
ABSTRACT
Introduction: Despite the recent advances in the treatment of malignant melanoma with immunotherapy and BRAF/MEK targeted agents, advanced disease still beholds a poor prognosis for a significant proportion of patients. Cyclin-dependent kinase (CDK) inhibitors have been investigated as novel melanoma therapeutics throughout a range of phase 1 and 2 trials, as single agents and in combination with established treatments.
Areas covered: This article summarizes the rationale for, and development of CDK inhibitors in melanoma, with their evolution from pan-CDK inhibitors to highly specific agents, throughout clinical trials and finally their potential future use.
Expert opinion: Whilst CDK inhibitors have been practice changing in breast cancer management, their efficacy is yet to be proven in melanoma. Combination with BRAF/MEK inhibitors has been hindered by dose-limiting toxicities, but their role may yet to be found within the spectrum of biomarker-derived personalized melanoma management. The effect that CDK inhibitors can have as an adjunct to immunotherapy also remains to be seen.
1. Introduction
The landscape of treatment for metastatic melanoma has improved significantly in recent years. Whilst previously associated with a dismal prognosis and very limited response to conventional chemotherapy, the advent of BRAF and subsequent MEK targeted therapy has led to significant survival improvement in patients with BRAF-mutant disease [Citation1]. Melanoma has also been at the forefront of the immunotherapy (IO) revolution, with anti-CTLA4 and anti -PD-1 agents leading to durable responses in some patient cohorts [Citation2].
Despite these recent advances, a significant proportion of patients fail to respond, or receive limited benefit from these treatment modalities. Only 40–50% of melanomas harbor BRAF mutations [Citation3,Citation4] and are therefore susceptible to BRAF targeted therapy. The majority of these patients invariably develop resistance to BRAF targeted therapy which is reflected with a median overall survival (OS) of 44% at 3 years with combination therapy [Citation5]. Combination immunotherapy with ipilimumab and nivolumab has dramatically improved outcomes irrespective of BRAF status with an OS of 52% at 5 years in recently published data [Citation2], although the need for improvement remains. Targeted treatment advances in other solid malignancies have resulted in novel agents and combinations being investigated in melanoma, in the hope that outcomes can be further improved. This review will focus on the recent advances with cyclin-dependent kinase (CDK) inhibitors in melanoma, including the rationale for their use, clinical trial outcomes and future direction.
1.1. The cell cycle and cyclin-dependent kinases 4 +6
The cell cycle is a tightly controlled process that eukaryotic cells undergo to facilitate cell division and is frequently dysregulated in cancer. The cell cycle consists of four phases; Gap 1 (G1); Synthesis (S); Gap 2 (G2) and Mitosis (M) [Citation6]. G1 is a metabolically active phase involving cell growth and preparation for division, with replication of organelles and other cellular components. S phase incorporates DNA replication, whilst G2 permits further cellular growth and preparation for the final phase, mitosis, where chromosome segregation and cellular division occurs [Citation6]. Cells can also exit the cell cycle from G1 and enter a 5th phase, G0, which can be either a transient or permanent state of cell cycle arrest. Within the cell cycle there are a number of transition points or ‘checkpoints’ that function to halt further cell cycle progression in the event of aberrant DNA replication or mitosis [Citation7].
CDKs are a family of serine/threonine kinases that act as key regulatory proteins at checkpoints during the cell cycle, in conjunction with cyclin proteins [Citation8]. They therefore play a vital role in control of the cell cycle [Citation9,Citation10,Citation11], with an array of CDK/Cyclin complexes and subsequently fluctuant levels of cyclin [Citation12]. CDK4 and CDK6 promote progression through the cell cycle through interaction with D-type cyclins () [Citation13]. The primary function of these complexes, in conjunction with cyclin E-cdk2 [Citation14], is to phosphorylate cell cycle pocket proteins such as retinoblastoma protein (Rb), which during cellular quiescence are dimerized with E2F [Citation15]. Phosphorylated Rb releases E2F transcription factors, including E2F1 which then dissociate, thus driving transition from G1 to S phase [Citation16]. The cyclin D-CDK4/6 complex also sequesters two CDK inhibitors, p21CIP1 and p27KIP1, which prevent activation of the cyclin E-CDK2 complex [Citation17,Citation18,Citation19]. It should also be noted that E2F1 despite its essential contribution to cell cycle transition may also play a role in apoptosis induction, via p53-mediated apoptosis [Citation20].
Figure 1. Image illustrating the phosphorylation of Rb by the CyclinD/CDK4/6 complex, releasing E2F transcription factors to dissociate, subsequently driving G1/S transition through the cell cycle. This also permits further phosphorylation of Rb by the Cyclin E/CDK2 complex. The image also demonstrates the integration of other oncogenic pathways (e.g. JAK/STAT and PI3K/AKT/mTOR) within the Cyclin D/CDK4/6/Rb/p16INK4A axis. CDK 4/6 inhibitors disrupt this oncogenic pathway. Other sites of action of alternative CDK/cyclin complexes are also demonstrated and have been investigated as potential targets for therapy
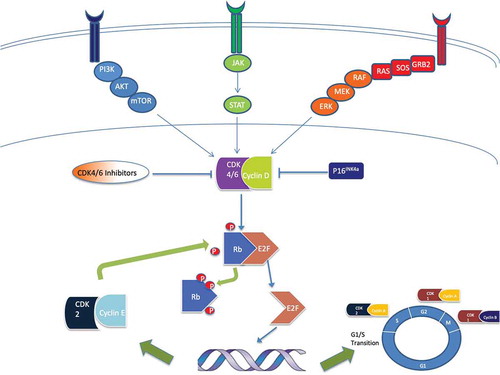
1.2. Dysregulation of CDK4/6-Rb-p16INK4A pathway in cancer
Hyper-activation of the CDK4/6-Rb-p16INK4A pathway () is common and implicated in approximately 90% of melanomas [Citation21–23–Citation23]. This can occur through deletion or mutation of Rb [Citation24], activating mutations in CDK4 [Citation25,Citation26] and CDK 6 [Citation27], increased Cyclin D1 expression [Citation21] or loss of p16INK4A (inhibitor of the CDK4/6-cyclinD1 complex) [Citation21,Citation28]. The Cyclin D-CDK4/6 complex is also involved in other oncogenic signaling pathways such as the mitogen-activated protein kinase (MAPK) pathway, the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway, the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway and wnt/β-catenin signaling [Citation27]. Copy number alterations of key cell cycle genes, such as CCND1 (cyclin-D1), CDKN2A (p16INK4A), and CDK4 are common and associated with worse outcomes in melanoma [Citation3,Citation29]. Sequential loss of p16INK4A expression from benign nevi (73%) to metastatic melanoma (14.7%) has also been demonstrated, purporting the significance of this pathway in melanomagenesis [Citation30,Citation31]. Genomic analysis in familial melanoma has shown germline loss of function mutations in CDKN2A, as well as activating mutations of CDK4, that are associated with a 40–50 fold increase in the risk of developing melanoma [Citation22,Citation32,Citation33].
In addition to forming a complex with cyclin D, CDK6 regulates a number of genes directly including EGR-1, FLT-3 and notably VEGF-A [Citation13], by acting as a transcriptional regulator. In knockout mice models, CDK4 and CDK6 have been shown to play a key role in melanoma cell proliferation and angiogenesis [Citation13], with CDK4 facilitating localization of CDK6 to the VEGF-A promoter. Additionally, CDK6 is thought to control the epigenetic regulator EZH2 [Citation13], which is important in melanoma progression and metastasis [Citation34]. CDK 4 expression has also been postulated to play a role in cell migration, cell proliferation and colony formation in lung cancer, as well as being an independent prognostic indicator [Citation35]. Given the spectrum of activity of CDK4 and CDK6, this has led to further interest in the potential use of CDK4/6 inhibitors (CDK4/6i) in solid tumors.
1.3. Senescence
Cellular senescence may be a powerful tumor suppression mechanism, preventing the uncontrolled proliferation of malignant cells and contributing to the clinical efficacy of radiotherapy, cytotoxic chemotherapy and targeted drug therapies, including CDK 4/6 inhibitors [Citation36]. This process often occurs in two stages, cell cycle arrest followed by gerogenic conversion (geroconversion) from reversible arrest to irreversible senescence. Senescence-associated growth arrest is orchestrated primarily by p16/Rb and p53 tumor suppression pathways [Citation37]. Thus, it is unsurprising that CDK 4/6 inhibition or overexpression of p16INK4A has been demonstrated to trigger cellular senescence within in vitro models [Citation38,Citation39]. Preclinical models have demonstrated maximal geroconversion occurs after 8 days of continuous palbociclib administration in melanoma cell lines [Citation40]. However, despite this finding, monotherapy with palbocicilb, abemociclib or ribocicilib does not uniformly lead to senescence in cancer cells [Citation36]. CDK 4/6 inhibition may induce a senescence permissive state with additional intrinsic and extrinsic factors required to determine cellular fate. Specifically, in melanoma, synergy between mTOR inhibition and palbociclib has been demonstrated in ex vivo models to be critical to triggering irreversible senescence [Citation40,Citation41].
The triggering of senescence may not always be beneficial. Studies have shown senescent cells within the tumor microenvironment may adopt a pro-inflammatory senescence-associated secretary phenotype (SASP) that may drive tumor proliferation and therapeutic resistance [Citation42]. Studies in melanoma cell lines, have demonstrated that fibroblast-induced SASP caused by palbociclib exposure can stimulate the growth of melanoma cells in a genotype-dependent manner [Citation43]. Furthermore, studies in human melanoma cell lines have demonstrated that triggering of geroconversion by palbociclib mediates vermurafenib resistance [Citation40].
2. Development of CDK inhibitors
Dysregulation of cell cycle control, specifically the G1 to S-phase transition is almost ubiquitous in cancer development [Citation44,Citation45]. This knowledge provides a strong rationale for considering the cell cycle and its complex regulation system as potential targets for therapeutic modulation. The fundamental role of CDKs in controlling the cell cycle, and specifically their role as regulators of cellular proliferation, apoptosis and senescence make them particularly appealing as pharmacological targets in cancer therapy [Citation9,Citation46]. However, the almost universal presence and essential function of CDKs within nearly all human cells makes this a challenging molecular target and may limit potential clinical utility. For example, CDK1 is essential for mitosis in all eukaryotic cells, thus has proven difficult to target without inflicting substantial toxicity on healthy cell populations [Citation9]. Alternative interphase CDKs are more appealing targets as have a more limited role, particularly in the proliferation of specialized cells and are frequently overexpressed in many cancer cell types [Citation46].
Preclinical data have demonstrated that certain CDKs may have significant influence in both the proliferation and spread of specific cancers [Citation46,Citation47]. Research over the past two decades has explored a wide range of compounds which affect various CDKs in a range of cancer subtypes with varying selectivity, targets and mechanisms of action. More recently, improved understanding of the role of specific CDKs in certain cancer types has led to the first global approvals of CDK 4/6 inhibitors in breast cancer [Citation44,Citation48]. Fueled by this breakthrough, these and other molecules are under investigation, both as monotherapy and in combination with other treatments in a number of other cancers, including melanoma [Citation49]. Greater understanding of the biology of CDKs in cancer pathology may lead to more rational and innovative drug development in this field.
2.1. First-generation CDK Inhibitors
The production of CDK inhibitors has proven challenging due to the high degree of sequence homology shown between different CDKs and also other intracellular protein kinases. Furthermore, significant functional redundancy is seen between different CDKs and therapeutic inhibition of one subtype may cause a compensatory rise in others, allowing resumption of cellular activity and proliferation [Citation46,Citation47]. Early CDK inhibitors developed were relatively nonspecific and often referred to as ‘pan-CDK’ inhibitors. Early drug discovery programs focused primarily on CDK2 inhibition as a target due to the observation of frequent dysregulation in a plethora of cancers subtypes [Citation44,Citation50]. Examples of first-generation inhibitors include flaviopiridol (alvocidib), olomucine, roscovitidine (seliciclib) and kenpaullone [Citation44,Citation51].
The most extensively studied first-generation drug is flaviopiridol, a semisynthetic flavonoid derived from rohitukine, an Indian shrub [Citation52]. It was the first potent CDK inhibitor to enter human studies and along with its analogue P276-00 (2nd generation CDK inhibitor) has been studied in over 60 clinical trials [Citation44,Citation53]. It has been shown to inhibit CDK1, CDK2, CDK4, CDK6, CDK7 and CDK9 and despite showing significant in vitro activity, clinical studies disappointed, showing substantially less activity in vivo [Citation54]. Clinical studies did show potential efficacy in a range of hematological malignancies, such as chronic lymphocytic leukemia and mantle cell lymphoma, however, despite extensive investment clinical development was discontinued in 2012 [Citation55].
2.2. 2nd generation CDK Inhibitors
Advances in drug design and development allowed increased selectivity for CDK1 and CDK2, and a reduction in dose-limiting toxicities [Citation44]. Second generation CDK inhibitors exhibit higher potency and greater selectivity for certain CDK subtypes. Numerous potential drug candidates entered development; the most notable examples include roniciclib, dinaciclib (MK-7965), voruciclib and riviciclib [Citation44,Citation51]. However, despite encouraging preclinical evaluation, relatively few progressed beyond phase 1 clinical trials.
One of the most promising was dinaciclib, specifically developed as a high potency inhibitor of CDK1, CDK2, CDK5 and CDK9 and showed significant efficacy in >100 tumor cell lines (including melanoma), administration triggering rapid induction of G2/M cell arrest and apoptosis [Citation56]. Several clinical trials evaluated clinical efficacy beyond phase 1, including a phase 2 clinical trial in melanoma (). However, they have not demonstrated significant clinical efficacy resulting in termination of clinical development in solid tumors [Citation44,Citation57]. No second-generation CDK inhibitors have been licensed but remain under evaluation primarily in hematological malignancies.
Table 1. Summary of CDK inhibitor trials in melanoma
2.3. Selective CDK inhibitors
The failure of nonselective CDK inhibitors in solid cancers treatment is multifactorial, including low CDK specificity, dose limiting toxicities and lack of understanding about in vivo mechanism of action. For example, flavopiridol showed significant promise in preclinical studies but disappointed in multiple clinical trials. Despite extensive investigation, its mechanism of action has not been fully elucidated and the potential significance of off target effects including transcriptional suppression, autophagy and endoplasmic reticulum stress are unknown [Citation54,Citation62]. This lack of understanding about mechanism of action is the case for many 1st and 2nd generation CDK inhibitors and has confounded further drug development. This situation has been further exacerbated by the lack of appropriate biomarkers.
The recognized disadvantages of 1st and 2nd generation CDK inhibitors suggest that more selective CDK inhibition may be the key to developing successful and tolerable anti-cancer agents. Developing specific CDK1 inhibitors may be challenging to develop due to toxicity [Citation45,Citation46]. Inhibition of CDK2 may offer a more appealing target, particularly in tumors such as melanoma, which may be driven by cyclin E amplification [Citation44]. Alternatively, selectively targeting CDK7, CDK8 and CDK9, which are associated with basal transcription, may also prove a successful strategy as cancer cells frequently harbor unique vulnerabilities to selective suppression [Citation50].
After the failure of multiple CDK inhibitors in clinical trials, development focus shifted toward more selective CDK targeting. The cancer genome atlas has identified CDK4 amplification and cyclin D1 overexpression in several subtypes of breast cancer [Citation63]. Discoveries in the early 1990s provided proof-of principal that CDK4 inhibition might retard cancer cell development and proliferation [Citation64]. High-throughput screening of chemical libraries focused on compounds that inhibited CDK4, resulting in chemists at Parke-Davis developing PD-0332991, a specific CDK4/6i that would eventually become palbociclib [Citation64].
2.4. CDK4/6 inhibitors
CDK4/6i bind to the ATP binding domain of CDK4/6 which competitively inhibit the kinase activity of these proteins, leading to G1/S arrest [Citation65]. In addition to enforcing cell arrest (and therefore causing cytostasis), CDK4/6i have been shown to enhance cancer cell immunogenicity in mice [Citation66], modulate MAPK signaling [Citation67] and induce a senescence-like phenotype [Citation68]. CDK4/6i also indirectly target protein arginine methyltransferase 5 (PRMT5), an epigenetic modifier, resulting in reduced expression of the oncogene MDM4 and subsequent p53 activation [Citation69].
The first licensed CDK inhibitor was palbociclib (PD-0332991), an oral reversible selective small-molecule inhibitor of CDK4 and CDK6 developed by Pfizer and licensed in estrogen receptor-positive (ER+) breast cancer [Citation48]. Parallel drug discovery at Novartis and Eli Lilly has led to the licensing of two further CDK4/6i ribociclib (LEE011) and abemaciclib (LY-2835219) respectively, also in ER+ breast cancer [Citation64]. All three licensed CDK4/6i have a similar mechanism of action but differ in pharmacokinetic properties (). The successful development of selective inhibitors of CDK4/6 has markedly changed the perception of CDK inhibition as a therapeutic target in cancer. These compounds are now under further investigation in a range of cancers including pancreatic, head & neck, ovarian and melanoma [Citation44].
Table 2. Comparative pharmacokinetics of palbociclib, ribociclib and abemaciclib
In vitro studies have shown that palbociclib has similar relative potency for cyclin D1/CDK4 and cyclin D2/CDK6 [Citation72], whereas ribociclib and abemaciclib have a greater potency for CDK4 compared to CDK6 [Citation73,Citation74]. Abemaciclib also inhibits CDK9 but at lower potencies compared to CDK4 and CDK6 and the clinical significance of this is not clear [Citation74].
3. Clinical trials
Increasing understanding of the importance of the CDK4/6 pathway in melanoma has led to clinical trials exploring the potential clinical utility of CDKi as monotherapy and in combination with other therapies. Trials to date can be seen in .
3.1. Pan-CDK inhibitors (first and second generation)
Flavopiridol was the first CDKi to show clinical activity in humans [Citation75] and its use in untreated melanoma was first assessed as part of a phase 2 trial [Citation76]. Sixteen patients with histologically proven malignant melanoma, who had progressed through standard therapy, were administered flavopiridol at a dose of 50 mg/m2 intravenously (IV) over 1 h daily for 3 days every 3 weeks. The toxicity profile was deemed acceptable with mostly mild (grade 1) toxicities (82% diarrhea, 47% nausea); however, despite 7 patients achieving stable disease (SD, 1.8 to 9.2 months), it was concluded that the drug failed to show any significant clinical activity with no objective responses.
Flavopiridol has also been assessed as part of a combination phase 1 trial with cisplatin or carboplatin, in 39 patients with advanced malignancies including melanoma [Citation77]. Within the flavopiridol/cisplatin cohort toxicities were deemed to be moderate with grade 3 nausea (30%) vomiting (19%) diarrhea (15%) and neutropenia (10%). Maximum tolerated dose (MTD) was established at 60 mg/m2 cisplatin and 100 mg/m2/24 hours flavopiridol. Within the carboplatin cohort toxicities were unexpectedly severe, with grade 4 anemia, grade 3 dehydration, and one intracranial bleed resulting in death. Again, there were no objective responses throughout, and due to the significant toxicities experienced there has been little enthusiasm for further CDK/chemotherapy combination studies.
Roniciclib, an oral pan-cyclin dependent kinase inhibitor of CDKs 1/2/4/6/7 and 9, has been the subject of a phase 1 dose escalation trial of 148 patients [Citation78]. This involved two dosing schedules: 3 days on/4 days off and 4 weeks on/2 weeks off, with 2 melanoma patients included in each schedule [Citation78]. Due to limited tolerability, the 4 weeks on/2 weeks off schedule was terminated, with the most common treatment-emergent toxicities being nausea (76%) fatigue (65%), diarrhea (63.1%) and vomiting (57.7%). Within the 3 days on/4 days off cohort, a recommended phase 2 dose (RP2D) of 5 mg twice daily (BD) was established. The investigators concluded that the agent displayed an acceptable toxicity profile, and notably the most significant disease control was observed in a refractory melanoma patient who experienced SD for 153 days. Expansion to further melanoma-specific trials with roniciclib however has not been pursued.
The intravenous CDK 1/2/5/9 inhibitor dinaciclib has been tested in three melanoma inclusive trials. A first-in-human multi-tumor-group phase 1 trial enrolled 48 patients, including 1 patient with melanoma, finding a MTD and RP2D of 12 mg/m2 as an intravenous infusion once weekly for 3 weeks [Citation79]. Adverse events (AEs) were reported in 98% of patients with dose-limiting toxicities (DLTs) including orthostatic hypotension and raised uric acid levels. A second trial, NCT01026324 assessed dinaciclib in a phase 1/2 melanoma-specific trial [Citation80] however this was terminated due to slow accrual. Finally, the agent was the subject of the stage IV melanoma-specific phase 2 trial, SWOG SO826 [Citation81]. 72 patients were enrolled and administered a fixed dose of 50 mg IV every 3 weeks. Results were not encouraging however, with 39% experiencing grade 4 toxicities (70% neutropenia) and no responses identified.
Whilst further pan-CDK inhibitors have been trialed in melanoma, robust assessment of their efficacy has been hindered by suspended and terminated trials. P1446A is a potent inhibitor of CDK 4/1 and 9 and was investigated in a multi tumor phase 1 trial [Citation82]. Twenty-nine patients across 5 centers in India were dosed using a modified Fibonacci scheme for dose escalation. A maximum-tolerated dose (MTD) of 600 mg/day was determined, with diarrhea being the DLT. This agent was taken on for further evaluation within a combination phase 1 trial with vemurafenib in patients with BRAF-mutant melanoma [Citation83], but the study was suspended due to sponsor decision.
3.2. Specific CDK inhibitors
Given the success of specific CDK inhibitors, such as palbociclib in breast cancer, multiple trials have attempted to replicate their success in melanoma. Such interest has been focussed on studies that have attempted single-agent regimens or alternatively in combination with other systemic anti-cancer therapies to optimize efficacy.
3.2.1. Single-agent CDK 4/6 inhibitors in melanoma
Although palbociclib is known for its practice-changing influence within estrogen positive breast cancer, clinical trials investigating its use in melanoma have been less profound. Upon development as a first in class oral CDK4/6i, palbociclib was tested in an advanced multi-tumor phase 1 trial [Citation84]. 41 patients (6 with melanoma) were enrolled and received palbociclib in six dose escalation cohorts using a standard 3 + 3 design. The MTD was established at 125 mg once daily (OD) with neutropenia being the sole DLT. This agent has not been further investigated as a single agent in melanoma-specific trials.
The most selective CDK 4/6 inhibitor to date is ribociclib [Citation49] and was first investigated in a first in human study in the USA and Europe, across multiple-advanced tumor types [Citation85]. One hundred and thirty-two patients were enrolled and received escalating doses of ribociclib (3 weeks on/1 week off or continuous), with neutropenia and thrombocytopenia being the most common DLTs. The recommended dose for expansion was established at 600 mg OD 3-weeks-on/1 week off. Intriguingly three partial responses (PR) were observed, one of which occurred in a patient with BRAF/NRAS wild type, CCND1 amplified melanoma.
Abemaciclib is another specific CDK4/6i but is of particular interest in melanoma due to pre-clinical studies demonstrating its ability to cross the blood–brain barrier, and the known propensity of melanoma to metastasize to the brain [Citation86]. The initial first-in-human phase 1 study involved 225 patients with multiple different tumors, including non-small cell lung cancer, glioblastoma, melanoma, breast and colorectal cancer [Citation87]. A 3 + 3 dose escalation was used to establish an oral MTD of 200 mg BD throughout a 28-day cycle. The most common adverse events across the study were diarrhea (52%), nausea (30%) and fatigue (21%). Within the melanoma cohort (n = 16) phamacodynamic inhibition of Rb phosphorylation was demonstrated. Notably, one patient with NRAS-mutant melanoma achieved a partial disease response.
A subsequent phase 2 study, evaluated abemaciclib in patients across multiple tumor types with brain metastases [Citation88]. Whilst the breast cohort results have been reported and suggest a significant OS benefit (8.4 months vs <4 months), the melanoma cohort results are yet to be reported.
3.3. Combination therapy
An area of high interest for CDK therapeutics in melanoma has been their potential role as an adjunct to BRAF and MEK inhibition. Preclinical models have suggested that the addition of ribociclib to the oral BRAF inhibitor (BRAFi) encorafenib delays development of BRAFi resistance leading to a number of phase 1b/2 clinical trials to evaluate potential utility [Citation89].
A phase 1b study has investigated palbociclib in combination with the reversible, highly selective MEK1/2 inhibitor, trametinib [Citation90]. The study evaluated palbociclib given OD 21 days on/7 days off (21/7), in combination with trametinib OD, in 28 patients with advanced malignancies. Two recommended combination regimens (RCR) were established; trametinib 2 mg + palbociclib 75 mg 21/7 (RCR1), and trametinib 1.5 mg + palbociclib 100 mg 21/7 (RCR2). Common adverse events throughout the study were diarrhea (67.9%), acneiform rash (64.3%) and fatigue (53.6%) with Grade 3 and 4 adverse events seen in 75% and 21.4% of patients, respectively. It was noted that one of two patients that achieved a PR at RCR1 had BRAF/NRAS wild type melanoma, with ongoing disease response at the time of publication (approximately 13 months).
NCT01777776 investigated the combination of ribociclib, given OD 21 days on/7 days off with encorafenib OD dosed continuously, in patients with melanoma and other skin tumors, within a phase 1b/2 trial [Citation89]. Dose escalation was guided by a Bayesian logistic regression model with cohorts of ribociclib and encorafenib dosed at 200/300, 300/200, 400/100 and 400 mg/200 mg, respectively. Eighteen patients received the combination at varying doses, with 78% having received prior BRAFi therapy. Study drug-related AEs at all grades included palmar-plantar hyperkeratosis (PPH 46%), flushing (36%) and rash (36%), with two DLTs (G3 myalgia and hyperbilirubinaemia) at the 200/300 dose level. Nine patients were evaluable for response at the time of publication, with 2 PRs and 6 SDs. Due to toxicity and higher than expected blood levels of encorafenib in the 200/300 and 300/200 cohorts, the 400/100 and 400/200 dose ratio regimens were predicted to be better tolerated. The trial was however terminated before completion by the sponsor in response to other clinical developments within the field.
The alternative combination of a CDK 4/6 inhibitor, ribociclib and MEK inhibitor, binimetinib in patients with NRAS-mutant melanoma, has also been investigated as a phase 1b/2 study [Citation91]. Phase 1b assessed a 28-day cycle of ribociclib OD for 21 days with binimetinib BD for 28 days, against a 21-day cycle of ribociclib OD and binimetinib BD for 14 days. A 28-day cycle of ribociclib 200 mg OD and binimetinib 45 mg BD was selected as the RP2D, with an unconfirmed PR of 35%. In phase 2 this schedule was received by 16 patients with a median exposure of 4 months. Common G3/4 AEs were transaminitis (19%/6%), nausea (19%/0), rash (19%/0) vomiting (6%/6%) and neutropenia (12%/0%). Four patients achieved PR, with SD in seven. Median progression-free survival (PFS) was 6.7 months, leading to the conclusion that this combination has favorable efficacy and manageable toxicity in this group of patients.
The triple combination of encorafenib, binimetinib and ribociclib has been evaluated in a phase 1b/2 dose escalation study in patients with BRAFV600 mutant disease [Citation92], aiming to enhance the synergistic effects already seen with combination BRAF/MEK inhibition. Phase 1b involved 21 patients with pan-tumor site BRAFV600 mutant advanced solid tumors, administering escalating doses of ribociclib OD 100–600 mg 3 weeks on/1 week off, in combination with encorafenib 200 mg OD and binimetinib 45 mg BD, in successive cohorts. Of note, the dose of encorafenib was lower than that of established in dual combination therapy (450 mg OD), due to suspected pharmacokinetic interactions with ribociclib. No DLTs were reported and the ribociclib RP2D was 600 mg. In the phase 2 expansion, 42 patients with confirmed BRAFV600 melanoma, naïve to prior BRAF inhibitor therapy were enrolled, with objective response rate (ORR) being the primary endpoint. The most common grade 3/4 toxicities were neutropenia (26%), raised ALT (14%), diarrhea (7%) and anemia (7%), with 23% of patients discontinuing treatment due to AEs. ORR was 52% with 4 CRs, 18 PRs and 15 with SD. Whilst the combination induced an objective response in over half the patients, there was also evidence of increased toxicity.
3.4. CDK4/6 inhibitors and immunotherapy
Given the success of IO in melanoma, there has been research into whether CDK4/6i could act as an adjunct to IO. There is some rationale for synergy with this combination. Preclinical models have shown that CDK4/6i trigger changes in the tumor microenvironment and tumor-secreted cytokines that could result in enhanced T-cell activity and a reduction in regulatory T cells [Citation93]. This could lead to a theoretical enhancement of immunotherapeutic effect. Some promising signs of efficacy have been demonstrated in vitro [Citation94] and several phase 1/2 clinical trials of immunotherapy and CDK4/6i combinations are underway in patients with breast cancer (NCT03294694, NCT03147287 and NCT02778685). Of most relevance for melanoma is the phase 1b/2 trial NCT02791334, investigating a novel anti- PDL-1 antibody, lodapolimab (LY-33000054), in combination with abemaciclib across multiple tumor groups including melanoma [Citation95].
4. Biomarkers
The majority of early clinical studies conducted with ‘pan CDK’ inhibitors involved unstratified patient cohorts [Citation44]. The lack of effective biomarkers in these studies is likely to have contributed to the unsatisfactory results achieved in many of these early trials. Despite the negative results overall, there were promising signs of potential activity in some cancers, such as flavopiridol in chronic lymphocytic leukemia and rare ‘super responders’ in other studies, suggesting efficacy in some undefined molecular phenotypes [Citation44,Citation96]. In recent years, improved understanding about the biology of the cell cycle and the therapeutic success of CDK 4/6 inhibitors has facilitated further work in biomarker development. Preclinical work has identified several potential biomarkers which could be used to predict efficacy and resistance to CDK 4/6 inhibition, but require further clinical validation [Citation97]. There has been significant interest in the CDK4/6-Rb-p16INK4A pathway and associated transcription factors, such as E2F [Citation49,Citation98].
Preclinical studies have identified Rb as a key marker of CDK 4/6 resistance [Citation45,Citation97,Citation99], with the Rb1 gene being thought to be mutated in around 2.5% of melanoma cases [Citation100]. Initial phase 1 clinical trials of licensed CDK 4/6 inhibitors (e.g. NCT01536743, NCT02334527, NCT01976169) mandated Rb preservation by immunohistochemistry for inclusion in these multi-tumor studies. Lack of Rb activity has shown potential as a biomarker of resistance to CDK 4/6 inhibitors [Citation101]. Loss of Rb results in increased p16INK4A expression due to intrinsic negative feedback, which similarly could be used to predict CDK 4/6 inhibitor resistance [Citation44,Citation101]. The p16INK4A protein encoded by the CDKN2A locus is a potent tumor suppresser and mutated in >90% of metastatic melanoma tumors [Citation49]. Whilst the mechanism of action of p16INK4A is comparable to that of specific CDK 4/6 inhibitors such as palbociclib (blocking CDK 4/6 kinase activity via the ATP binding domain), it is likely that p16INK4A has a multitude of CDK 4/6 independent functions thus giving it a more complex role in cell cycle regulation [Citation102]. The potent function of p16INK4A in supressing melanoma progression likely explains why germline mutations in CDKN2A are strongly associated with an increased risk of developing the disease [Citation103]. Mutation and hypermethylation of CDKN2A are also being explored as potential biomarkers in melanoma and a plethora of other cancers. Of note recent studies have shown that mutations in CDKN2A and Rb are mutually exclusive, which may impact the individual use of these biomarkers in future clinical practice [Citation97].
Another potential biomarker of CDKi resistance that has been investigated is a result of the indirect action of CDK4/6i on the PMRT5-MDM4-p53 axis [Citation69]. Studies have suggested that the indirect suppression of PMRT5 by pablociclib is vital for subsequent p53 activation, and the loss of this action could be a measurable driver for Palbociclib resistance [Citation69]. In addition, work by Romano and colleagues [Citation104] has looked to assess the mechanism of resistance in one patient who initially responded but then progressed within the aforementioned phase 1b ribociclib/binimetinib combination trial. Whole exome sequencing of longitudinal biopsies identified an acquired PIK3CAE545K mutation. They have suggested that these specific resistance mutations may be identifiable pre-treatment to detect drug-resistant cohorts, although this remains to be further investigated.
A number of basket and umbrella trials using CDK 4/6i are underway that utilize genetic features of tumors to assign molecular therapies across multiple tumors types, including melanoma. The diagnostic-agnostic NCI-MATCH (NCT02465060) identifies tumors with cyclin D1 amplification for treatment with palbociclib. The SIGNATURE trial (NCT02187783) enrolled patients with tumors which display cyclin pathway abnormalities (e.g. CDK4, CDK6, CCND1, CCND3, P16INK4A) for treatment with ribociclib, this study has been completed but not yet reported. A further clinical trial (NCT02065063) has explored the efficacy and safety of palbociclib with trametinib (MEK inhibitor) in a range of cancers, including BRAFv600 melanoma with NRAS mutations. This investigated the effects on potential tumor biomarkers including pERK, Rb, Ki67, FoxM1, p16INK4A and CCDN1, however has not been reported.
Specifically, in melanoma several trials are underway which explore the use of CDK 4/6i with concomitant use of exploratory biomarkers. The LOGIC (NCT01820364) & LOGIC-2 (NCT02159066) trials utilizes an adaptive trial design which focuses on BRAFv600 mutant melanoma patients who have progressed on encorafenib and binimetinib. This drug combination is continued beyond progression with a third agent added based on tumor molecular phenotyping, including ribociclib, buparlisib (phosphoinositide-3-kinase inhibitor), infigratinib (fibroblast growth factor receptor inhibitor) or capmatinib (MET inhibitor). The LOGIC trial has been terminated due to scientific and business considerations, however LOGIC 2 is still recruiting at the date of censoring. A further clinical trial based on molecular profiling and matched targeted therapy, called MatchMel (NCT02645149) is underway for patients with melanoma and BRAF or NRAS mutations. This includes palbociclib as a potential therapy, in addition to other targeted therapies for patients with abnormalities in cyclin D1, cyclin D3 and p16INK4A.
Clinical trials exploring the use of CDK inhibitors and the predictive utility of effective biomarkers in a range of cancers, including melanoma are ongoing. However, it remains unclear if any single or combination of biomarker(s) will prove effective. In melanoma, the CDK4/6-Rb-p16INK4A pathway is the most promising candidate based on preclinical observations. Further exploratory clinical work in biomarker-enriched cohorts is necessary to validate potential predictive biomarkers against treatment response.
5. Conclusion
Despite the practice-changing influence of CDK 4/6 inhibitors in the field of breast cancer, these agents are yet to show significant benefit in malignant melanoma. Multiple trials have attempted to test their efficacy across phase 1 and 2 trials, from 1st generation pan-CDK inhibitors to highly specific CDK/BRAF/MEK inhibitor combinations. However, even with the lack of discernible progress to date, the high prevalence of CDK4/6-Rb-p16INK4A axis dysregulation in melanoma has fueled further hope that repurposing existing licensed CDK 4/6 inhibitors may prove an effective strategy. Several clinical trials incorporating biomarkers and different melanoma molecular subtypes are underway to explore this, and CDK inhibitors may still prove to be a useful targeted therapy for this aggressive malignancy.
6. Expert opinion
Melanoma is an often-aggressive disease affecting mostly younger patients. CDK inhibition within melanoma has been a significant focus of research. Transition from broad acting to specific CDK inhibitors has allowed us to target the G1-S transition with greater specificity, with excellent results seen thus far with CDK4/6i in breast cancer. There is hope that similar successes can be achieved across other solid tumors.
Trials of nonspecific CDKi have not been particularly successful in melanoma, with lack of efficacy or significant toxicity (in particularly when combined with chemotherapy) hindering further expansion into larger trials. There is evidence of some modest efficacy with single agent CDK4/6i [Citation85–87], though robust assessment within the context of randomized phase 3 trials is awaited. The results of the melanoma cohort treated with abemiciclib with brain metastases in NCT02308020 will be of particular interest given the predilection for CNS penetration in melanoma.
CDK4/6i have showed most promise thus far in combination therapy with BRAF/MEKi; however toxicity has been of significant concern with cutaneous rash, neutropenia and gastrointestinal disturbance of particular significance. The 1b/phase 2 trials of CDK/BRAFi or CDK/MEKi have displayed only modest signs of efficacy [Citation89–91] and therefore have not been taken forward into subsequent phase 3 trials. Despite this, positive responses seen in some BRAF/KRAS wild type patient groups gives some hope that CDK4/6i may be of future benefit in treating this aggressive subtype. Similarly, triple therapy BRAFi/MEKi and CDK4/6i has shown promising efficacy but has been unfortunately hindered by dose-limiting toxicities which has halted further development. Given the early phase nature of these trials and consequent absence of comparator arms, assessment of efficacy above and beyond BRAF/MEKi in BRAF mutant, treatment naive patients, has not been possible.
The novel combination of CDK4/6i and immunotherapy has some promise given the immune-modulatory effects of CDK4/6i [Citation85–87]. This may be of particularly use in IO refractory disease and provide further options within the melanoma treatment armamentarium for melanoma patients, particularly those with BRAF wild type disease whose treatment options are already limited to immunotherapy.
One common theme throughout CDKi combination trials is the need for dose reduction of one or more agents in order to preserve tolerability, in turn impacting on efficacy. The reduced encorafenib dose of 300 mg used in CDK inhibitor combination trials would be considered subtherapeutic in BRAF/MEKi therapy [Citation89], and the same can be said for the need to reduce ribociclib doses compared to that used in breast cancer [Citation91]. To our knowledge there has not been significant research into sequencing of CDK4/6i with BRAF/MEKi and whether introduction of CDK4/6i at the point of resistance could have a similar effect to that seen in breast cancer when overcoming endocrine resistance. Similarly, sequencing of immunotherapy and CDK4/6i is yet to be attempted, likely in part due to immaturity of data regarding this novel combination.
Whilst there are reasons for optimism with CDK4/6 inhibition in melanoma, robust assessment of clear efficacy above that demonstrated by BRAF/MEK inhibition or immunotherapy has not yet been seen. Further clinical trials are warranted, and data are awaited especially in combination with immunotherapy. In addition, further exploration of predictive and prognostic biomarkers to identify those who may benefit from treatment would permit greater tailoring of treatment to optimize benefit for patients. Genetic analysis and molecular profiling of those patients who develop resistance to conventional treatment could help identify those who may benefit from CDK4/6 inhibition, whilst also expanding our knowledge of a disease that despite significant treatment advances thus far, can still portend a poor prognosis.
Article highlights
With malignant melanoma still beholding a poor prognosis in a significant proportion of patients, the high prevalence of CDK4/6-Rb-p16INK4A pathway dysregulation means CDK inhibitors may have the potential to improve patient outcomes.
The development of CDK inhibitors has evolved from ‘pan-CDK’ inhibitors to highly specific agents.
A number of phase 1&2 clinical trials have investigated CDK inhibitors as single agents and in combination with BRAF and MEK inhibitors, but outcomes have been limited by dose-limiting toxicities.
CDK inhibitor use in biomarker lead personalized melanoma treatment is under investigation and could potentially result in therapeutic options for patients who fail to respond to currently licensed therapies.
Immunotherapy combinations are currently in development but are yet to be reported.
This box summarizes key points contained in the article.
Declaration of intereset
M Julve and M Lythgoe have both received educational grants from Bayer outside of this work. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Correction Statement
*MJ and JJC contributed equally to this work as first authors.This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015 Jan 1;372(1):30–39.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019 Oct 17;381(16):1535–1546.
- Cancer Genome Atlas N. Genomic classification of cutaneous melanoma. Cell. 2015 Jun 18;161(7):1681–1696.
- Colombino M, Capone M, Lissia A, et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol. 2012 Jul 10;30(20):2522–2529.
- Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017 Jul 1;28(7):1631–1639.
- Alberts BJA, Johnson A, Lewis J, et al. An overview of the cell cycle. Molecular biology of the cell. 4th ed. New York: Garland Science; 2002.
- Morgan DO. Principles of CDK regulation. Nature. 1995 Mar 9;374(6518):131–134.
- Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003 Jun;36(3):131–149.
- Nurse P, Masui Y, Hartwell L. Understanding the cell cycle. Nat Med. 1998 Oct;4(10):1103–1106.
- Nurse PM. Nobel Lecture. Cyclin dependent kinases and cell cycle control. Biosci Rep. 2002 Oct-Dec;22(5–6):487–499.
- Hunt T. Nobel lecture. Protein synthesis, proteolysis, and cell cycle transitions. Biosci Rep. 2002 Oct-Dec;22(5–6):465–486.
- Pines J. Cyclins: wheels within wheels. Cell Growth Differ. 1991 Jun;2(6):305–310.
- Kollmann K, Briand C, Bellutti F, et al. The interplay of CDK4 and CDK6 in melanoma. Oncotarget. 2019 Feb 15;10(14):1346–1359.
- Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998 Feb;18(2):753–761.
- Lees JA, Saito M, Vidal M, et al. The retinoblastoma protein binds to a family of E2F transcription factors. Mol Cell Biol. 1993 Dec;13(12):7813–7825.
- Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013 Aug;14(8):518–528.
- Polyak K, Kato JY, Solomon MJ, et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994 Jan;8(1):9–22.
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995 May 15;9(10):1149–1163.
- Reynisdóttir I, Polyak K, Iavarone A, et al. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. 1995 Aug 1;9(15):1831–1845.
- Ertosun MG, Hapil FZ, Osman Nidai O. E2F1 transcription factor and its impact on growth factor and cytokine signaling. Cytokine Growth Factor Rev. 2016 Oct;31:17–25.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005 Nov 17;353(20):2135–2147.
- Griewank KG, Scolyer RA, Thompson JF, et al. Genetic alterations and personalized medicine in melanoma: progress and future prospects. J Natl Cancer Inst. 2014 Feb;106(2):djt435.
- Walker GJ, Flores JF, Glendening JM, et al. Virtually 100% of melanoma cell lines harbor alterations at the DNA level within CDKN2A, CDKN2B, or one of their downstream targets. Genes Chromosomes Cancer. 1998 Jun;22(2):157–163.
- Sellers WR, Kaelin WG Jr. Role of the retinoblastoma protein in the pathogenesis of human cancer. J Clin Oncol. 1997 Nov;15(11):3301–3312.
- Zuo L, Weger J, Yang Q, et al. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet. 1996 Jan;12(1):97–99.
- Sotillo R, Garcia JF, Ortega S, et al. Invasive melanoma in Cdk4-targeted mice. Proc Natl Acad Sci U S A. 2001 Nov 6;98(23):13312–13317.
- Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. 2016 Apr;45:129–138.
- Fountain JW, Karayiorgou M, Ernstoff MS, et al. Homozygous deletions within human chromosome band 9p21 in melanoma. Proc Natl Acad Sci U S A. 1992 Nov 1;89(21):10557–10561.
- Nathanson KL, Martin AM, Wubbenhorst B, et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res. 2013 Sep 1;19(17):4868–4878.
- Karim RZ, Li W, Sanki A, et al. Reduced p16 and increased cyclin D1 and pRb expression are correlated with progression in cutaneous melanocytic tumors. Int J Surg Pathol. 2009 Oct;17(5):361–367.
- Sanki A, Li W, Colman M, et al. Reduced expression of p16 and p27 is correlated with tumour progression in cutaneous melanoma. Pathology. 2007 Dec;39(6):551–557.
- Eliason MJ, Larson AA, Florell SR, et al. Population-based prevalence of CDKN2A mutations in Utah melanoma families. J Invest Dermatol. 2006 Mar;126(3):660–666.
- Goldstein AM, Chan M, Harland M, et al. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet. 2007 Feb;44(2):99–106.
- Mahmoud F, Shields B, Makhoul I, et al. Role of EZH2 histone methyltrasferase in melanoma progression and metastasis. Cancer Biol Ther. 2016 Jun 2;17(6):579–591.
- Wu A, Wu B, Guo J, et al. Elevated expression of CDK4 in lung cancer. J Transl Med. 2011 Apr 11;9:38.
- Wagner V, Gil J. Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene. 2020 Jul;39(29):5165–5176.
- Blagosklonny MV. Geroconversion: irreversible step to cellular senescence. Cell Cycle. 2014;13(23):3628–3635.
- Coppé JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008 Dec 2;6(12):2853–2868.
- Zou X, Ray D, Aziyu A, et al. Cdk4 disruption renders primary mouse cells resistant to oncogenic transformation, leading to Arf/p53-independent senescence. Genes Dev. 2002 Nov 15;16(22):2923–2934.
- Yoshida A, Lee EK, Diehl JA. Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended inhibition of CDK4/6. Cancer Res. 2016 May 15;76(10):2990–3002.
- Damsky W, Micevic G, Meeth K, et al. mTORC1 activation blocks BrafV600E-induced growth arrest but is insufficient for melanoma formation. Cancer Cell. 2015 Jan 12;27(1):41–56.
- Coppé JP, Rodier F, Patil CK, et al. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011 Oct 21;286(42):36396–36403.
- Guan X, LaPak KM, Hennessey RC, et al. Stromal senescence by prolonged CDK4/6 INHIBITION POTENTIATES TUMOR GROWTH. Mol Cancer Res. 2017 Mar;15(3):237–249.
- Asghar U, Witkiewicz AK, Turner NC, et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015 Feb;14(2):130–146.
- Xu W, McArthur G. Cell cycle regulation and melanoma. Curr Oncol Rep. 2016 Jun;18(6):34.
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009 Mar;9(3):153–166.
- Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001 Dec;1(3):222–231.
- Dhillon S. Palbociclib: first global approval. Drugs. 2015 Apr;75(5):543–551.
- Lee B, McArthur GA. CDK4 inhibitors an emerging strategy for the treatment of melanoma. Melanoma Manag. 2015 Aug;2(3):255–266.
- Chohan TA, Qayyum A, Rehman K, et al. An insight into the emerging role of cyclin-dependent kinase inhibitors as potential therapeutic agents for the treatment of advanced cancers. Biomed Pharmacother. 2018;107:1326–1341.
- Whittaker SR, Mallinger A, Workman P, et al. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther. 2017;173:83–105.
- Sedlacek H, Czech J, Naik R, et al. Flavopiridol (L86 8275; NSC 649890), a new kinase inhibitor for tumor therapy. Int J Oncol. 1996 Dec;9(6):1143–1168.
- Joshi KS, Rathos MJ, Joshi RD, et al. In vitro antitumor properties of a novel cyclin-dependent kinase inhibitor, P276-00. Mol Cancer Ther. 2007 Mar;6(3):918–925.
- Carlson BA, Dubay MM, Sausville EA, et al. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res. 1996 Jul 1;56(13):2973–2978.
- Blum KA, Ruppert AS, Woyach JA, et al. Risk factors for tumor lysis syndrome in patients with chronic lymphocytic leukemia treated with the cyclin-dependent kinase inhibitor, flavopiridol. Leukemia. 2011 Sep;25(9):1444–1451.
- Parry D, Guzi T, Shanahan F, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010 Aug;9(8):2344–2353.
- Stephenson JJ, Nemunaitis J, Joy AA, et al. Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer. 2014 Feb;83(2):219–223.
- Efficacy study of P276-00 in subjects of malignant melanoma positive for cyclin D1 expression. [cited 2020 May 31]. Available from: https://ClinicalTrials.gov/show/NCT00835419.
- AT7519M in treating patients with advanced or metastatic solid tumors or refractory non-Hodgkin’s Lymphoma. [cited 2020 May 31]. Available from: https://ClinicalTrials.gov/show/NCT00390117.
- Phase Ii trial of the cyclin-depedent kinase inhibitor Pd 0332991 in patients with cancer. [cited 2020 May 31]. Available from: https://ClinicalTrials.gov/show/NCT01037790.
- A study of LY2835219 in Japanese participants with advanced cancer. [cited 2020 May 31]. Available from: https://ClinicalTrials.gov/show/NCT02014129.
- Mahoney E, Byrd JC, Johnson AJ. Autophagy and ER stress play an essential role in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Autophagy. 2013 Mar;9(3):434–435.
- Koboldt D, Fulton R, McLellan M, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012 Oct 4;490(7418):61–70.
- Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from Discovery to Therapy. Cancer Discov. 2016 Apr;6(4):353–367.
- O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016 Jul;13(7):417–430.
- Goel S, DeCristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017 Aug 24;548(7668):471–475.
- Yadav V, Burke TF, Huber L, et al. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol Cancer Ther. 2014 Oct;13(10):2253–2263.
- Yoshida A, Diehl JA. CDK4/6 inhibitor: from quiescence to senescence. Oncoscience. 2015;2(11):896–897.
- AbuHammad S, Cullinane C, Martin C, et al. Regulation of PRMT5-MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. Proc Natl Acad Sci U S A. 2019 Sep 3;116(36):17990–18000.
- Syed YY. Ribociclib: first Global Approval. Drugs. 2017 May;77(7):799–807.
- Kim ES. Abemaciclib: first global approval. Drugs. 2017 Dec;77(18):2063–2070.
- Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004 Nov;3(11):1427–1438.
- Tripathy D, Bardia A, Sellers WR. Ribociclib (LEE011): mechanism of action and clinical impact of this selective cyclin-dependent kinase 4/6 inhibitor in various solid tumors. Clin Cancer Res. 2017 Jul 1;23(13):3251–3262.
- Gelbert LM, Cai S, Lin X, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs. 2014 Oct;32(5):825–837.
- Wang LM, Ren DM. Flavopiridol, the first cyclin-dependent kinase inhibitor: recent advances in combination chemotherapy. Mini Rev Med Chem. 2010;Oct;10(11):1058–1070.
- Burdette-Radoux S, Tozer RG, Lohmann RC, et al. Phase II trial of flavopiridol, a cyclin dependent kinase inhibitor, in untreated metastatic malignant melanoma. Invest New Drugs. 2004 Aug;22(3):315–322.
- Bible KC, Lensing JL, Nelson SA, et al. Phase 1 trial of flavopiridol combined with cisplatin or carboplatin in patients with advanced malignancies with the assessment of pharmacokinetic and pharmacodynamic end points. Clin Cancer Res. 2005;11(16):5935.
- Bahleda R, Grilley-Olson JE, Govindan R, et al. Phase I dose-escalation studies of roniciclib, a pan-cyclin-dependent kinase inhibitor, in advanced malignancies. Br J Cancer. 2017 Jun;116(12):1505–1512.
- Nemunaitis JJ, Small KA, Kirschmeier P, et al. A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J Transl Med. 2013;11:259.
- Dinaciclib in treating patients with stage III-IV melanoma. [cited 2020 May 31]. Available from: https://ClinicalTrials.gov/show/NCT01026324.
- Lao CD, Moon J, Fruehauf JP, et al. SWOG S0826: A phase II trial of SCH 727965 (NSC 747135) in patients with stage IV melanoma. J clin oncol. 2012 May 20;30(15_suppl):8521.
- Gupta S, Jain MM, Maru A, et al. A phase I study of selective cyclin dependent kinase inhibitor P1446A-05 administered on an intermittent schedule in patients with advanced refractory tumors. J clin oncol. 2012 May 20;30(15_suppl):3011.
- Study of an oral cdk inhibitor administered with an oral BRAF inhibitor in patients with advanced or inoperable malignant melanoma with BRAF mutation. [cited 2020 May 31]. Available from: https://ClinicalTrials.gov/show/NCT01841463.
- Flaherty KT, Lorusso PM, Demichele A, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012 Jan;18(2):568–576.
- Infante JR, Cassier PA, Gerecitano JF, et al. A phase I study of the cyclin-dependent kinase 4/6 inhibitor ribociclib (LEE011) in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2016 Dec;22(23):5696–5705.
- Nguyen LV, Searle K, Jerzak KJ. Central nervous system-specific efficacy of CDK4/6 inhibitors in randomized controlled trials for metastatic breast cancer. Oncotarget. 2019 Oct;10(59):6317–6322.
- Shapiro G, Rosen LS, Tolcher AW, et al. A first-in-human phase I study of the CDK4/6 inhibitor, LY2835219, for patients with advanced cancer. J clin oncol. 2013 May 20;31(15_suppl):2500.
- Tolaney SM, Sahebjam S, Le Rhun E, et al. Abstract P1-19-01: A phase 2 study of abemaciclib in patients with leptomeningeal metastases secondary to HR+, HER2- breast cancer. Cancer Res. 2019;79(4Supplement):P1-19-01.
- Taylor M, Sosman J, Gonzalez R, et al. Phase Ib/Ii study of Lee011 (Cdk4/6 inhibitor) and Lgx818 (Braf inhibitor) in Braf-mutant melanoma. Ann Oncol. 2014;25:Iv374.
- Sullivan RJ, Amaria RN, Lawrence DP, et al. Abstract PR06: phase 1b dose-escalation study of trametinib (MEKi) plus palbociclib (CDK4/6i) in patients with advanced solid tumors. Mol Cancer Ther. 2015;14(12 Supplement 2):PR06.
- Schuler MH, Ascierto PA, FYFL DV, et al. Phase 1b/2 trial of ribociclib+binimetinib in metastatic NRAS-mutant melanoma: safety, efficacy, and recommended phase 2 dose (RP2D). J clin oncol. 2017 May 20;35(15_suppl):9519.
- Ascierto PA, Bechter O, Wolter P, et al. A phase Ib/II dose-escalation study evaluating triple combination therapy with a BRAF (encorafenib), MEK (binimetinib), and CDK 4/6 (ribociclib) inhibitor in patients (Pts) with BRAF V600-mutant solid tumors and melanoma. J clin oncol. 2017 May 20;35(15_suppl):9518.
- Teh JLF, Aplin AE. Arrested developments: CDK4/6 inhibitor resistance and alterations in the tumor immune microenvironment. Clin Cancer Res. 2019 Feb 1; 25(3):921–927.
- Schaer DA, Beckmann RP, Dempsey JA, et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. 2018 Mar 13;22(11):2978–2994.
- A study of anti-PD-L1 checkpoint antibody (LY3300054) alone and in combination in participants with advanced refractory solid tumors. [cited 2020 May 31]. Available from: https://ClinicalTrials.gov/show/NCT02791334.
- Byrd JC, Lin TS, Dalton JT, et al. Flavopiridol administered using a pharmacologically derived schedule is associated with marked clinical efficacy in refractory, genetically high-risk chronic lymphocytic leukemia. Blood. 2007 Jan 15;109(2):399–404.
- Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer. 2017 Jan;3(1):39–55.
- Bruyère C, Meijer L. Targeting cyclin-dependent kinases in anti-neoplastic therapy. Curr Opin Cell Biol. 2013 Dec;25(6):772–779.
- Dean JL, Thangavel C, McClendon AK, et al. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010 Jul 15;29(28):4018–4032.
- Biomarkers/Rb1: my cancer genome; 2017. [cited 2020 Sep 17]. Available from: https://www.mycancergenome.org/content/gene/rb1/
- Young RJ, Waldeck K, Martin C, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014 Jul;27(4):590–600.
- LaPak KM, Burd CE. The molecular balancing act of p16(INK4a) in cancer and aging. Mol Cancer Res. 2014 Feb;12(2):167–183.
- Helgadottir H, Höiom V, Tuominen R, et al. Germline CDKN2A mutation status and survival in familial melanoma cases. J Natl Cancer Inst. 2016 Nov;108(11). DOI: 10.1093/jnci/djw135.
- Romano G, Chen P-L, Song P, et al. A preexisting rare PIK3CA E545K subpopulation confers clinical resistance to MEK plus CDK4/6 inhibition in NRAS melanoma and is dependent on S6K1 signaling. Cancer Discov. 2018;8(5):556.