
ABSTRACT
Introduction: Parkinson’s disease is a neurodegenerative disorder which is characterized by the combination of motor and non-motor symptoms. As yet, there is no curative treatment. The gold standard for symptom control is levodopa. Two years after the start of substitution therapy, around 50% of patients experience some degree of fluctuation in motor performance. Catechol-O-methyltransferase (COMT) inhibitors are important agents in treating these fluctuations.
Areas covered: This article summarizes our knowledge about a new third-generation COMT inhibitor, namely opicapone (OPC) (Search period: 2016–2019). The authors detail the pharmacological profile of OPC and summarize the results of completed clinical trials. In addition, they briefly summarize the achievements of the past few years.
Expert opinion: Based on clinical trials conducted so far, OPC is an effective and safe new drug. In comparison to entacapone and tolcapone, it does not require close laboratory monitoring or multiple oral administrations, which may result in better adherence. No serious adverse event was reported during the drug development phases. Dyskinesia was the most common complaint. Further comparative studies and broader trial inclusion criteria are needed to help the decision between COMT inhibitors and to expand the patient spectrum where this drug can be applied.
1. Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder, which is characterized by the combination of motor (bradykinesia, rest tremor and rigidity) and non-motor symptoms (fatigue, anxiety, leg pain, sleep disturbance, urinary problems, concentration difficulties) [Citation1,Citation2]. The estimated prevalence is around 1–2 per 1000, and increases with age, 1% of the population is affected above the age of 60 [Citation3]. In Hungary there is a high prevalence (age-standardized prevalence: 471/100,000) and incidence (age-standardized incidence: 56/100,000/year) [Citation4]. The main characteristic neuropathological feature of PD is the loss of dopaminergic neurons in the substantia nigra (pars compacta) [Citation5]. There is no curative treatment according to our current knowledge. The gold standard of symptomatic therapy is levodopa (L-dopa; LD) [Citation6]. The most prominent debilitating complication of long-lasting LD treatment is the development of motor complications [Citation7]. Two years after the start of substitution therapy, around 50% of patients sense some degree of fluctuation in motor performance [Citation8]. The pathophysiological background of fluctuations is the pulsatile and decreased dopaminergic stimulation of the striatal neurons [Citation9].
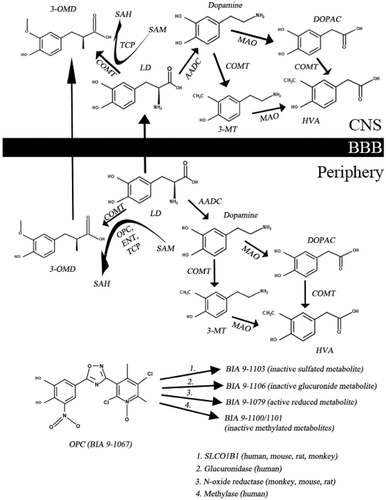
Dopamine (DA) is not able to cross the blood-brain barrier (BBB), therefore we use its precursor, LD. However, LD is rapidly converted by DOPA decarboxylase (DDC) and catechol-O-methyltransferase (COMT) enzymes to its metabolites in the periphery (; [Citation10]). If we administer LD orally, only 1% will penetrate into the central nervous system (CNS [Citation11];). To increase CNS availability of LD, we use DDC and COMT inhibitors. The function of the COMT enzyme is to transfer a methyl group to catecholamines. During the catalytic process S-adenosyl methionine (SAM) is converted to S-adenosyl homocysteine (SAH) [Citation10]; the detailed metabolism is presented in . The most common COMT inhibitors are entacapone (ENT) and tolcapone (TLC) [Citation12]. Since 24 June 2016 a third COMT inhibitor, namely opicapone (OPC) is also available, which was approved by the European Medicine Agency for the treatment of end-of-dose motor fluctuations in adult patients whose symptoms are not controllable by LD/DOPA decarboxylase inhibitor (DDCI) combination [Citation13]. Probably the most important advantages of OPC in comparison with second generation COMT inhibitors are the following: there is a less frequent need to administer and there is no real risk of hepatotoxicity [Citation12].
Figure 1. The main steps of levodopa metabolism and important therapeutic targets and drugs. (3-MT – 3-methoxy-tyramine; 3-OMD – 3-O-methyldopa; AADC – aromatic amino acid decarboxylase; BBB – blood-brain barrier; COMT – catechol-O-methyltransferase; DA – dopamine; DOPAC – 3,4-dihydroxy-phenylacetic acid; ENT – entacapone; HVA – homovanillic acid; LD – levodopa; MAO – monoamine oxidase; OPC – opicapone; SAH – S-adenosyl homocysteine; SAM – S-adenosyl methionine; SLCO1B1 – solute carrier organic anion transporter family, member 1B1; TCP – tolcapone).
The aim of this drug evaluation article is to summarize our previous knowledge about the pharmacological profile and clinical studies of OPC and to demonstrate the achievements of the past few years.
1.1. Review data
The aim of this review article is to summarize our current knowledge about on OPC. Relevant articles were collected through the most important databases (PubMed (MEDLINE); Web of Science; ScienceDirect; Wiley Online Library; Scopus). The search period was from October 2016 to August 2019. (The article also includes the most relevant data before October 2016. For more information see our previous article [Citation10].) The key terms of the search were ‘opicapone’ and ‘BIA 9-1067ʹ. We included the most comprehensive reviews as further evaluation. The purpose of this review article was primarily to summarize human results, therefore preclinical articles were excluded. After collecting the most relevant articles, we synthesized the most important data and tried to provide an update on OPC.
1.2. Overview of the market
Currently two COMT inhibitors are in regular use: ENT and TLC [Citation7]. ENT is a safe product to be given several times a day [Citation10]. Although TLC is more potent compared to ENT, TLC requires close monitoring due to the potential risk of hepatotoxicity [Citation13]. Nebicapone was another COMT inhibitor under development, but had to be terminated due to the risk of hepatotoxicity [Citation14]. Currently OPC is in phase 4 trial (OPTIPARK, European Union) to examine the safety and tolerability of a 50 mg OPC dose in a selected PD population [Citation7]. Currently OPC is marketed under the Ongentys® brand name in the United Kingdom, Germany, Spain, Italy and North America [Citation7,Citation10].
1.3. Introduction to the compound
OPC (BIA 9–1067; chemical name: 2,5-dichloro-3-(5-[3,4-dihydroxy-5-nitrophenyl]-1,2,4-oxadiazol-3-yl)-4,6-dimethylpyridine-1-oxide) is a third-generation COMT inhibitor molecule (; ) [Citation15]. This drug is currently approved by the European Medication Agency as an adjunctive therapy for the treatment of those elderly PD patients, whose symptoms (end-of-dose motor fluctuations) are not controllable with LD/DDCI treatment. Ongentys® is the market name of OPC [Citation15].
Box 1. Drug summary box.
1.4. Chemistry
From a chemical perspective, OPC is a peripherally acting compound, which has very high protein-binding affinity (in the subpicomolar range) to the COMT enzyme [Citation7,Citation16]. The pyridine N-oxide residue (position 3) is responsible for this inhibitory potential and cell toxicity avoidance [Citation17].
1.5. Pharmacodynamics
After oral administration, OPC shows a significant and long-lasting COMT inhibitory effect [Citation18–Citation21]. The extent of inhibition depends on the applied dose (5 mg – 50%; ≥ 200 mg – 100%) [Citation14]. Thanks to the slow complex dissociation rate OPC has a long-lasting effect (24 h after the last dose (5–30 mg) – 50–70%) [Citation20,Citation21]. If opicapone (25–100 mg) is given concomitantly with immediate-/controlled-release 100/25 mg LD/carbidopa or with immediate-/controlled-release 100/25 mg LD/benserazide drug, LD and benserazide Cmax and AUC are elevated [Citation22]. Previous studies showed that ENT and TLC have shorter effect durations (8 and 18 h) [Citation23,Citation24]. Co-administration of OPC with another drug(s) used in the treatment of PD (DA agonists, monoamine oxidase B inhibitors) did not influence the pharmacological action mechanism [Citation25,Citation26].
1.6. Pharmacokinetics and metabolism
Oral administration of OPC to healthy volunteers in different doses (10 to 1200 mg) was performed by Almeida et al [Citation18]. They found the terminal elimination half-life of OPC between 0.8 (50 mg) to 3.2 hours (1200 mg) [Citation18]. The COMT inhibitory effect seems to be dose-dependent [Citation18]. The peak inhibitory effect was 36.1% by 10 mg OPC and 100% at doses greater than 200 mg [Citation18]. Interestingly, despite the relatively short half-life time, the COMT inhibitory effect of OPC can be detected even after 24 hours, due to the slow dissociation rate [Citation18]. Therefore, a once-daily dosing regimen is sufficient [Citation7,Citation10].
As shown in , there are many metabolites of OPC, of which only BIA 9–1079 (reduced metabolite) shows any metabolic activity [Citation18,Citation20]. The main elimination route is sulfation via the hepato-biliary system (BIA 9–1103, SULT1A1) [Citation27]. In humans, BIA 9–1103 is the most important metabolite. The effect of BIA 9–1103 on various transporters was examined in vitro using CPMP/EWP/560/95/Rev. OPC is transported by P-gp and BCRP and BIA 9–1103 is transported by OATP1B1 and -B3. There is evidence from in vitro studies that OPC and BIA 9–1103 have inhibitory effects on OAT1, −3, OATP1B1 and -B3, BSEP [Citation28]. We know from previous studies, that there is an increase in OPC levels in patients with moderate liver impairment (Child-Pugh B) [Citation27]. Today there is no data about OPC pharmacokinetics in severe liver dysfunction (Child-Pugh C),that is the reason, why this drug is not recommended for this population [Citation10]. A high-fat meal could delay the time of peak plasma drug levels [Citation29]. Sex, age and ethnicity do not influence the drug effect [Citation7]. Falcao et al. reported an RCT dose study (10 days: OPC (5, 25, 50 mg) or PLC), which resulted in no significant difference in pharmacokinetics and pharmacodynamics between Caucasian and Japanese populations [Citation30].
Regarding the relationship of LD availability and OPC, it was demonstrated that OPC at 25, 50 and 75 mg significantly increases the LD area under the curve (AUC) compared to placebo (PLC) and ENT (in a latter case, 50 and 75 mg OPC) [Citation19]. Rocha et al. administrated LD/carbidopa (LC) or LD/benserazide (LB) in combination with OPC according to two different study design protocols [Citation31]. They showed that all applied doses of OPC (5, 15, 30 and 50 mg) resulted in higher extents of LD exposure (AUC) [Citation31].
OPC should be taken at bedtime once-daily after the last administered LD/AADCI dose, to avoid any of possible pharmacokinetic interaction [Citation32]. Clinical trials are investigating (currently no results are available) the pharmacokinetic effect of OPC on repaglinide (NCT01536366), rasagiline (NCT01532141, NCT01532128) and warfarin (NCT02169440, NCT02305030) [Citation33].
1.7. Clinical efficacy
Two phase II and two phase III studies provide information on clinical efficacy of OPC () [Citation17,Citation21,Citation34–Citation36]. Thoroughly selected PD patients were involved in the two randomized, double-blind, controlled phase II studies (> 1.5 hours OFF time daily; Hoehn and Yahr (HY) scale < 5; etc.) [Citation21,Citation34]. Rocha et al. included 10 patients with PD in a study, in which they had an improvement in ON time after a single oral OPC administration compared to placebo (18–25% ON-time increase (25–50 mg OPC); 73% increase in ON-time without dyskinesia (50 mg) [Citation34]. Ferreira et al. also performed a randomized controlled trial (RCT) with 35 PD patients (they used a similar inclusion/exclusion criteria as Rocha et al), who received 28-day long 5, 15 or 30 mg OPC [Citation21]. All treatment regimens resulted in a decrease in OFF time (5 mg – 15.6 min reduction and 30 mg – 145 min reduction) and significant increase in ON time without troublesome dyskinesia compared to PLC [Citation21]. No serious adverse events were detected in the two studies mentioned above [Citation21,Citation34].
Table 1. Clinical trials with OPC (AUC – area under the curve; BIPARK – efficacy and safety of BIA 9–1067 in idiopathic Parkinson’s disease patients; BZ – benserazide; CD – carbidopa; Cmin – minimum blood plasma concentration; COMT – catechol-O-methyltransferase; ENT – entacapone; LD – levodopa; OPC – opicapone; PD – Parkinson’s disease; PLC – placebo). Adapted from [Citation14] with permission of Taylor & Francis. Data also derived from [Citation7] and [Citation10].
The BIPARK-I study was a phase III study, in which 600 patients with PD were involved in a randomized, double-blind, controlled way with the aim to estimate the clinical efficacy of 5, 25 and 50 mg orally administrated OPC for 14–15 weeks compared to placebo or ENT [Citation15]. The most important criteria for patient selection were the following: disease duration ≥ 3 years, stage 1–3 in HY scale and ≥ 1.5 hours OFF time (excluding early morning akinesia) [Citation15]. Furthermore, patients with a score greater than 3 on item 33 (disability) of the UPDRS and severe or unpredictable periods in the off state were excluded as well [Citation15]. The patients were divided into five different groups: 5, 25 and 50 mg OPC (1 h after the last LD dose, evening), PLC and 200 mg ENT [Citation15]. The major improvements were found in the 50 mg OPC group, where OFF time reduction was significant in comparison to PLC (−116.8 vs. −56 minutes) and this dose was non-inferior to the ENT group as well (−116.8 vs. – 96.3 minutes) [Citation15]. In the 5 and 25 mg groups there were no significant OFF time reductions [Citation15]. In terms of ON time, in the 50 mg OPC group, there was an increase in ON time without troublesome dyskinesia compared to the PLC group [Citation15]. From the perspective of non-motor symptoms of PD, despite the seeming amelioration of NMSS total score, no significant alteration was observed (The 39-Item Parkinson’s Disease Questionnaire (PDQ-39); Non-motor Symptoms Scale (NMSS); Parkinson’s Disease Sleep Scale (PDSS) [Citation15]. Adverse reactions occurred in all five groups, mostly dyskinesia [Citation15]. Dyskinesia occurrence was most prominent in the 50 mg OPC group [Citation15]. No death was reported during the study [Citation15].
In the open label extension of the BIPARK-I study (495 patients were involved), the switch from PLC or ENT to OPC resulted in a further decrease of OFF time (−65 min (PLC), −39 min (ENT) and increase of ON time without dyskinesia (43 min/PLC/, 45/ENT/) [Citation35,Citation36]. 25 mg was the applied OPC dose in the first week, which was followed by an individual response-dependent modification of dose [Citation35,Citation36]. 68.1% of the patients reported mild to moderate adverse reactions. Dyskinesia was the most common side effect, which was more frequent in the 1–3 weeks. In summary, the OL extension of the BIPARK-I study confirmed that the effect of opicapone remained durable and safe for at least 1 year [Citation37].
BIPARK-II was an RCT study, very similar to the BIPARK-I study, with similar patient selection criteria [Citation15,Citation38]. The difference between the two studies was the dose of OPC (in the BIPARK-II study the 5 mg dose was omitted) and the number of patients included (in this study it was lower, 427 PD patients) [Citation17,Citation38]. The main result of this study was also similar to the BIPARK-I: 25 and 50 mg OPC reduced the OFF time in comparison with PLC group (- 47 and – 54 minutes) [Citation17,Citation38]. Increase of ON time was also detectable in OPC groups (1.4 h (25 mg), 1.43 h (50 mg) and 0.8 h (PLC) [Citation38]. Similar to the above-mentioned clinical trial, no significant alteration (but non-significant improvement in the NMSS total score) was observed in non-motor scales (PDQ-39, NMSS, PDSS, Unified Huntington’s Disease Rating Scale (UHDRS), global assessment scale) [Citation38]. No serious adverse event occurred during the trial [Citation38].
376 patients continued the treatment in the open label phase, where the OFF time reduction seemed to be long-lasting and the ON time increased by an additional 24.9 minutes [Citation36].
Interestingly the overall analysis of BIPARK-I and -II trials also examined the OFF time reduction effect above and below 2,5 HY stages and regarding disease duration time (more or less than 8 years) [Citation7,Citation38]. The effect of 50 mg OPC on OFF time reduction was independent of HY stage or disease duration [Citation7,Citation38].
Lees et al. analyzed the onset and the stabilization time of the OPC over a 14–15 week study time period (BIPARK-I, -II). They analyzed the data of 750 patients (PLC: 255; OPC (25 mg): 241; OPC (50 mg): 262). They found that there was an improvement in the motoric performance already in the first week of treatment (at week 1 mean OFF-time reduction was 23 min in the PLC group, in contrast, 61 min (OPC (25 mg) and 75 min (OPC (50 mg) in the treatment groups), which stabilized after 2–3 weeks [Citation39].
Currently, a phase IV clinical trial (NCT02847442, OPTIPARK) is ongoing [Citation7]. A 50 mg dose of OPC is applied orally over a time period of 3–6 months with the involvement of 518 PD patients [Citation7].
1.8. Safety and tolerability
Several clinical trials assessed the safety (e.g. hepatotoxicity) or tolerability problems regarding OPC [Citation18–Citation20]. In the conducted phase I studies (single and repeated oral OPC doses) no remarkable side effects occurred [Citation18–Citation20]. However, in light of the fact that OPC is excreted via the hepatobiliary system, there is a possibility of plasma concentration increase in hepatopathy [Citation27]. That hypothesis led to test the pharmacological profiles of OPC in patients with moderate liver disease which showed a mildly elevated drug concentration level [Citation27]. Therefore, dose reduction is an issue to consider in this population [Citation10]. From a cardiological perspective OPC does not prolong the QTc value [Citation40,Citation41].
In the pooled analysis of two phase III trials (BIPARK-I and – II) the following adverse events were revealed on the treated arms: dyskinesia, insomnia, dry mouth, dizziness, constipation, blood creatine phosphokinase elevation [Citation7,Citation36]. In the open label phases, no relevant new potentially treatment-related adverse event occurred [Citation7,Citation36]. In the post-marketed period, 100 adverse events were reported (most frequently gastrointestinal- and psychiatric complaints) [Citation42].
1.9. Regulatory affairs
As mentioned above, currently OPC is marketed in the United Kingdom, Germany, Italy, Spain, North America (Bial-Portela & Ca. S.A. has license agreement with Ono Pharmaceutical CO., Ltd., Neurocrine Biosciences, Inc. and with Jiangsu Wanbang Biopharmaceutical Group Co., Ltd.).
2. Conclusion
OPC is a novel third-generation COMT inhibitor, which has been shown to be an effective and safe drug based on clinical trials conducted so far [Citation15,Citation18–Citation21,Citation34–Citation36,Citation38]. Compared to ENT, the necessity of one daily dose may increase the adherence of patients [Citation15]. From previous studies we know that TLC requires close laboratory monitoring for potential hepatotoxic effect [Citation7,Citation10]. In contrast, OPC use did not result in detectable liver function impairment [Citation7].
This new peripherally acting, once daily drug, could be an effective tool when treating PD with motor fluctuations. The recommended dose is 50 mg daily orally, but the personalized dose depends on the application of other antiparkinsonian drugs. No dose modification is needed in mild liver impairment (Child-Pugh B) or in renal failure [Citation14,Citation27].
The performed clinical trials did not register fatal outcome after OPC treatment [Citation7]. The most common adverse event was dyskinesia [Citation7,Citation10,Citation14].
3. Expert opinion
OPC is a novel third-generation, peripherally acting, reversible COMT inhibitor, requiring only once daily administration compared to ENT, and has no documented hepatotoxic effect as compared to TLC, thus no close laboratory monitoring is required. Accordingly, OPC may yield an add-on therapy with improved efficacy to the gold standard medication, LD, in advanced stages of PD. However, direct comparative trials between COMT inhibitors are lacking. The BIPARK-I study was the only trial, which reported the non-inferiority of OPC to ENT [Citation15]. Furthermore, the switch from ENT to OPC may also be meaningful in relation to OFF time reduction [Citation35]. From the perspective of non-motor symptoms there was a non-significant improvement in the NMSS total score in the BIPARK-I and II studies (with a sustained trend in the OL phase) [Citation7]. This effect was more evident in the 50 mg OPC group [Citation7]. Sleep and fatigue domains showed a positive response to OPC [Citation7]. Morning akinesia, OFF dystonia and unpredictable end-of-dose fluctuations may also be important treatment aspects, about which we have no direct information from studies. Furthermore, trials are lacking regarding the comparison of OPC with other possible add-on therapies (e.g. rasagiline) in advanced stages of PD, so the decision should be based on good clinical practice at the individual level.
Nevertheless, OPC could serve as an effective therapeutic tool in the treatment of ENT non-responder patients. Although the data from studies are impressive, the global availability of OPC may limit its world-wide application. Accordingly, the increase of clinical experience and further studies are needed to find the optimal place for this new drug in the treatment of PD.
Article highlights
Since 24 June 2016 a third COMT inhibitor, namely opicapone (OPC) is also available, which was approved by the European Medicine Agency for the treatment of end-of-dose motor fluctuations in adult patients whose symptoms are not controllable by LD/DOPA decarboxylase inhibitor combination.
OPC is a peripherally acting, reversible COMT inhibitor, requiring only once daily administration compared to ENT, and has no documented hepatotoxic effect as compared to TLC, thus no close laboratory monitoring is required.
The performed clinical trials did not register fatal outcome after OPC treatment. The most common adverse event was dyskinesia.
Further comparative studies and broader trial inclusion criteria are needed to help the decision between COMT inhibitors and to expand the patient spectrum where this drug can be applied.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
One referee was an investigator for one of the two phase III trials with opicapone and served as a consultant for Bial in opicapone’s development program. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386:896–912.
- Barone P, Antonini A, Colosimo C, et al. The PRIAMO study: A multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov Disord. 2009;24:1641–1649.
- Tysnes OB, Storstein A. Epidemiology of Parkinson’s disease. J Neural Transm. 2017;124:901–905.
- Szatmári S Jr, Ajtay A, Bálint M, et al. Linking individual patient data to estimate incidence and prevalence of Parkinson’s disease by comparing reports of neurological services and pharmacy prescription refills at a nationwide level. Front Neurol. 2019;10:640.
- Dickson DW. Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med. 2012;2:1–15.
- Rogers G, Davies D, Pink J, et al. Parkinson’s disease: summary of updated NICE guidance. BMJ. 2017;358:j1951.
- Fabbri M, Ferreira JJ, Lees A, et al. Opicapone for the treatment of Parkinson’s disease: A review of a new licensed medicine. Mov Disord. 2018;33:1528–1539.
- Olanow CW, Kieburtz K, Rascol O, et al. Factors predictive of the development of Levodopa-induced dyskinesia and wearing-off in Parkinson’s disease. Mov Disord. 2013;28:1064–1071.
- Olanow CW, Schapira AH. Therapeutic prospects for Parkinson disease. Ann Neurol. 2013;74:337–347.
- Annus Á, Vécsei L. Spotlight on opicapone as an adjunct to levodopa in Parkinson’s disease: design, development and potential place in therapy. Drug Des Devel Ther. 2017;11:143–151.
- Khor SP, Hsu A. The pharmacokinetics and pharmacodynamics of levodopa in the treatment of Parkinson’s disease. Curr Clin Pharmacol. 2007;2:234–243.
- Müller T. Catechol-O-methyltransferase inhibitors in Parkinson’s disease. Drugs. 2015;75:157–174.
- Scott LJ. Opicapone: a review in Parkinson’s disease. Drugs. 2016;76:1293–1300.
- Rodrigues FB, Ferreira JJ. Opicapone for the treatment of Parkinson’s disease. Expert Opin Pharmacother. 2017;18:445–453.
- Ferreira JJ, Lees A, Rocha JF, et al. Opicapone as an adjunct to levodopa in patients with Parkinson’s disease and end-of-dose motor fluctuations: a randomised, double-blind, controlled trial. Lancet Neurol. 2016;15:154–165.
- Palma N, Bonifacio MJ, Loureiro AI, et al. Computation of binding affinity of catechol-O-methyltransferase-opicapone complexes. Parkinsonism Relat Disord. 2012;18:S125.
- Kiss LE, Ferreira HS, Torrão L, et al. Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem. 2010;53:3396–3411.
- Almeida L, Rocha JF, Falcão A, et al. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel catechol-O-methyltransferase inhibitor, in healthy subjects: prediction of slow enzyme-inhibitor complex dissociation of a short-living and very long-acting inhibitor. Clin Pharmacokinet. 2013;52:139–151.
- Rocha JF, Falcão A, Santos A, et al. Effect of opicapone and entacapone upon levodopa pharmacokinetics during three daily levodopa administrations. Eur J Clin Pharmacol. 2014;70:1059–1071.
- Rocha JF, Almeida L, Falcão A, et al. Opicapone: a short lived and very long acting novel catechol-O-methyltransferase inhibitor following multiple dose administration in healthy subjects. Br J Clin Pharmacol. 2013;76:763–775.
- Ferreira JJ, Rocha JF, Falcão A, et al. Effect of opicapone on levodopa pharmacokinetics, catechol-O-methyltransferase activity and motor fluctuations in patients with Parkinson’s disease. Eur J Neurol. 2015;22:815–825.
- Falcão A, Santos A, Ferreira J, et al. Decision-making process for opicapone’s bedtime regimen [abstract]. Mov Disord. 2017;32 (suppl 2).
- Dingemanse J, Jorga KM, Schmitt M, et al. Integrated pharmacokinetics and pharmacodynamics of the novel catechol-O-methyltransferase inhibitor tolcapone during first administration to humans. Clin Pharmacol Ther. 1995;57:508–517.
- Keränen T, Gordin A, Karlsson M, et al. Inhibition of soluble catechol-O-methyltransferase and single-dose pharmacokinetics after oral and intravenous administration of entacapone. Eur J Clin Pharmacol. 1994;46:151–157.
- Lopes N, Ferreira J, Lees A, et al. Efficacy of opicapone in combination with dopamine agonists or MAO-B inhibitors on the treatment of motor fluctuations in Parkinson’s disease. Eur J Neurol. 2015;22:439.
- Lopes N, Ferreira J, Lees A, et al. Exploratory efficacy of opicapone in combination with dopamine agonists or MAO-B inhibitors on the treatment of motor fluctuations in Parkinson’s disease. Mov Disord. 2015;30:S101.
- Rocha JF, Santos A, Falcão A, et al. Effect of moderate liver impairment on the pharmacokinetics of opicapone. Eur J Clin Pharmacol. 2014;70:279–286.
- Committee for Medicinal Products for Human Use (CHMP). Ongentys assessment report; 28 April 2016. EMA/343011/2016.
- Santos A, Falcao A, Rocha J, et al. Influence of food on opicapone pharmacokinetics and pharmacodynamics. Eur J Neurol. 2017;24:S123–444.
- Falcão A, Rocha JF, Santos A, et al. Opicapone pharmacokinetics and pharmacodynamics comparison between healthy Japanese and matched white subjects. Clin Pharmacol Drug Dev. 2016;5:150–161.
- Rocha JF, Sicard É, Fauchoux N, et al. Effect of opicapone multiple-dose regimens on levodopa pharmacokinetics. Br J Clin Pharmacol. 2017;83:540–553.
- Svetel M, Tomić A, Kresojević N, et al. Pharmacokinetic drug evaluation of opicapone for the treatment of Parkinson’s disease. Expert Opin Drug Metab Toxicol. 2018;14:353–360.
- Fabbri M, Rosa MM, Ferreira JJ. Clinical pharmacology review of opicapone for the treatment of Parkinson’s disease. Neurodegener Dis Manag. 2016;6:349–362.
- Rocha JF, Ferreira JJ, Falcão A, et al. Effect of 3 single-dose regimens of opicapone on levodopa pharmacokinetics, Catechol-O-methyltransferase activity and motor response in patients with Parkinson disease. Clin Pharmacol Drug Dev. 2016;5:232–340.
- Ferreira J, Lees A, Rascol O, et al. Switching entacapone or placebo to opicapone open-label: efficacy results of the BIPARK-I 1-year extension study. J Neurol Sci. 2017;381:561–756.
- Ferreira JJ, Lees A, Rocha JF, et al. Long-term efficacy of opicapone in fluctuating Parkinson’s disease patients: a pooled analysis of data from two phase 3 clinical trials and their open-label extensions. Eur J Neurol. 2019;26:953–960.
- Lopes N, Ferreira J, Lees A, et al. Efficacy of opicapone in Parkinson’s disease patients with motor fluctuations at different stages of symptom progression. Presented at the 21st International Congress of Parkinson’s Disease and Movement Disorders, Vancouver; 2017 June 4–8; British Columbia, Canada. Meeting Abstract; 2017.
- Lees AJ, Ferreira J, Rascol O, et al. Opicapone as adjunct to levodopa therapy in patients with Parkinson disease and motor fluctuations: a randomized clinical trial. JAMA Neurol. 2017;74:197–206.
- Lees AJ, Ferreira JJ, Reichmann H, et al. Onset and stabilization of opicapone treatment effects in fluctuating Parkinson’s disease patients: exploratory by-week efficacy analysis of pooled phase III studies [abstract]. Mov Disord. 2016;31(suppl):2).
- Pinto R, l’Hostis P, Patat A, et al. Evaluation of opicapone on cardiac repolarization in a thorough QT/QTc study. Clin Pharmacol Drug Dev. 2015;4:454–462.
- Pinto R, Vaz-da-Silva M, Lopes N, et al. Cardiac safety of opicapone in patients with Parkinson’s disease: analysis of the centralized phase III ECG dataset. Mov Disord. 2015;30:S112.
- Caldas CA, Teodoro T, Ferreira JJ. The launch of opicapone for Parkinson’s disease: negatives versus positives. Expert Opin Drug Saf. 2018;17:331–337.