320.
- Kaku K, Honda D, Tsubamoto Y, et al. Efficacy and safety of anagliptin add to insulin therapy in Japanese patients with type 2 diabetes: a multi-center, randomized, placebo-controlled, double-blind, parallel-group study with an open-label, long-term extension. Jpn Pharmacol Ther. 2015;43(12):1663–1686.
- Tajiri Y, Kawano S, Hirao S, et al. Adding of sitagliptin on insulin therapy effectively and safely reduces a hemoglobin A1c level and glucose fluctuation in Japanese patients with type 2 diabetes. Int Sch Res Notices. 2014;2014:639489.
- Takai M, Ishikawa M, Maeda H, et al. Safety and efficacy of adding sitagliptin to insulin in patients with type 2 diabetes: the ASSIST-K study. Diabetes Res Clin Pract. 2014;103:e30–3.
- Kadowaki T, Kondo K, Sasaki N, et al. Efficacy and safety of teneligliptin add-on to insulin monotherapy in Japanese patients with type 2 diabetes mellitus: a 16-week, randomized, double-blind, placebo-controlled trial with an open-label period. Expert Opin Pharmacother. 2017;18:1291–1300.
- Konya H, Yano Y, Matsutani S, et al. Profile of saxagliptin in the treatment of type 2 diabetes: focus on Japanese patients. Ther Clin Risk Manag. 2014;10:547–558.
- Kadowaki T. Effcacy and safety of a novel DPP-4 inhibitor saxagliptin in patients with type 2 diabetes mellitus — the results of clinical trials in Japan. Jpn Pharmacol Ther. 2013;41(8):803–815.
- Seino Y. Efficacy and safety of saxagliptin in Japanese patients with type 2 diabetes — two multi-centre, randomized, double-blind, placebo-controlled studies. Jpn Pharmacol Ther. 2014;42(7):503–518.
- Seino Y. Efficacy and safety of long-term saxagliptin monotherapy and add-on therapy in Japanese patients with type 2 diabetes. Jpn Pharmacol Ther. 2014;42:519–534.
- Barnett AH, Charbonnel B, Donovan M, et al. Effect of saxagliptin as add-on therapy in patients with poorly controlled type 2 diabetes on insulin alone or insulin combined with metformin. Curr Med Res Opin. 2012;28(4):513–523.
- Barnett AH, Charbonnel B, Li J, et al. Saxagliptin add-on therapy to insulin with or without metformin for type 2 diabetes mellitus: 52-week safety and efficacy. Clin Drug Investig. 2013;33(10):707–717.
- Rosenstock J, Sankoh S, List JF. Glucose-lowering activity of the dipeptidyl peptidase-4 inhibitor saxagliptin in drug-naive patients with type 2 diabetes. Diabetes Obes Metab. 2008;10:376–386.
- Rosenstock J, Aguilar-Salinas C, Klein E, et al. Effect of saxagliptin monotherapy in treatment-naive patients with type 2 diabetes. Curr Med Res Opin. 2009;25(10):2401–2411.
- Frederich R, McNeill R, Berglind N, et al. The efficacy and safety of the dipeptidyl peptidase-4 inhibitor saxagliptin in treatment-naive patients with type 2 diabetes mellitus: a randomized controlled trial. Diabetol Metab Syndr. 2012;4:36.
- Goto Y, Kikuchi H. Application of the M-value, an index of blood sugar control, for the true blood sugar value. J Japan Diabetes Soc. 1972;15(5):319–323.
- Hovorka R, Soons PA, Young MA. ISEC: a program to calculate insulin secretion. Comput Methods Programs Biomed. 1996;50(3):253–264.
- Leibowitz G, Cahn A, Bhatt DL, et al. Impact of treatment with saxagliptin on glycaemic stability and β-cell function in the SAVOR-TIMI 53 study. Diabetes Obes Metab. 2015;17(5):487–494.
- Nowicki M, Rychlik I, Haller H, et al. Long-term treatment with the dipeptidyl peptidase-4 inhibitor saxagliptin in patients with type 2 diabetes mellitus and renal impairment: a randomised controlled 52-week efficacy and safety study. Int J Clin Pract. 2011;65(12):1230–1239.
- Nowicki M, Rychlik I, Haller H, et al. Saxagliptin improves glycaemic control and is well tolerated in patients with type 2 diabetes mellitus and renal impairment. Diabetes Obes Metab. 2011;13(6):523–532.
- Boulton DW, Li L, Frevert EU, et al. Influence of renal or hepatic impairment on the pharmacokinetics of saxagliptin. Clin Pharmacokinet. 2011;50(4):253–265.