
ABSTRACT
Introduction
Autism, like other neurodevelopmental disorders (NDDs), has a strong association with epilepsy. There are known common genetic pathways in both autism and epilepsy. There are also specific genetic syndromes associated with both complex epilepsy and the autism phenotype.
Areas covered
This review explores the evidence for common genetic etiologies and pathophysiological pathways in relation to both epilepsy and autism. Autism with comorbid epilepsy are associated with a high prevalence of medical and psychiatric comorbidities. This paper discusses how this influences assessment, treatment, and outcomes. The evidence for the treatment of specific seizure types in the context of NDDs is also examined alongside clinical commentary.
Expert opinion
Despite the strong association, there is a limited evidence base to support the efficacy and tolerability of anti-seizure medications specifically in autism, with no Level 1 evidence or National Guidance available. Autism and epilepsy should be approached under a NDD model with cautious introduction and titration of anti-seizure medication. Alongside this, there is evidence to support a move toward precision medicine in specific genetic syndromes such as Tuberous Sclerosis Complex and other genetic seizure disorders. The first-line treatments that should be considered for focal seizures include carbamazepine, lamotrigine, and levetiracetam.
1. Introduction
Epilepsy, in particular, the genetic epilepsies and epileptic encephalopathies have a strong positive association with the family of neurodevelopmental disorders (NDDs) including autism and intellectual disability (ID) [Citation1]. The evidence suggests a common etiology, and that epilepsy in the context of NDDs should be considered as part of a complex conundrum and not approached as a discrete condition [Citation2]. This review will explore the NDD approach to pharmacological therapy for people with epilepsy and autism and , examine the emerging evidence base for precision medicine, where treatment can be targeted based upon an individual’s genomic variation. The main focus of this review will be a clinical guide to successful treatment using a neurodevelopmental approach.
1.1. Epilepsy
A seizure is a transient presentation of neurological symptoms resulting from abnormal or excessive neuronal activity. The origin of the neuronal activity will influence the semiology of a seizure and may include sensory, motor, emotional, or behavioral changes [Citation3]. A seizure may be provoked by a broad range of physiological insults including fever, sleep deprivation, hypoglycemia, hypernatremia, hypocalcemia, toxic substances. However, a provoked seizure in context does not meet the clinical criteria for a diagnosis of epilepsy. Epilepsy is conceptually a disorder where the brain has a predisposition to further seizure activity. The practical definition of epilepsy defined by the International League Against Epilepsy (ILAE) Task Force includes the clinical situations in which there is a high probability of seizure recurrence after just one unprovoked or reflex seizure (>60%) [Citation4].
The ILAE classification system of the epilepsies is designed to help clarify communication between clinicians and improve diagnostic accuracy for research questions. The multilevel system considers seizure type, followed by epilepsy type, and finally epilepsy syndrome [Citation5]. Seizure type is categorized by nature of onset-focal, generalized, or unknown. Within each category a seizure should be described based upon the semiological features considering motor manifestations (tonic, tonic-clonic, atonic, myoclonic, other), non-motor manifestations (e.g. sensory), and impact upon a person’s awareness during an event.
1.2. Autism
Autism is diagnostically classified under the umbrella of NDDs. An NDD is a condition that emerges during childhood with deficits in a number of core domains including language development, cognition, reciprocal social skills, and motor function. With improvements in service provision, autism is now commonly diagnosed before the age of 3 years but can be diagnosed at any age including in adults where the symptoms were missed during their development due to lower recognition levels. Autism is a conceptual diagnosis based upon social constructs of a constellation of symptoms, informed by subjective experiences and objective behavioral observations. As a result, the assessment process is usually performed by specialist services and can be a complex. There are numerous validated diagnostic assessment tools including observational models, each of which requires specific training to implement.
The working diagnostic criteria for autism have broadened over time, but the core features of impaired communication, inhibited social interaction, and restrictive repetitive behaviors remain consistent. The Diagnostic and Statistical Manual of Mental Disorders fifth edition (DSM5) apply a spectrum approach to diagnosis and those previously meeting the criteria for Asperger’s or pervasive developmental disorder – not otherwise specified, fulfilling the criteria for autism spectrum disorder [Citation6].
1.3. Epidemiology
A systematic review and meta-analysis of international studies found the point prevalence of active epilepsy overall was 6.38 per 1,000 persons (95% confidence interval [95% CI: 5.57–7.30] [Citation7]. The Center for Disease Control and Prevention (CDC) in the United States (US) reports the prevalence of autism as being 1 in 54 children by 2016 [Citation8]. However, the reason for this remains under scrutiny due to methodological challenges including a lack of consistency in case ascertainment. A systematic review of the co-occurrence of epilepsy and autism identified 74 studies (283,549 cases) and found the median period prevalence of epilepsy in people with autism was 121 per 1000. The median point prevalence of autism in people in epilepsy was 90 per 1000 [Citation9].
Seizures may also present before a diagnosis of autism particularly in association with genetic epilepsy syndromes. The presence of seizures in early life (less than 1 year) particularly infantile spams significantly increases the likelihood of a future autism diagnosis [Citation10]. A systematic review and meta-analysis investigating the prevalence and risk factors for autism in epilepsy identified a pooled prevalence of 63 per 1000 [Citation11]. Studies included with a majority ID population (>50%) were almost five times more likely to be diagnosed with autism compared to those studies with a minority ID population. A population-based study investigating the incidence of epilepsy found 23.6 cases per 1000 person years (95% CI: 21.3, 26.2) in adults with autism with ID, 7.7 cases per 1000 person years (95% CI: 6.6, 8.9) in adults with autism without intellectual disability, and 15.9 incident cases per 1000 person years (95% CI: 15.2, 16.7) in adults with ID alone. This finding is supported by results from a pooled data analysis (n = 2112) finding the point prevalence of epilepsy in ASD with comorbid ID as 214 per 1000, compared to 80 per 1000 in those with autism without ID [Citation12].
1.4. Electroencephalogram changes and seizure types
In a retrospective cohort review, 33 of 130 people with autism developed seizures in adolescence [Citation13]. Over two-thirds of those who developed seizures had epileptiform changes on electroencephalogram (EEG) before the first clinical seizure. The most common seizure type observed in the group were focal seizures with impaired awareness. The onset of epilepsy in people with autism has two distinct peaks, in early childhood and in adolescence [Citation14].
There is a growing body of evidence showing that subclinical EEG changes are more common in seizure-free people with autism compared to seizure-free controls without autism [Citation15]. Epileptiform EEG changes are more common than non-epileptiform abnormalities in autism and are more common in association with intellectual impairment and behavioral challenges. These epileptiform changes include a wide range of generalized and focal or multifocal discharges in different regions of the brain. However, discharges in the temporal region (right/left/bilateral) are the most commonly observed [Citation1,Citation16]. These findings postulate a theory of pathogenesis in autism etiology, which at present remains unproven, although there is some evidence that treatment before seizure onset can improve a range of behavioral symptoms [Citation16]. This is an evolving evidence base requiring clearer robust data before any firm recommendations can be made for clinical application.
1.5. Common genetic pathways
There is a high rate of de novo mutations and copy number variants (CNVs) associated with developmental disabilities including autism, epilepsy, and ID. There is developing understanding of the impact upon phenotype of the genomic region affected and the subsequent functional change, however this remains a highly heterogeneous population. For example, many of the known developmental and epileptic encephalopathies () have a wide phenotypic spectrum. This arises from dozens of known de novo mutations (duplications, deletions, rearrangements) associated with gene disorders, which all inform the spectrum of clinical presentations [Citation17]. The single-gene disorders and CNVs with a high frequency of co-occurring autism and epilepsy demonstrate common biological pathways from gene transcription to synaptic function [Citation18,Citation19]. A retrospective review of children with autism and epilepsy (n = 88) identified pathogenic or likely pathogenic variants in over half of the sample. The most common gene mutations identified were SCN1A (14.9%) and MECP2 (10.6%) [Citation20].
Table 1. Genomic variants associated with autism phenotype and epilepsy [Citation18,Citation19,Citation36–45,Citation64,Citation65,Citation68–72]
In the future, we will identify more candidate genes strengthening the possibility of a shared genetic etiology and neuropathological processes for specific epilepsy syndromes and autism. By grouping specific genetic mutations with similar functional effects known to impact upon epilepsy (ion transmembrane transport) and autism (synaptic signaling), it has been possible to identify common genes associated with both profiles. Peng et al. extrapolated 205 known genes associated with both epilepsy and autism. These findings can be used to identify new candidate genes for epilepsy and autism [Citation21]. Studies in mouse models indicate syndromes characterized by both epilepsy and autism likely have multiple genomic variations that influence the excitatory and inhibitory balance at synapses during the neurodevelopmental period [Citation22]. This suggests that as our understanding of these neurodevelopmental phenotypes improves, we will be in a position to tailor therapeutic intervention [Citation23].
2. Complexity
2.1. Comorbidities
A systematic review and meta-analysis explored the prevalence of co-occurring neurological disorders in autism. Aside from epilepsy, people with autism were significantly more likely to have macrocephaly, hydrocephalus, cerebral palsy, migraine, and neurological congenital abnormalities [Citation24]. A large population study (n = 6649) in Scotland based on census data including 6649 people with autism over the age of 25 years found high levels of comorbid medical and psychiatric conditions with significantly higher risk than in controls without autism [Citation25]. These findings are consistent with a United States population-based study including 1507 people with autism [Citation26]. Insight into the prevalence of multiple morbidities in autism emphasizes the complexity of assessment and treatment in this population, often impacted by diagnostic overshadowing. For example, the point prevalence of any physical disability in people with autism has been observed at 29.4%, odds ratio of 94.5 (95% CI: 89.4–100.0) compared to controls without autism. The point prevalence of any mental illness has been observed at 33.0%, odds ratio 8.6 (95% CI: 8.2–9.0) compared to controls [Citation25].
2.2. Assessment
The first stage in management is to know when to start treatment, which first requires a clear rational and accurate diagnosis. Unfortunately, due to the complexity of the clinical picture, misdiagnosis is not uncommon [Citation27]. The presence of an ID, its severity, and the degree of communication impairment will influence the accuracy of assessment by limiting the understanding of subjective experiences. As discussed people with autism have high rates of comorbid psychiatric illness, behavioral challenges, and repetitive stereotyped movements. The most common paroxysmal events in autistic children are staring events that are often mistaken for seizure activity [Citation28]. Neuropsychological testing has consistently found in 25–35% of cases, there is no EEG correlation with repetitive movements that had previously been diagnosed as epileptic seizures [Citation28,Citation29].
Generalized onset, bilateral tonic clonic motor seizures are stereotyped and readily recognizable. However, more commonly observed in autism is focal seizures with impaired awareness. These events may be difficult to identify if there is minimal motor involvement. Where there is motor involvement, it can be difficult to distinguish between epileptic events and non-epileptic stereotyped movements or tics. The presentation of a focal seizure in association with neurodevelopmental disorders can be complex and difficult to distinguish from behavioral changes, particularly with adolescent onset where focal seizures may be associated with pre-ictal or post-ictal aggression or complex motor movements. The fundamental principle is therefore to have a high level of suspicion for seizure activity based upon the epidemiologic population data indicating the significantly higher prevalence rates. In such scenarios it is important to pursue specialist assessment, diagnosis, and subsequently treatment as would be done if considering an alternative diagnosis such as mental illness.
The assessment must be informed by a reliable collateral witness, and the use of home video recordings has real clinical utility. Any video recording will require appropriate consent or a multi-disciplinary best interest decision before proceeding. The video should only be attempted when safe to do so and it is most useful to attempt to capture the entire event including the beginning (including pre-ictal/aura phase) the event or ictal phase itself, and the post-ictal recovery period. Following the first event, it is appropriate to consider neurophysiological testing (EEG) and brain imaging (MRI) to rule out any structural abnormality [Citation30]. The gold standard investigation is video telemetry (preferably overnight), but this is not readily available outside of specialist centers. As we have discussed, the use of standard EEG may not always be helpful as even a finding of epileptiform activity is not specific to epilepsy when associated with autism. In addition, such investigations require a reasonable degree of co-operation, and in-depth consideration may be required to enhance the chances of success.
Considering the complexity, people with autism in general but especially those with intellectual disability and neuropsychiatric comorbidities would perhaps benefit most from detailed assessment and thorough investigation. However, it is these complexities that also make routine investigation a significant challenge to the person, clinicians, and services. Those who are most vulnerable, with the highest level of need, face the biggest challenges in accessing appropriate services. People with epilepsy and developmental disabilities receive fragmented care provision with far longer waits for investigation such as MRI, which in most cases may require a general anaesthetic [Citation31].
2.3. Treatment choice
The evidence base for pharmacological treatment currently available consists of observational studies with limited participants making any inferences limited [Citation32]. Many larger investigations will inherently include a population of people with autism, but they are not usually defined. Investigations with treatment-resistant populations in particular will include a high proportion of people with ID and autism with a range of epilepsy syndromes. Data from high quality, appropriately powered randomized, double-blind, placebo-controlled trials should be extrapolated to help inform decision-making, but it is important to recognize that generalizability must be considered with caution.
There is no robust evidence to support a clear positive outcome with the treatment of epileptiform EEG changes in autism, even when associated with regression. There may be tangential benefits from anti-seizure medication with a positive neuropsychiatric profile such as the impact upon affective lability, aggression, and other behavioral challenges [Citation33]. The autistic phenotype can be present in a number of epileptic encephalopathies, and draws particular comparison with Laundau–Kleffner syndrome. Therefore, the key to treatment success is accurate diagnosis, and identifying the specific epilepsy syndrome will provide valuable information into the most appropriate treatment choice. The ILAE classification of seizure’s and seizure syndromes provides the framework to epilepsy management including anti-seizure drug choice [Citation5].
The evidence base for the initial treatment of seizures, including focal seizures in childhood is detailed in the updated ILAE evidence review for anti-seizure medicines as initial monotherapy and the Cochrane Review and network meta-analysis of anti-seizure drug monotherapy () [Citation34,Citation35]. It should be noted that this evidence applies to the treatment of the described seizure type for any individual and is not autism specific. Having an understating of the most appropriate anti-seizure drugs to consider by seizure type should inform treatment choice. However, as we have discussed, autistic people are a complex and heterogeneous population. Clinical response may not necessarily align with the evidence base for treatment of focal seizures arising from a different etiology. For a more detailed discussion on the approach to treatment in epilepsy, consider the Expert Opinion section of this review.
Table 2. Treatment choice based on evidence and clinical opinion [Citation34,Citation35,Citation46]
Epileptic encephalopathies in association with the autism phenotype are a more robustly researched area and treatment should be aligned to epilepsy syndrome. There is an evidence base to support treatment pathways and ASM classification choice when clinical epilepsy syndromes are identified. First-line, adjunctive treatment options, and ASMs to avoid for some of the epileptic encephalopathies do have a limited evidence base, but with limited success and high levels of treatment resistance.
Lennox–Gastaut Syndrome (LGS): Sodium Valproate may be considered 1st line treatment, with Lamotrigine, clobazam, corticosteroids, and Levetiracetam as adjunctive ASMs.
Continuous Spikes and Wave during Sleep (CSWS): Corticosteroids and clobazam may be considered first line with sodium valproate, ethosuximide, and sulthiame as adjunctive.
Landau–Kleffner Syndrome (LKS): Corticosteroids may be considered first line, with valproate, clobazam, and sulthiame as adjunctive [Citation30,Citation47,Citation48].
However, we are now increasingly identifying genetic etiology in association with epileptic encephalopathies and developmental and epilepsy encephalopathies; therefore, more specific targeted treatment choices can be made. The ASM of choice can be narrowed by the mechanism of action when we understand the functional impact of the genetic variant. This will lead to the development of novel treatment options tailored to protein change, e.g. the impact on the function of certain voltage gated sodium channels in Dravet syndrome. Furthermore, the future of treatment is likely to shift toward adopting a gene therapy approach [Citation49].
2.3.1. Commonly prescribed anti-seizure medicines
A systematic review of randomized, double-blind, placebo-controlled evidence suggest that valproate (VPA) containing medicines may have a beneficial impact upon neuropsychiatric symptoms associated with autism including irritability, aggression, and repetitive behaviors [Citation32]. However, this improvement is not always observed and this is likely related to inherent weaknesses in study design including low number of participants. The most likely contributor is the heterogeneity of the autism population, which will influence response to psychotropic medication. VPA is associated with a range of potential side effects from weight gain to encephalopathy. However, in general it is well tolerated and efficacious for a wide range of seizure types and epilepsy syndromes. VPA should be avoided for women or girls of childbearing age because of its teratogenicity [Citation50]. There is evidence from observational studies that lamotrigine may improve cognition and neuropsychiatric symptoms of autism. However, this change is likely related to the anti-seizure effect even if there is no tangible seizure reduction. In a randomized, double-blind, placebo control setting, any impact upon behavioral symptoms was equivocal [Citation51]. Like valproate, lamotrigine has mood-stabilizing properties, and the limited data suggest that the impact upon cognitive and behavioral domains is neutral at worst.
2.3.2. Cannabidiol (CBD) – lack of evidence to support efficacy
The evidence base for specialist prescriptions including the range of cannabinoids has not yet developed to the threshold of demand. There is reasonable evidence to support the use of cannabidiol (CBD)-based medicines (Epidiolex) in the treatment of refractory epilepsy in Lennox–Gastaut syndrome and Dravet syndrome. There is also evidence to support consideration of adjunctive use in other genetic epileptic encephalopathies such as CDKL5 deficiency disorder, Doose syndrome, and TSC from open-label observational studies and post -hoc analysis of phase 3 trials [Citation52,Citation53]. There is also now emerging evidence for positive long-term safety outcomes in Lennox–Gastaut and Dravet syndromes [Citation54,Citation55].
However, there is still limited evidence to support the use of CBD or other cannabinoids for autism and associated neuropsychiatric symptoms. A recent scoping review suggests that there are observational data from small cohorts demonstrating a positive impact upon a number of behavioral domains including repetitive behaviors, aggression, sleep disturbance, cognition, and mood dysregulation [Citation56,Citation57]. The use of medicinal cannabis was also associated with a reduction in other concomitant psychotropic prescriptions. However, the level of adverse events observed was significant. At present, there is a lack of high quality, long-term, well-powered investigations to evaluate benefit and any potential impact upon neurodevelopment.
2.3.3. Drug profile
When considering which anti-seizure medication to choose, the adverse effect profile of the drug should be considered against the patient profile () [Citation73]. Some of the anti-seizure medications discussed have a negative cognitive profile (phenobarbital, phenytoin, valproate, carbamazepine {therapeutic windows}, topiramate, zonisamide). Whereas there is some evidence to support the positive role of lamotrigine and levetiracetam on cognitive function [Citation73]. The positive mood-stabilizing effect of some anti-seizure medications (valproate, lamotrigine, and carbamazepine) supports their utility for the treatment of major mood disorders such as recurrent depressive disorder and bipolar disorder, as well as other neuropsychiatric presentations. Conversely, other anti-seizure medications have a recognized negative behavioral profile (phenytoin, phenobarbital, zonisamide, levetiracetam, perampanel) which should be considered where these symptoms are prominent or there is comorbid or history of mental illness or significant distress behavior [Citation73]. Other anti-seizure medication options have little to no evidence for guidance and these considerations need to be observed in this context. For example, contradictory effects can be observed when comparing one observational study to another, such as the positive neuropsychiatric impact of perampanel observed in a Japanese cohort of people with autism with refractory epilepsy as compared to studies in the UK or Europe [Citation58–61].
Table 3. Neuropsychiatric profile of commonly prescribed anti-seizure medications based upon clinical opinion
As stipulated, the drug profile should be considered in context of the patient profile including the current pharmacological load (particularly anticholinergic burden as people age). Increased caution is advised when multiple anti-seizure medications are prescribed, particularly if they have a similar neuropsychiatric profile as the clinical effect may be more than cumulative.
2.3.3.1. Drug Interactions
We know that people with autism with epilepsy have more physical and psychiatric comorbidities. This leads to higher rates of polypharmacy increasing the risk of drug-to-drug interactions and a higher drug burden. There are drug-to-drug interactions between many of the ASM when used in combination. A useful example is the common synergistic paring of valproate and lamotrigine, when used in combination serum lamotrigine levels increase significantly. Before prescribing, it is important to consider the impact upon other existing medications. Many of the older and some newer ASMs are hepatic enzyme inducers. Enzyme inducing ASM like the potent carbamazepine, phenytoin, topiramate, rufinamide, and many more will reduce the effectiveness of the combined Oral Contraceptive Pill (OCP) for example [Citation62].
2.3.3.2. Drug and galenic formulation
In general, there is no or limited research in the understanding of how autism influences response to maximum tolerable doses, impact dose selection and potential drug hazards. There is a lack of safety markers specific to this population.
Another essential but often overlooked consideration is the formulation of the medicine prescribed. It is not unusual for people with autism to have particular sensory needs that will impact what medicines they can tolerate (including taste, texture, size, look, smell). Understanding a person’s needs in this area may indicate the requirement of a particular formulation (tablet, oral solution, granules). If this is not considered there is a risk of non-concordance with pharmacological treatment. Furthermore, people with NDDs have higher rates of swallowing problems [Citation63]. This again may lead to the requirement of particular formulations and even the administration via gastronomy tube. All of these factors should be considered closely when making prescribing choices including possible long-term outcomes. It is sensible to avoid switching between formulations and manufacturing brands without necessity. The Medicines and Healthcare products Regulatory Agency (MHRA) categorizes ASM based on the likely effect changing ASM formulation and/or branding will have upon bioavailability. For any ASM to be effective, it relies on good concordance and this is influenced by a range of factors that should be considered at every review and prescribing decision [Citation62].
2.4. Progress toward precision therapy
We are moving away from symptom-based pharmacological treatments and toward medicines specifically targeted at genetic disorders. At present, this precision-based medicine approach is a specialist intervention for those who do not readily respond to conventional ASM. However, as the evidence base improves and further tailored treatment options are developed, this will likely become the mainstay of treatment for genetic epilepsies before progression to gene therapy is achieved.
2.4.1. Tuberous sclerosis complex
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder caused by mutations in the TSC1 and TSC2 genes. TSC affects multiple systems and is associated with the early onset of polymorphic seizures that are more often than not treatment resistant. Early onset seizures is also associated with autism and developmental disabilities [Citation64]. The functional effect of the TSC mutations is over activation of mammalian target of rapamycin (mTOR) protein complex [Citation65]. A phase 3, randomized, double-blind, placebo-controlled study has demonstrated the efficacy and tolerability of adjunctive use of an mTOR Inhibitor (Everolimus) for treatment-resistant focal-onset seizures in TSC [Citation66,Citation67].
2.4.2. SCN2A-related disorders
The severe pathological end of the SCN2A phenotype spectrum is associated with encephalopathy and a range of distinct epilepsy syndromes [Citation68]. SCN2A mutations have also been associated with the etiology of autism and ID without epilepsy [Citation69]. The SCN2A gene encodes for one of the most common voltage-gated sodium channels and variances in the functional impact may account for the phenotypic diversity. A heterogeneous phenotype exists even when the genotype is similar. Therefore, there are other influencing factors to consider and the limited treatment data available underpins the necessity for greater understanding of the mutation type and the natural history. For example, treatment outcomes for early infantile epilepsy with onset before 3 months are positive with sodium channel blockers (particularly phenytoin). However, with later childhood onset epilepsy, the response to sodium channel blockers is poor, aligned to the observations in Dravet syndrome [Citation68,Citation70].
2.4.3. Protocadherin 19 (PCDH19)-related disorders
The onset of PCDH19-related epilepsy is usually in the first few years of life (age <3 years) presenting with febrile and afebrile seizures of a generalized and/or focal onset. The seizures are treatment resistant in the early years, but control tends to improve through adolescence. Seizures are characteristically focal, occurring in clusters, and associated with fever between the ages of 2 and 10 years. PCDH19 is also associated with cognitive and behavioral changes including psychiatric disorders and autism [Citation71]. At present, no one anti-seizure drug demonstrates good efficacy in PCDH19 and most people require a polytherapy regime. On a cellular level, PCDH19 likely plays a role in GABA-A receptor function [Citation72]. Ganaxolone is a molecule trialed in a number of severe seizure disorders and plays a role in GABA-A modulation. There is some positive evidence that this may be a useful treatment adjunct for at least a part of the PCDH19 spectrum. However, a role as a precision treatment encompassing the disorder’s neuropsychiatric symptoms remains unproven [Citation71].
3. Conclusion
The treatment of epilepsy specifically in autism is under researched. The approach to management should be person-centered considering epilepsy-related factors and wider individual factors. Specific epilepsy syndromes should be approached in accordance with the evidence base and guidelines for that syndrome or by seizure type. Where genetic etiology is identified, there may be specific targeted treatments available. For people with autism with ID, there are clinical guidelines for anti-seizure medication prescribing available based on current evidence and expert clinical opinion [Citation73,Citation74]. People with neurodevelopmental disorders may be more susceptible to the adverse effects of anti-seizure medication and have greater difficulty communicating these symptoms. It is also important to consider the impact of treatment on comorbid conditions, particularly neuropsychiatric symptoms and psychiatric illness. Some anti-seizure medication will improve neuropsychiatric symptoms; however, for some people, certain anti-seizure medications may worsen symptoms including psychosis. In addition the treatment of comorbid psychiatric conditions with psychotropic medication including dopamine antagonists may lower seizure threshold. However, treatment of psychiatric illness should not be withheld, a holistic approach is required. Epilepsy in association with developmental disabilities should be considered as part of a complex neurodevelopmental model as opposed to discrete conditions [Citation73,Citation74].
Treatment choice has historically been largely based upon clinical assessment of seizure type and seizure syndrome (Levels 2 and 3) and this is has led to how people are categorized for research and therefore where the evidence base exists that ultimately informs clinical guidelines (). For example, targeting treatment toward an epileptic encephalopathy such as Lennox–Gastaut syndrome is based upon meeting the diagnostic criteria, or following national guidance on the treatment of focal seizures. As we have discussed, this evidence is not specific to people with autism with epilepsy and so must be interpreted cautiously. The evidence base should not be ignored; however, we are now starting to progress toward treatments targeting the functional changes caused by genetic variants of significance (Levels 4 and 5). In this move toward more precise medicine, we must continue to go further to personalize medicine and ensure all interventions ultimately consider the person on an individual basis (Level 1). All efforts should be made to support people with autism to make choices regarding their treatment.
Table 4. Targeting treatment more effectively
4. Expert opinion
There is very limited Level 1 evidence to support the efficacy of any anti-seizure medications specifically in autism. There are no high-quality randomized, double-blind, placebo-controlled trails investigating the efficacy and safety of anti-seizure medication for epilepsy in autism. The approach to treatment should be guided by the evidence base following a robust diagnosis of seizure type and seizure syndrome. The evidence base for treatment by seizure type outlined by the ILAE is informative and should be used as a guide for clinicians. However, we advocate a move toward individualized treatment plans in taking the early steps toward personalized medicine. Identification of a genetic etiology will inform treatment choice. For example, those conditions outlined in the move toward precision medicine, where particular therapies are targeted at the functional effect and resulting phenotype caused by the genotypic variant.
Clinicians should consider epilepsy in association with autism under a wider neurodevelopmental disorder model including ID and other developmental disabilities. A neurodevelopmental approach to assessment and treatment will improve outcomes and can be guided by a still limited, but more substantial evidence base () [Citation62].
Table 5. A neurodevelopmental approach to epilepsy care
People with autism are a heterogeneous group. The outcome of treatment will depend upon individual characteristics. The approach to prescribing should be person-centered and tailored to the individual profile of the person including etiology of epilepsy, genetic syndrome, EEG changes, seizure type, comorbid medical conditions, comorbid psychiatric conditions, concomitant medications (including those which lower the seizure threshold or interact with anti-seizure medicines), and individual risk factors. The individual and their family or caregivers should be fully involved in the decision-making process. This may require reasonable adjustments such as increased clinic time and the use of accessible information to facilitate collaborative working.
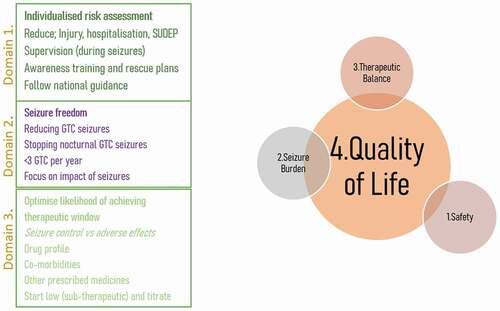
The first standard of epilepsy treatment should be to reduce risk and help keep the individual safe (). Risk should be assessed individually for each risk factor in the context of a person’s lifestyle and environment. Reducing seizure burden through treatment with anti-seizure medication will help reduce risk of injury, hospitalization, and sudden unexpected death in epilepsy (SUDEP) [Citation75]. The goal of treatment should be seizure freedom. If this is not possible then reducing seizure frequency is the next best option, with a focus on generalized, bilateral, tonic-clonic motor seizures. Using tools such as the SUDEP and Seizure safety Checklist could help mitigate exacerbations of seizures across a lifetime [Citation76,Citation77]. Treatment should be targeted at accurately diagnosed seizure type and epilepsy syndrome in accordance with the ILAE guidelines. However, there must be a balance between seizure control and the impact of treatment on quality of life, such as adverse effects of anti-seizure medications. Prescribing new anti-seizure medications or changing between anti-seizure medications should be approached cautiously. It is advised to make one change at a time so that it is easier to attribute the effect of change. Dosing should be started at low sub-therapeutic doses and titration should be slow to maximize the likelihood of achieving the optimal therapeutic window [Citation73].
Figure 1. Key clincial standards for the four domains of epilepsy care.
4.1. Neuropsychiatric and behavioral changes
The management of seizures and the management of neuropsychiatric behavioral changes may be viewed as antagonistic on the surface. The pharmacological treatment options for each may in theory exacerbate the other. It is therefore fundamental that the ‘right hand is aware of what the left hand is doing.’ This is of specific importance in this complex population and expert holistic oversight is required. This is achieved through coordinated specialist care with open communication between services. The Step Together report is a blue print toward achieving holistic service provision for people with epilepsy and associated neurodevelopmental disorders including autism [Citation78].
Behavioral disorders are multifactorial, and therefore anyone prescribed medication with the potential to impact upon behavior (anti-seizure medications) ideally requires a detailed neuropsychiatric profile and functional analysis of behavior before considering pharmacological intervention. People with autism and epilepsy should have access to psychological assessment and therapy as appropriate. Collaborative working with a specialist community team including a Psychiatrist is essential to ensuring a holistic approach. Treatment of comorbid psychiatric illness should not be avoided for fear of impact upon seizure control (e.g. SSRI, neuroleptics) [Citation79]. The approach to treatment is no different to guidelines for treatment of the psychiatric illness in the general population. Approach to diagnosis should be done with rigor in accordance with an appropriate diagnostic classification system such as the DSM 5. Where there is a level of complexity this should be led by an appropriately skilled psychiatrist and multidisciplinary team. In further complex situations including neuropsychiatric disturbances related to seizure activity (pre-ictal, ictal, post ictal), treatment approach may focus upon seizure control. This again should be done collaboratively with good communication between the specialists involved. Prescribing psychotropic medication for behavior should never be a first-line consideration outside of emergencies where risk outweighs any negative effects. Where there are complex behaviors that pose challenges to people and services, a specialist psychiatric and psychology service should be involved to monitor effects closely. There may be a role for non-pharmacological intervention including specific psychological therapies such as cognitive-behavioral therapy (CBT). If an accurate diagnosis proves elusive, any psychotropic medication should be targeted at specific symptoms, reviewed regularly, and only used for as short a duration as possible [Citation62,Citation80,Citation81].
4.2. The future
Rapid advances in our understanding of genomic variants associated with autism, epilepsy and other NDDs have identified syndromes where there can be due consideration of a precision medicine approach. However, despite the progress, any clinical application is still in its infancy. The heterogeneous autism population identified through the broad diagnostic criteria makes generic treatment recommendations challenging. Recruiting people with autism with epilepsy into clinical trials is complex, and we are often reliant upon later subgroup analysis. Therefore, progress from the development of a new pharmacological therapy to its clinical use in people with autism is prolonged and often far later than for those without autism or NDDs. In an ideal scenario, robust clinical intervention trails would be established with populations clearly defined by genetic profile and symptomatology. However, in reality, given constraints the focus for clinical trial designs in this population should be pragmatic observational methodologies that are applicable to real-word practice. Five years from now, it is far more likely that treatment will be targeted based on genetic profiles and the functional impact of genomic variation than based upon a combination of symptoms fulfilling certain neurodevelopmental diagnostic criteria. However, this does not change the core principles of the approach to treatment considering epilepsy in association with autism under the umbrella of NDDs.
Article highlights
There is a strong correlation between epilepsy and all the neurodevelopmental disorders (NDDs).
Epilepsy and autism have known common genetic etiologies and pathophysiological pathways.
There is no Level 1 evidence supporting the efficacy and/or tolerability of commonly prescribed anti-seizure medications specifically for autistic.
The best evidence available can be used to define first- and second-line anti-seizure medication choices for focal and absence seizures, the most common presentations in autism.
There is increasing evidence of a move toward precision medicine with specific treatment available for genetic syndromes associated with both autism and epilepsy.
Epilepsy and autism should be approached as a complex neurodevelopmental model, and not as discrete conditions.
The approach to treatment should be personalized based upon seizure type, epilepsy syndrome, genetic variant, physical and psychiatric comorbidities, and risk level.
Future research should focus on pragmatic study designs considering the risk factors that influence seizure outcomes and neuropsychiatric comorbidities.
This box summarizes key points contained in the article.
Declaration of Interest
R Shankar has received institutional and research support from LivaNova, UCB, Eisai, Veriton Pharma, Bial, Averelle, and GW Pharmaceuticals outside the submitted work. He was the author/lead of the Royal College of Psychiatrists reports CR203, CR206, and the National Step Together Report discussed significantly in the current paper. He is also the medical lead of the freely available SUDEP and Seizure safety Checklist, which is referenced in the paper. He is the developmental disabilities representative in the recent NICE revision/update of the epilepsies (2022), the NHS RightCare report (2020), the NHS England specialist commissioning report for epilepsy (2021) and the National Confidential Inquiry into epilepsy deaths. R Shankar is also supported by the following grants: an National Institute for Health Research (NIHR) AI grant entitled DECODE: mapping the challenges and requirements for Data-driven, machinE learning aided stratification and management of multiple long-term COnditions in adults with intellectual DisabilitiEs (Award ID: NIHR202627); an NIHR AI grant entitled BioEP: From prototype to clinical evaluation (Award ID: AI_AWARD01646); A Research for Patient Benefit (RdPB) NIHR grant entitled what works best for people with epilepsy and their clinicians (Award ID: NIHR200763); Humanising the Healthcare of People with Learning Disabilities (including people with autism who also have learning disabilities) A Co-produced research project funded by the Economic and Social Research Council (Award ID ES/W003406/1) and a Small Business Research Initiative (SBRI) Healthcare award – Brain in Hand. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Reilly C, Atkinson P, Das KB, et al. Features of autism spectrum disorder (ASD) in childhood epilepsy: a population-based study. Epilepsy Behav. 2015;42:86–92.
- Shankar R, Perera B, Thomas RH. Epilepsy, an orphan disorder within the neurodevelopmental family. J Neurol Neurosurg. 2020;91(12):1245–1247.
- Fisher RS, Boas WVE, Blume W, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia. 2005;46(4):470–472.
- Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55(4):475–482.
- Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: position Paper of the ILAE Commission for classification and terminology. Epilepsia. 2017;58(4):522–530.
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington VA: American Psychiatric Association; 2013. p. DSM–5.
- Fiest KM, Sauro KM, Wiebe S, et al. Prevalence and incidence of epilepsy: a systematic review and meta-analysis of international studies. Neurology. 2017;88(3):296–303.
- Maenner MJ, Shaw KA, Baio J, et al. Prevalence of autism spectrum disorder among children aged 8 Years — autism and developmental disabilities monitoring network, 11 sites, United States, 2016. MMWR Surveill Sum. 2020;69(No. SS–4):1–12.
- Lukmanji S, Manji SA, Kadhim S, et al. The co-occurrence of epilepsy and autism: a systematic review. Epilepsy Behav. 2019;98:238–248.
- Saemundsen E, Ludvigsson P, Rafnsson V. Risk of autism spectrum disorders after infantile spasms: a population‐based study nested in a cohort with seizures in the first year of life. Epilepsia. 2008;49(11):1865–1870.
- Strasser L, Downes M, Kung J, et al. Prevalence and risk factors for autism spectrum disorder in epilepsy: a systematic review and meta‐analysis. Dev Med Child Neurol. 2018;60(1):19–29.
- Amiet C, Gourfinkel-An I, Bouzamondo A, et al. Epilepsy in autism is associated with intellectual disability and gender: evidence from a meta-analysis. Biol Psychiatry. 2008;64(7):577–582.
- Hara H. Autism and epilepsy: a retrospective follow-up study. Brain Dev. 2007;29(8):486–490.
- Volkmar FR, Nelson DS. Seizure disorders in autism. J Am Acad Child Adolesc Psychiatry. 1990;29(1):127–129.
- Precenzano F, Parisi L, Lanzara V, et al. Electroencephalographic abnormalities in autism spectrum disorder: characteristics and therapeutic implications. Medicina (B Aires). 2020;56(9):419.
- Chez MG, Chang M, Krasne V, et al. Frequency of epileptiform EEG abnormalities in a sequential screening of autistic patients with no known clinical epilepsy from 1996 to 2005. Epilepsy Behav. 2006;8(1):267–271.
- Chow J, Jensen M, Amini H, et al. Dissecting the genetic basis of comorbid epilepsy phenotypes in neurodevelopmental disorders. Genome Med. 2019;11(1):1–14.
- Marini C, Scheffer IE, Nabbout R, et al. The genetics of Dravet syndrome. Epilepsia. 2011;52:24–29.
- Lee BH, Smith T, Paciorkowski AR. Autism spectrum disorder and epilepsy: disorders with a shared biology. Epilepsy Behav. 2015;47:191–201.
- Long S, Zhou H, Li S, et al. The clinical and genetic features of co-occurring epilepsy and autism spectrum disorder in Chinese children. Front Neurol. 2019;10:505.
- Peng J, Zhou Y, Wang K. Multiplex gene and phenotype network to characterize shared genetic pathways of epilepsy and autism. Sci Rep. 2021;11(1):1–16.
- Bozzi Y, Provenzano G, Casarosa S. Neurobiological bases of autism–epilepsy comorbidity: a focus on excitation/inhibition imbalance. Eur J Neurosci. 2018;47(6):534–548.
- Tuchman R, Cuccaro M. Epilepsy and autism: neurodevelopmental perspective. Curr Neurol Neurosci Rep. 2011;11(4):428–434.
- Pan PY, Bölte S, Kaur P, et al. Neurological disorders in autism: a systematic review and meta-analysis. Autism. 2021;25(3):812–830.
- Rydzewska E, Hughes-mccormack LA, Gillberg C, et al. Prevalence of long-term health conditions in adults with autism: observational study of a whole country population. BMJ open. 2018 Aug 1;8(8):e023945.
- Croen LA, Zerbo O, Qian Y, et al. The health status of adults on the autism spectrum. Autism. 2015 Oct;19(7):814–823.
- Chapman M, Iddon P, Atkinson K, et al. The misdiagnosis of epilepsy in people with intellectual disabilities: a systematic review. Seizure. 2011;20(2):101–106.
- Uldall P, Alving J, Hansen LK, et al. The misdiagnosis of epilepsy in children admitted to a tertiary epilepsy centre with paroxysmal events. Arch Dis Child. 2006;91(3):219–221.
- Donat JF, Wright FS. Episodic symptoms mistaken for seizures in the neurologically impaired child. Neurology. 1990;40(1):156.
- National Institute for clinical Excellence (CG 137), 2012. The Epilepsies: the diagnosis and management of the epilepsies in adults and children in primary and secondary care. [cited 2021 Aug 20]. Available Online: http//:www.nice.org.uk/cg137.
- Kerr MP, Watkins LV, Angus-Leppan H, et al. R2018. The provision of care to adults with an intellectual disability in the UK. A special report from the intellectual disability UK chapter ILAE. Seizure. 2018;56:41–46.
- Frye RE, Rossignol D, Casanova MF, et al. A review of traditional and novel treatments for seizures in autism spectrum disorder: findings from a systematic review and expert panel. Front Public Health. 2013;1:31.
- Peake D, Notghi LM, Philip S. Management of epilepsy in children with autism. Current Paediatrics. 2006;16(7):489–494.
- Glauser T, Ben‐Menachem E, Bourgeois B, et al., ILAE Subcommission on AED Guidelines. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2013;54(3):551–563.
- Nevitt SJ, Sudell M, Weston J, et al. Antiepileptic drug monotherapy for epilepsy: a network meta-analysis of individual participant data. Cochrane Database Syst Rev. 2017 Jun 29;66: CD011412 https://doi.org/10.1002/14651858.CD011412.pub2.
- Darnell JC, Richter JD. Cytoplasmic RNA-binding proteins and the control of complex brain function.Cold spring. Harb Perspect Biol. 2012;4:a012344.
- Berry-Kravis E. Epilepsy in fragile X syndrome. Dev Med Child Neurol. 2002;44(11):724–728.
- McCary LM, Roberts JE. Early identification of autism in fragile X syn-drome: a review. J Intellect Disabil Res. 2013;57(9):803–814.
- Seltzer LE, Ma M, Ahmed S, et al. Epilepsy and outcome in FOXG1‐related disorders. Epilepsia. 2014;55(8):1292–1300.
- Kortüm F, Das S, Flindt M, et al. The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J Med Genet. 2011 Jun;48(6):396–406.
- Ip JP, Mellios N, Sur M. Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci. 2018;19(6):368–382.
- Operto FF, Mazza R, Pastorino GMG, et al. Epilepsy and genetic in Rett syndrome: a review. Brain Behav. 2019;9(5):e01250.
- Percy AK. Rett syndrome: exploring the autism link. Arch Neurol. 2011;68(8):985–989.
- Wolff M, Johannesen KM, Hedrich U, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017;140(5):1316–1336.
- Bassani S, Cwetsch AW, Gerosa L, et al. The female epilepsy protein PCDH19 is a new GABAAR-binding partner that regulates GABAergic transmission as well as migration and morphological maturation of hippocampal neurons. Hum Mol Genet. 2018;27(6):1027–1038.
- Compagno Strandberg M, Söderberg‐Löfdal K, Kimland E, et al. Evidence‐based anti‐seizure monotherapy in newly diagnosed epilepsy: a new approach. Acta Neurol Scand. 2020;142(4):323–332.
- McTague A, Cross JH. Treatment of epileptic encephalopathies. CNS Drugs. 2013;27(3):175–184.
- Vigevano F, Arzimanoglou A, Plouin P, et al. Therapeutic approach to epileptic encephalopathies. Epilepsia. 2013;54:45–50.
- Myers KA, Scheffer IE. Precision medicine approaches for Infantile-Onset developmental and epileptic encephalopathies. Annu Rev Pharmacol Toxicol. 2021;62(1):641–662.
- Tomson T, Battino D, Bonizzoni E, et al. Dose-dependent teratogenicity of valproate in mono-and polytherapy: an observational study. Neurology. 2015;85(10):866–872.
- Belsito KM, Law PA, Kirk KS, et al. Lamotrigine therapy for autistic disorder: a randomized, double-blind, placebo-controlled trial. J Autism Dev Disord. 2001;31(2):175–181.
- Devinsky O, Verducci C, Thiele EA, et al. Open-label use of highly purified CBD (Epidiolex®) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and doose syndromes. Epilepsy Behav. 2018;86:131–137.
- Saneto R, Sparagana S, Kwan P, et al. Efficacy of Add-on Cannabidiol (CBD) Treatment in Patients with Tuberous Sclerosis Complex (TSC) and a History of Infantile Spasms (IS): post Hoc Analysis of Phase 3 Trial GWPCARE6 (1534). Neurology Apr. 2021;96(15 Supplement):1534.
- Patel AD, Mazurkiewicz-Bełdzińska M, Chin RF, et al. Long-term safety and efficacy of add-on cannabidiol in patients with Lennox–Gastaut syndrome: results of a long-term open-label extension trial. Epilepsia;. 2021;62(9):2228–2239.
- Scheffer IE, Halford JJ, Miller I, et al. Add-on cannabidiol in patients with Dravet syndrome: results of a long-term open-label extension trial. Epilepsia. 2021;00:1–13.
- Fletcher S, Pawliuk C, Ip A, et al. Medicinal cannabis in children and adolescents with autism spectrum disorder: a scoping review. Child Care Health Dev. 2021;1–12. https://doi.org/10.1111/cch.12909
- Upadhya D, Castro OW, Upadhya R, et al. Prospects of cannabidiol for easing status Epilepticus-Induced epileptogenesis and related comorbidities. Mol Neurobiol. 2018 Aug;55(8):6956–6964.
- Kanemura H, Sano F, Hoshino H, et al. Efficacy of perampanel in epilepsy patients with autism spectrum disorder. Epilepsy Res. 2021;170:106550.
- Shankar R, Henley W, Wehner T, et al. Perampanel in the general population and in people with intellectual disability: differing responses. Seizure. 2017 Jul;49:30–35.
- Allard J, Henley W, Snoeijen-Schouwenaars F, et al. European perspective of perampanel response in people with intellectual disability. Acta Neurol Scand. 2020 Sep;142(3):255–259.
- Wehner T, Mannan S, Turaga S, et al. Retention of perampanel in adults with pharmacoresistant epilepsy at a single tertiary care center. Epilepsy Behav. 2017 Aug;73:106–110.
- Watkins L, O’Dwyer M, Kerr M, et al. Quality improvement in the management of people with epilepsy and intellectual disability: the development of clinical guidance. Expert Opin Pharmacother. 2020;21(2):173–181.
- Robertson J, Chadwick D, Baines S, et al. Prevalence of dysphagia in people with intellectual disability: a systematic review. Intellect Dev Disabil. 2017;55(6):377–391.
- Curatolo P, Moavero R, de Vries PJ. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015;14(7):733–745.
- Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37(1):19–24.
- French JA, Lawson JA, Yapici Z, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016;388(10056):2153–2163.
- Sabitha KR, Shetty AK, Upadhya D. Patient-derived iPSC modeling of rare neurodevelopmental disorders: molecular pathophysiology and prospective therapies. Neurosci Biobehav Rev. 2021 Feb;121:201–219.
- Sanders SJ, Murtha MT, Gupta AR, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–241.
- Wolff M, Brunklaus A, Zuberi SM. Phenotypic spectrum and genetics of SCN 2A -related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia. 2019;60(S3):S59–S67.
- Samanta D. PCDH19-related epilepsy syndrome: a comprehensive clinical review. Pediatr Neurol. 2020;105:3–9.
- Jakimiec M, Paprocka J, Śmigiel R. CDKL5 deficiency disorder—A complex epileptic encephalopathy. Brain Sci. 2020;10(2):107.
- Scheffer IE, Nabbout R. SCN1A‐related phenotypes: epilepsy and beyond. Epilepsia. 2019;60(S3):S17–S24.
- Royal college of psychiatrists management of epilepsy in adults with intellectual disability 2017. [cited 2021 Sep 04]. Available from https://www.rcpsych.ac.uk/improving-care/campaigning-for-better-mental-health-policy/college-reports/2017-college-reports/prescribing-anti-epileptic-drugs-for-people-with-epilepsy-and-intellectual-disability-cr206-oct-2017
- Royal college of psychiatrists management of epilepsy in adults with intellectual disability 2017. [cited 2021 Sep 04]. http://www.rcpsych.ac.uk/usefulresources/publications/collegereports/cr/cr203.aspx
- Watkins L, Shankar R. Reducing the Risk of Sudden Unexpected Death in Epilepsy (SUDEP). Curr Treat Options Neurol. 2018 Aug 22;20(10):40. PMID: 30136125
- Watkins L, Shankar R, Sander JW. Identifying and mitigating Sudden Unexpected Death in Epilepsy (SUDEP) risk factors. Expert Rev Neurother. 2018;18(4):265–274.
- Shankar R, Ashby S, McLean B, et al. Bridging the gap of risk communication and management using the SUDEP and Seizure Safety Checklist. Epilepsy Behav. 2020 Feb;103(Pt B):106419.
- The national Step Together Report. [cited 2021 Sep 04]. Available from: https://www.bild.org.uk/wp-content/uploads/2020/11/Step-Together-17-November-2020-Download-Link-.pdf
- Watkins LV, Pickrell WO, Kerr MP. Treatment of psychiatric comorbidities in patients with epilepsy and intellectual disabilities: is there a role for the neurologist? Epilepsy Behav. 2019;98:322–327.
- Kerr MP, Mensah S, Besag F, et al. International consensus clinical practice statements for the treatment of neuropsychiatric conditions associated with epilepsy. Epilepsia. 2011;52(11):2133–2138.
- Challenging behaviour and learning disabilities: prevention and interventions for people with learning disabilities whose behaviour challenges, 2015. Nice Guideline (NG11). National Institute for Health and Care Excellence.