
1. Introduction
Proximal spinal muscular atrophy (SMA) is a neurodegenerative disorder and the leading genetic cause of infant death. SMA is recessively inherited and has a carrier frequency of 1:35 to 1:50 and an incidence of 1:6,000 to 1:10,000 live births [Citation1,Citation2]. The disease is caused by the homozygous loss of the survival of motor neuron 1 (SMN1) gene. Typical SMA patients show progressive degeneration of the α-motor neurons in the anterior horns of the spinal cord, eventually leading to muscle atrophy [Citation3]. Based on the severity and age of onset, SMA is classified in five subtypes ranging from type 0, the most severe form, to type IV, the mildest form. 50% of patients suffer from severe SMA type I. Without treatment, SMA type I patients never learn to sit or walk and usually die within the first two years of life, with mean life expectancy of seven months [Citation3]. On the other end of the spectrum, SMA type IV is a slow progressing disease, with an onset later in life (>30 years) [Citation4]. The major underlying reason for this heterogeneity of disease expression is the unique genetic background of SMA. While homozygous loss of the disease gene SMN1 causes SMA, the gene copy number of the main modifying gene SMN2 correlates with the severity. This correlation (the more SMN2 copies, the milder the phenotype) can be explained by the genetic similarity of SMN1 and SMN2 (reviewed in [Citation5]). During evolution, the SMN1 gene was duplicated in primates and later mutated into SMN2. Therefore, SMN1 and SMN2 differ only in 5 nucleotide exchanges, all of which do not affect the protein translation. However, the crucial difference, the translationally silent C to T transition in exon 7, disrupts an exonic splicing enhancer and creates an exonic splicing silencer, resulting in mis-splicing of SMN2 exon 7 [Citation6,Citation7]. In fact, only 10% of SMN2 transcripts produce a full-length protein that is functional, while 90% lack exon 7, encoding a dysfunctional protein that undergoes rapid degradation [Citation8]. This unique genetic setting in which every patient carries at least one (usually more) copy of the major genetic modifier provides an interesting therapeutic target for SMA.
2. Developing therapies for SMA
Besides general neuroprotective and gene replacement therapies, therapeutic approaches that target the endogenous SMN2 gene have been evaluated with varying success in clinical trials (reviewed in [Citation9]). In the following, this editorial will give a short summary of the SMN2-related therapies and focus on the most promising approach, which is antisense oligonucleotide (ASO)-mediated splicing correction of SMN2. Since SMN2 typically generates only 10% of the fully functional full-length (FL) SMN protein, elevating the level of expression results in increased SMN protein levels. Epigenetic modification of the SMN2 locus by histone deacetylase inhibitor (HDACi) treatment has been shown to increase the SMN protein level up to 10-fold in vitro. Other HDACis have been proven to ameliorate the SMA phenotype in animal models [Citation10,Citation11]. Eventually, the FDA-approved HDACi valproic acid (VPA) was evaluated in clinical trials, but with limited success [Citation12]. Other experimental drugs that have been shown to elevate the SMN levels in vitro and in vivo never made it into clinical trials [Citation9]. The most promising experimental approaches, based on their capability to upregulate the SMN protein level and to ameliorate the SMA phenotype in SMA animal models, were splice correction approaches. Academia and pharmaceutical companies performed SMN2 minigene-based high-throughput screening programs to identify small molecules that are capable of correcting the splicing pattern of the SMN2 pre-mRNA transcript. The most potent candidates were analyzed and some showed significant upregulation of FL-SMN protein in both cells and animals (reviewed in [Citation9]). Recently Roche, in collaboration with the SMA Foundation and PTC Therapeutics, developed the orally available drug, risdiplam. Although the molecular mechanism of splicing correction has not been described, risdiplam shows relative selectivity towards SMN2 splicing. Other advantages include the benefit of oral delivery, and the ability to reach peripheral organs by systemic distribution [Citation13]. Risdiplam has been evaluated in the clinical trials FIREFISH (open-label trial in infants aged 1–7 months, type I SMA), SUNFISH (double-blind, placebo-controlled trial in children and young adults with type II or III SMA) and JEWELFISH (open-label exploratory trial in people aged 12–60 with type II or III SMA). Preliminary data from the first two studies showed motor function improvements and no severe adverse effects [Citation14]. These results appear to be quite promising and follow-up studies are currently ongoing. Moreover, the oral drug might be advantageous with respect to administration and systemic availability. However, these drugs are not FDA-approved yet. Currently, the only FDA-approved drug for SMA therapy is a highly efficient synthetic antisense oligonucleotide (ASO) molecule called nusinersen, which will be discussed in the following section.
3. Antisense splicing correction therapy
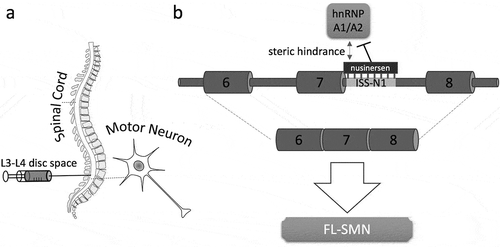
In 2006, Singh et al. identified the intronic splicing silencer ISS-N1, a 15 nucleotide sequence located in intron 7 of SMN2 that encompasses two binding sites for the splicing repressor hnRNP A1/A2 (reviewed in [Citation15]). The researchers demonstrated that treatment with specific synthetic oligonucleotides restored the SMN2 pre-mRNA splicing with a significant increase of exon 7 inclusion [Citation16]. ASO binding to ISS-N1 in the SMN2 pre-mRNA sterically blocks the binding of the splicing repressor hnRNP A1/A2 to ISS-N1, which in turn promotes correct splicing and exon 7 inclusion (). In a subsequent in vivo study, A. Krainer’s laboratory in collaboration with F. Bennett from Isis Pharmaceuticals (now Ionis Pharmaceuticals) showed unprecedented benefits of splicing correction in a severe mouse model of SMA [Citation17]. Together, these findings laid the foundation for the development of a novel therapeutic approach for the treatment of SMA: ASO-mediated splicing correction (reviewed in [Citation15]). Only a few years after the identification of ISS-N1, a controlled study was started in order to assess the clinical efficacy and safety of the ASO (now called nusinersen). This study was a phase 3, randomized, double-blind sham procedure that was administered intrathecally in patients with infantile-onset SMA (ENDEAR). A few months later, a similar phase 3 trial was started with later-onset SMA patients between 2 and 12 years of age (CHERISH). Both clinical trials were enormously successful. In fact, in the interim analyses, a significantly higher percentage of patients in the treatment group had a motor-milestone response, which prompted early termination of the trials. In follow-up open label studies, the results of the CHERISH trial were confirmed in extended treatment over almost three years [Citation18]. Nusinersen treatment in SMA type II patients improved Revised Upper Limb Module (RULM) and Hammersmith Functional Motor Scale Expanded (HFMSE)-scores. SMA type III patients presented a stabilized HFMSE-score over the duration of the study [Citation19,Citation20]. Two treated patients regained their ability to walk. Based on the excellent outcomes of the clinical studies, nusinersen was approved by the FDA to treat children and adults with SMA in 2016. The drug is an injection administered into the fluid surrounding the spinal cord (). To further evaluate the optimal timepoint for treatment, babies (6 weeks or younger) with genetic predispositions for SMA were included in a phase 2 clinical trial (NURTURE). In this study, the infants were pre-symptomatically treated with nusinersen. Based on the SMN2 gene copy number, the infants were expected to develop SMA type I or II. However, the splice correction treatment rescued their SMA phenotype so that some infants showed an almost normal motoric development. This study provides evidence that the best therapeutic strategy for nusinersen treatment is to administer the drug as early as possible, before motor neurons start to degenerate. To ensure pre-symptomatic treatment, doctors have to be aware of the genetic background of babies, especially for those with family history of SMA. Therefore, genetic newborn screening programs will be necessary. Fortunately, nine US states have already adopted the recommendation by the federal Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC) to implement SMA newborn screenings, three more states have begun pilot screening and many more are considering implementation.
Figure 1. Overview of the administration and the mode of action of nusinersen. (a) The drug is administered into the fluid surrounding the spinal cord in the lumbar region usually between the disc space of L3–L4. The compound can then reach the target cells, the motor neurons. (b) In the nuclei of the motor neurons the synthetic antisense oligonucleotide nusinersen binds specifically to the intronic splicing silencer in intron 7 (ISS-N1) of SMN2 pre-mRNA and prevents the splicing repressor hnRNP A1/A2 from binding (without steric hindrance, binding of hnRNPA1/A2 would cause exon 7 exclusion). Nusinersen leads therefore to the inclusion of SMN2 exon 7, producing the stable, fully functional, full-length SMN (FL-SMN) protein. (only relevant exons are shown, not to scale).
4. Expert opinion
It is an exciting time for SMA researchers and especially for SMA patients and their families. The rapid development of novel therapeutic strategies for this devastating genetic disorder and their positive outcomes in clinical trials, is beyond compare. The recent accelerated FDA-approval of nusinersen as the first drug to treat SMA crowned the success of SMA research. However, there are currently three main caveats to consider for nusinersen-based SMA therapy. First, although nusinersen showed enormous improvements in some patients, there is a wide variation in response. Identification of biomarkers would be desirable in order to predict the outcome of treatment in each patient and to identify responders and non-responders (reviewed in [Citation21]). The second aspect to consider is the timepoint at which to begin the therapy, which holds true for all therapeutic approaches for SMA. Although nusinersen has been shown to be beneficial in older patients, preliminary studies showed that pre-symptomatic treatment generated optimal results [Citation22]. Maintaining the survival of motor neurons before their loss occurs is preferable to starting treatment after the onset of neurodegeneration. However, newborn genetic screening to identify SMA patients before the onset of symptoms would be crucial to ensure pre-symptomatic treatment and the best possible outcome. The third unknown of nusinersen therapy is the long-term effects of the treatment. Even if it were possible to maintain high levels of SMN in the main target tissue, the motor neurons, it remains unanswered what the long-term effects of low levels of SMN in other cell types would be. Since SMN is a known housekeeping protein with crucial biological functions in every single cell, it may become necessary to boost SMN levels throughout the whole body later in life. These effects must be considered, since the small synthetic compounds that are currently in clinical trials are taken orally and have a systemic effect on SMN levels. Therefore, a future combination therapy using nusinersen together with small splicing correctors may be an option. Importantly, this option would require further understanding of the molecular mode of action of the small splicing correctors, in order to prevent unwanted interference/competition with nusinersen. Recently, some research progress has been made in that direction [Citation23]. Furthermore, genetic modifiers of SMA have been identified and manipulation of these modifiers was beneficial in SMA animal models [Citation24,Citation25]. Pharmaceutical manipulation of these modifiers in addition to SMN2 splicing correction might further improve the clinical outcome. It remains unclear if patients with only one SMN2 copy would need additional boost of gene transcription. In summary, there has been unprecedented success in the development of splicing correction therapy for SMA, but there is still some need for further characterization of the disease (e.g. non-motor neuron function) and additional drug development. Finally, given the rapid development of functional therapies, the answer to the question posed in the title of this article is yes, there is hope for the development of an effective SMA therapy. But more importantly, for patients and their families, there is more hope than ever before.
Declaration of interest
The author holds a patent on antisense oligonucleotide regulation of neurocalcin delta (NCALD), a genetic modifier of SMA. He has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Feldkotter M, Schwarzer V, Wirth R, et al. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002 Feb;70(2):358–368.
- Munsat TL, Davies KE. International SMA consortium meeting (26–28 June 1992, Bonn, Germany). Neuromuscul Disord. 1992;2(5–6):423–428.
- Rudnik-Schoneborn S, Berg C, Zerres K, et al. Genotype-phenotype studies in infantile spinal muscular atrophy (SMA) type I in Germany: implications for clinical trials and genetic counselling. Clin Genet. 2009 Aug;76(2):168–178.
- Zerres K, Rudnik-Schoneborn S, Forkert R, et al. Genetic basis of adult-onset spinal muscular atrophy. Lancet. 1995 Oct 28;346(8983):1162.
- Wirth B, Garbes L, Riessland M. How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr Opin Genet Dev. 2013 Jun;23(3):330–338.
- Lorson CL, Hahnen E, Androphy EJ, et al. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999 May 25;96(11):6307–6311.
- Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003 Aug;34(4):460–463.
- Lim SR, Hertel KJ. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3ʹ splice site pairing. J Biol Chem. 2001 Nov 30;276(48):45476–45483.
- Kaczmarek A, Schneider S, Wirth B, et al. Investigational therapies for the treatment of spinal muscular atrophy. Expert Opin Investig Drugs. 2015;24(7):867–881.
- Garbes L, Riessland M, Wirth B. Histone acetylation as a potential therapeutic target in motor neuron degenerative diseases. Curr Pharm Des. 2013;19(28):5093–5104.
- Riessland M, Ackermann B, Forster A, et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet. 2010 Apr 15;19(8):1492–1506.
- Kissel JT, Elsheikh B, King WM, Freimer M, Scott CB, Kolb SJ, et al. SMA valiant trial: a prospective, double-blind, placebo-controlled trial of valproic acid in ambulatory adults with spinal muscular atrophy. Muscle Nerve. 2014 Feb;49(2):187–192.
- Ratni H, Ebeling M, Baird J, et al. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem. 2018 Aug 9;61(15):6501–6517.
- Roche PR Roche announces new data for risdiplam in spinal muscular atrophy (SMA) at the world muscle society congress. Media Release. [cited 2019 Jan 28]. Available from: https://www.roche.com/media/releases/med-cor-2018-10-03.htm. 2018.
- Singh NN, Howell MD, Androphy EJ, et al. How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy. Gene Ther. 2017 Sep;24(9):520–526.
- Singh NK, Singh NN, Androphy EJ, et al. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006 Feb;26(4):1333–1346.
- Hua Y, Sahashi K, Rigo F, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011 Oct 5;478(7367):123–126.
- Biogen. Biogen later-onset study results. [cited 2019 Mar 03]. Available from: https://wwwspinrazacom/en_us/home/results/later-onsethtml. 2019.
- Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus Sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017 Nov 2;377(18):1723–1732.
- Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus Sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018 Feb 15;378:(7)625–635.
- Gidaro T, Servais L. Nusinersen treatment of spinal muscular atrophy: current knowledge and existing gaps. Dev Med Child Neurol. 2019 Jan;61(1):19–24.
- Hwu W, De D, Bertini E, et al. Outcomes after 1-year in presymptomatic infants with genetically diagnosed spinal muscular atrophy (SMA) treated with nusinersen: interim results from the NURTURE study. Neuromuscul Disord. 2017;27:212.
- Garcia-Lopez A, Tessaro F, Jonker HRA, et al. Targeting RNA structure in SMN2 reverses spinal muscular atrophy molecular phenotypes. Nat Commun. 2018 May 23;9(1):2032.
- Oprea GE, Krober S, McWhorter ML, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008 Apr 25;320(5875):524–527.
- Riessland M, Kaczmarek A, Schneider S, et al. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am J Hum Genet. 2017 Feb 02;100(2):297–315.