
1. Introduction
Macular edema is a leading cause of moderate to severe visual impairment in the developed world. Diabetes causes 7 million cases [Citation1], exudative macular degeneration results in 4 million cases [Citation2], retinal vein occlusions produces 3 million cases [Citation3], and 40% of ocular inflammation/uveitis-related vision loss is due to macular edema [Citation4]. Different diseases cause macular edema by activating different inflammatory pathways, the predominant pathways are difficult to identify with current clinical biomarkers. The inability to mechanistically stratify macular edema poses the greatest challenge for pharmacotherapy selection and targeted drug development.
2. Macular edema pathogenesis
Macular edema is the abnormal accumulation of fluid localized to the macular region of the posterior pole. It can be identified on funduscopic examination or documented and quantified with optical coherence tomography. While medical history and fluorescein angiography further differentiate the causative disease (for example, posterior uveitis [)] versus diabetic macular edema [)]), clinical assessment is a poor guide for therapeutic selection. Aside from identifying the presence of edema, clinical assessment does not differentiate the predominant inflammatory mechanism of disease.
Figure 1a. Fluorescein angiogram of a patient with macular edema from posterior uveitis.
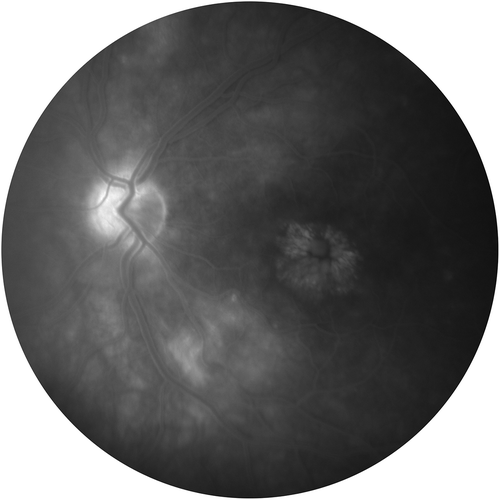
Figure 1b. Fluorescein angiogram from a patient with diabetic macular edema.
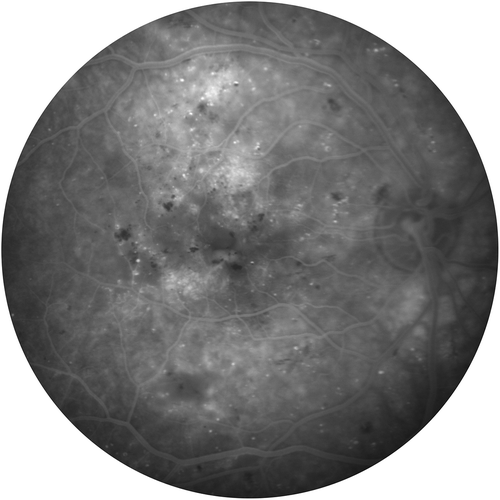
Figure 2a. Cytokine-induced mechanisms of macular edema. VEGF activates active transport of caveolae, TNF redistributes occludin with a relaxation of tight junctions, TGF beta induces MMP-9 which degrades occludins.
VEGF-Vascular endothelial growth factor VEGF- Vascular endothelial growth factor TNF- Tumor Necrosis Factor TNFR-Tumor Necrosis Factor ReceptorMMP-9- Matrix Metallo Proteinase −9TGFB-Transforming Growth factor Beta TGFBR-Transforming Growth factor Beta Receptor.
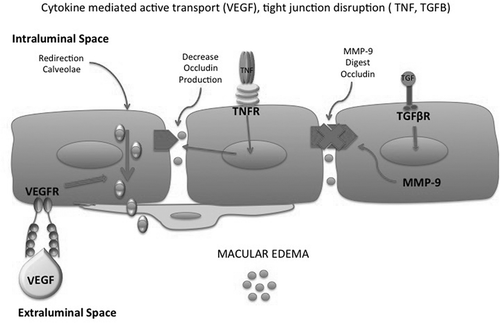
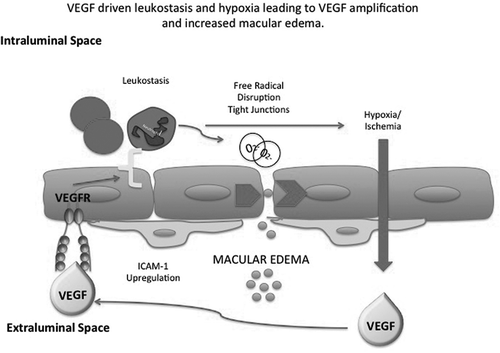
Figure 2b. VEGF causes chemoattraction of leukocytes and up-regulation of ICAM-1. This causes capillary non-perfusion and ischemia and amplification of VEGF production resulting in an increase in macular edema.
Inflammatory stage determines the predominant mechanism of macular edema [Citation5]. Early inflammation is driven by upregulation of cytokine (tumor necrosis factor-alpha [TNF-alpha], Il-1, Il-6, etc.), chemokine (Monocyte chemotactic protein 1(MCP-1)), growth factor (vascular endothelial growth factor (VEGF), and biogenic amine (histamine) [Citation6]. These early mediators, stimulated by oxidative, hypoxic, and metabolic stressors activate the immune system. These mediators also have a direct and indirect effect on the blood-retinal barrier (BRB). VEGF, for example, immediately reverses caveolae-mediated fluid transport [Citation7]. [)] TNF-alpha affects the distribution of tight junction proteins creating openings between endothelial cells [Citation8]. TGF-Beta up-regulates matrix metalloproteinase-9 expression which degrades occludins [Citation9]. [)]
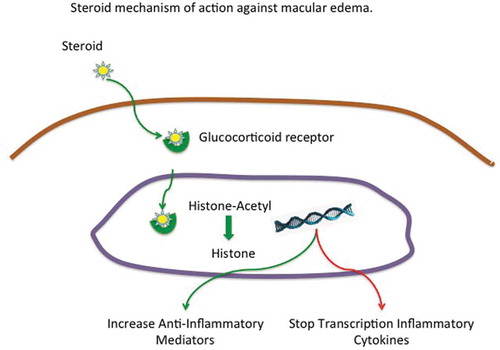
Figure 3. Steroids bind to intracytoplasmic glucocorticoid receptor which translocates to the nucleus and deacetylates histone turning of inflammatory genes and transcribing anti-inflammatory genes.
Amplification stage, result from chemo-attraction, recruitment, and activation of inflammatory cells by these initiating inflammatory mediators (VEGF, C3a anaphylotoxins, MCP-1). The type of inflammatory cells recruited and activated changes the predominant mediators driving macular edema.
The constant inflammatory cell recruitment and activation directly damage the BRB via the complement pathway [Citation10] and inflammatory up-regulation of free radicals [Citation11]. This cytokine-mediated damage disrupts the BRB and endothelial cell/pericytes tight junctions [Citation12]. This disruption allows passive diffusion of fluid and egress of proteins into the extraluminal space. The direct destruction of the BRB attracts the innate immune system to eliminate damaged cellular material. Chronic inflammation eventually results in irreversible disruption of BRB, retinal/RPE cell death, apoptosis, phagocytosis (atrophy), and fibrosis (disciform scarring).
Diabetic and retinal vein occlusion associated macular edema is initiated by VEGF. VEGF when injected into a normal nonhuman primate eye, results in the activation of vascular endothelial cell caveolae. Caveolae increase intraluminal fluid transport into the extravascular space. This effect is instantaneous and immediate [Citation7]. Increased doses of VEGF promote leukocyte attraction and up-regulation of ICAM-1 leading to leukocyte plugging of small capillaries with clinically visible vasculitis and macular/retinal ischemia [Citation13,Citation14]. Resultant ischemia produces hypoxia stimulated up-regulation of VEGF driving a VEGF amplification cycle leading to increased leukocyte accumulation in the retina and macula.
The recruitment and accumulation of activated leukocytes, directly and indirectly, disrupt the BRB. Direct disruption is mediated by free radical production and cytokine reduction of tight junction proteins [Citation15]. Indirect damage is mediated by complement activation. This accumulation of leukocytes signals a transition in mechanism from VEGF driven active transport to retinal barrier breakdown and cytokine-induced tight junction disruption. [)]
The persistent chronic accumulation of inflammatory cells eventually results in photoreceptor cell death, RPE cell atrophy, and macular fibrosis. The loss of retinal and RPE cell and permanent damage to the vascular endothelium results in the inability to drain accumulated fluid. This stage is irreversible and overwhelms the retinas reparative mechanisms. Avoiding this end stage serves as the main goal of therapy.
3. Current therapeutic choice for macular edema
The limited choice of therapies and the inability to identify predominant mechanism clinically is a true challenge in treating patients with macular edema. Each disease state can produce macular edema using one or all these mechanisms simultaneously or in sequence. Anti-VEGF, the most widely used therapy for macular edema, targets only a single growth factor. Corticosteroids inhibit inflammatory transcription factors, reduce the production of multiple cytokines including VEGF without inactivating the downstream action or inhibiting the cytokines that have already been produced. Non-steroidal anti-inflammatories can also reduce inflammatory mediators but have not been developed for intravitreal administration and have limited bioavailability when used as eyedrops [Citation16]. There is no current treatment for cellular loss.
Therapeutic choice is currently guided by disease etiology and FDA drug approval. FDA approval as a therapeutic guide is flawed because FDA indication is predominantly influenced by economically not scientific factors. Fluocinolone Acetonide intravitreal implant (Iluvien), an intravitreal 3-year sustained release device is an example of this flaw. Currently, it is only approved for diabetic retinopathy and is very effective. Scientifically it should be useful in other diseases such as retinal vein occlusion and exudative macular degeneration. The lack of corporate funding to perform clinical trials limit Iluviens ability to be tested and used in other indications.
The only FDA approved class of drugs to treat macular edema for exudative macular degeneration are anti-VEGF therapies. For diabetic retinopathy and retinal vein occlusions, anti-VEGF therapies and sustained release of steroids have undergone FDA clinical trials and approval. For post-surgical inflammation steroidal and nonsteroidal anti-inflammatory drops are indicated. For uveitis and primary ocular inflammation sustained release steroids and steroidal eyedrops have been approved. FDA approval limits therapeutic options a difficult challenge to surmount.
Polyunsaturated fatty acids such as Omega-3 fatty acids and their metabolites lipoxin, resolvins, and protectins have been postulated as a potential anti-inflammatory treatment for diabetic macular edema. While there is very little clinical evidence for their efficacy, the intake of omega-3 fatty acids, and the potential production of these anti-inflammatory metabolites could be used in macular edema cases without the opposition of the FDA or physicians.
4. Conclusion
The inability to identify pathomechanism clinically, the lack of useful biomarkers, the limited choice of drugs, and the regulatory limitations for use of clinically available drugs make drug choice for the treatment of macular edema difficult. These hurdles necessitate further research to develop better diagnostics and therapeutic drugs for macular edema.
5. Expert opinion
The ideal method for therapeutic selection is to stratify mechanism in individual patients and apply the appropriate targeted treatment. Currently, anti-VEGF therapy is the only targeted approach, while corticosteroids represent the broad non-specific approach. Steroids bind to glucocorticoid receptors in the cytoplasm which initiates complex translocation to the nucleus. In the nucleus, the complex reverses histone acetylation leading to a deactivation of inflammatory genes. The complex can also bind glucocorticoid receptor element that activates transcription of anti-inflammatory molecules [Citation17]. This broad action, especially the reversal of histone acetylation reduces transcription of several pro-inflammatory genes [Citation18]. This broad transcriptional inhibition also inhibits genes such as osteocalcin which accelerates osteoporosis, keratin which leads to dermal thinning, and corticotropin releasing factor −1 which leads to potential adrenal insufficiency all well-described side effect of steroids [Citation19]. []
Macular edema resulting predominantly from innate immune activation such as exudative macular degeneration does not respond robustly to steroids. Steroids can alter surface markers of classical (M1) alternative (M2) macrophage activity but shift the macrophage polarization toward M2 [Citation20]. Since M2d VEGF producing macrophages drives exudative AMD associated macular edema [Citation21], steroids are theoretically and clinically less effective than anti-VEGF intravitreal injections for exudative AMD. The ability to resolve activated macrophages is an active area of research and potentially effective in treating and preventing macular edema in exudative macular degenerations [Citation22].
Diabetic, vein occlusion and ischemic retinopathy related macular edema poses a greater challenge since these retinopathies can transition through all stages of inflammation and involve innate and adaptive immune activation simultaneously or in sequence. Steroids predominantly inhibit the adaptive immune reaction and explain why steroids work better in ischemic retinopathies than in exudative AMD.It is in these forms of macular edema that a mechanistically correlated biomarker is needed to choose or develop effective therapies. The potential for broad inflammatory activation in these patients necessitates combination therapy with anti-VEGF and steroidal therapy administered within 2 weeks of each other. Currently, this form of macular edema is treated with a sequential trial and error approach. Anti-VEGF therapy is utilized first, if nonresponsive 4–8 weeks after injection, then treatment with steroidal sustained delivery such as Ozurdex or Iluvien is attempted. Combination therapy is hindered by the expense of injecting expensive medicines in close proximity.
The majority of patients cannot be stratified mechanistically into predominantly VEGF-driven or inflammatory cell/cytokine-mediated and invariably need combination therapy. Sequential therapy may prove harmful in patients where all mechanisms are activated. If there is a delay in resolving VEGF or inflammation induced macular edema the disease transitions to the inflammatory-mediated structural damage to the BRB leading to irreversible vision loss.
Logically, it is optimal to start with a steroid implant and quickly add anti-VEGF therapy if retinal thickness does not decrease. Mechanistically, steroids inhibit VEGF transcription, not mRNA stabilization or the release of VEGF protein from hematologic cells such as platelets and leukocytes a major component of VEGF up-regulation. Steroid protects against direct and indirect damage to the BRB. If macular edema persists after steroid therapy, then the BRB is protected and anti-VEGF therapy should provide additional benefit.
Non-infectious uveitis or post-surgical Irvine-Gass syndrome is an inflammatory mediated form of macular edema. In these conditions, VEGF plays a tertiary role. The use of steroids is the current therapy of choice. VEGF does not play a major role in this form of macular edema. Systemically administered TNF alpha inhibitor adalimumab has shown promise in reducing the inflammation in non-infectious uveitis although its effect on macular edema was not a primary endpoint for approval of this uveitis treatment [Citation23].
The ideal therapy for macular edema would be a molecule that deactivates inflammatory cells profoundly and specifically. Current therapies just inhibit production of inflammatory mediators produced by activated inflammatory cells. Because of the mechanistic limitations of current therapies and the ability of all stages of inflammation to cause macular edema with differing mechanisms, combining steroids, and anti-VEGF therapies reassures that macular edema is maximally treated. Future therapies should be focused on deactivating inflammatory cells [Citation24,Citation25].
Declaration of interest
MJ and AJ Tolentino are both employees of Blue Ocean Clinical Research. MJ Tolentino has also received grant support from the Tolentino Eye Research Foundation and has served as a speaker and/or consultant for Alimera Sciences, Regeneron, Genentech, Bayer Healthcare, Allergan and Bausch and Lomb. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
One referee consults and performs research for Allergan, Regeneron, and Genentech. They also perform research for Alimera Sciences. Another referee has an employment relationship with Clearside Biomedical. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Yau JW, Rogers SL, Kawasaki R, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35(3):556–564. [published Online First: Epub Date]
- Pennington KL, DeAngelis MM. Epidemiology of age-related macular degeneration (AMD): associations with cardiovascular disease phenotypes and lipid factors. Eye Vis (Lond). 2016;3:34. [published Online First: Epub Date].
- Rogers S, McIntosh RL, Cheung N, et al. The prevalence of retinal vein occlusion: pooled data from population studies from the United States, Europe, Asia, and Australia. Ophthalmology. 2010;117(2):313–9e1. [published Online First: Epub Date]
- Rothova A, Suttorp-van Schulten MS, Frits Treffers W, et al. Causes and frequency of blindness in patients with intraocular inflammatory disease. Br J Ophthalmol. 1996;80(4):332–336.
- Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18(12):1450–1452. [published Online First: Epub Date]
- Daruich A, Matet A, Moulin A, et al. Mechanisms of macular edema: beyond the surface. Prog Retin Eye Res. 2018:63:20–68. [published Online First: Epub Date].
- Hofman P, Blaauwgeers HG, Tolentino MJ, et al. VEGF-A induced hyperpermeability of blood-retinal barrier endothelium in vivo is predominantly associated with pinocytotic vesicular transport and not with formation of fenestrations. Vascular endothelial growth factor-A. Curr Eye Res. 2000;21(2):637–645.
- Aveleira CA, Lin CM, Abcouwer SF, et al. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59(11):2872–2882. [published Online First: Epub Date].
- Behzadian MA, Wang XL, Windsor LJ, et al. TGF-beta increases retinal endothelial cell permeability by increasing MMP-9: possible role of glial cells in endothelial barrier function. Invest Ophthalmol Vis Sci. 2001;42(3):853–859.
- Schraufstatter IU, Trieu K, Sikora L, et al. Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J Immunol. 2002;169(4):2102–2110.
- Shojaee N, Patton WF, Hechtman HB, et al. Myosin translocation in retinal pericytes during free-radical induced apoptosis. J Cell Biochem. 1999;75(1):118–129.
- Betts-Obregon BS, Mondragon AA, Mendiola AS, et al. TGFbeta induces BIGH3 expression and human retinal pericyte apoptosis: a novel pathway of diabetic retinopathy. Eye (Lond). 2016;30(12):1639–1647. [published Online First: Epub Date]
- Tolentino MJ, Miller JW, Gragoudas ES, et al. Intravitreous injections of vascular endothelial growth factor produce retinal ischemia and microangiopathy in an adult primate. Ophthalmology. 1996;103(11):1820–1828.
- Joussen AM, Poulaki V, Qin W, et al. Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol. 2002;160(2):501–509. [published Online First: Epub Date].
- Joussen AM, Murata T, Tsujikawa A, et al. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158(1):147–152. [published Online First: Epub Date].
- Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002;16(3):438–440. [published Online First: Epub Date].
- Lasa M, Abraham SM, Boucheron C, et al. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol. 2002;22(22):7802–7811.
- Kracht M, Saklatvala J. Transcriptional and post-transcriptional control of gene expression in inflammation. Cytokine. 2002;20(3):91–106.
- Saklatvala J. Glucocorticoids: do we know how they work?. Arthritis Res. 2002;4(3):146–150. [published Online First: Epub Date].
- Tedesco S, Bolego C, Toniolo A, et al. Phenotypic activation and pharmacological outcomes of spontaneously differentiated human monocyte-derived macrophages. Immunobiology. 2015;220(5):545–554. [published Online First: Epub Date]
- Ferrante CJ, Leibovich SJ. Regulation of macrophage polarization and wound healing. Adv Wound Care (New Rochelle). 2012;1(1):10–16. [published Online First: Epub Date].
- Yang Y, Liu F, Tang M, et al. Macrophage polarization in experimental and clinical choroidal neovascularization. Sci Rep. 2016;6:30933. . [published Online First: Epub Date].
- Suhler EB, Adan A, Brezin AP, et al. Safety and efficacy of adalimumab in patients with noninfectious uveitis in an ongoing open-label study: VISUAL III. Ophthalmology. 2018. DOI:10.1016/j.ophtha.2017.12.039. [published Online First: Epub Date].
- Zanin-Zhorov A, Flynn R, Waksal SD, et al. Isoform-specific targeting of ROCK proteins in immune cells. Small GTPases. 2016;7(3):173–177. [published Online First: Epub Date].
- Zandi S, Nakao S, Chun KH, et al. ROCK-isoform-specific polarization of macrophages associated with age-related macular degeneration. Cell Rep. 2015;10(7):1173–1186. [published Online First: Epub Date]