
ABSTRACT
Introduction
Ruxolitinib is the most commonly used JAK-inhibitor (JAKi) for the management of symptoms related to splenomegaly and cytokine-mediated inflammation in patients with myelofibrosis (MF), but is limited by variable durability of response with most patients experiencing failure after 2–3 years. Long-term data on other approved JAKi, fedratinib and pacritinib, are not available due to the clinical hold put on pivotal trials for toxicity concerns.
Areas covered
Following the initial hold for concern of Wernicke’s encephalopathy, fedratinib was approved by the Food and Drug Administration (FDA) in 2019 for MF. We review the data available from early, and late phase critical trials, outline a role for fedratinib in the current treatment landscape of MF, and highlight the knowledge gaps in optimizing use of fedratinib.
Expert opinion
The JAKARTA and JAKARTA2 trials established efficacy in spleen volume response (SVR) and symptom reduction in JAKi-naïve and ruxolitinib-exposed MF patients, respectively. Further trials, FREEDOM and FREEDOM2, are in progress to understand long-term effects of fedratinib; and include strategies to mitigate gastrointestinal toxicity, monitor thiamine levels and surveil for encephalopathy. We use fedratinib for symptomatic MF following ruxolitinib failure in patients without significant cytopenias; with practical strategies for monitoring and managing potential toxicity.
1. Introduction
The myeloproliferative neoplasms are a group of clonal disorders of hematopoiesis that include essential thrombocythemia (ET), polycythemia vera (PV), and myelofibrosis (MF). Myelofibrosis may exist as its own entity (primary MF), or following a preceding history of PV, or ET (secondary MF). Current National Comprehensive Cancer Network (NCCN) and European LeukemiaNet (ELN) management guidelines recommend clinical and molecular risk stratification, and consideration for curative hematopoietic stem cell transplantation (HCT) in fit patients with predicted survival ≤5 years [Citation1–4]. Medical therapy with JAK-inhibitors (JAKi) is recommended for patients with symptomatic splenomegaly or cytokine-mediated-constitutional symptoms including fever, night-sweats, and weight loss [Citation5–7]; with ongoing controversy as to whether JAKi-therapy improves survival in patients with symptomatic MF [Citation8,Citation9].
2. Overview of the market
The discovery of JAK2V617F mutation paved the way for development of small molecular inhibitors in MF, and at least 11 JAKi have entered clinical trials (). Following its approval in 2011, ruxolitinib has become the mainstay of symptom-directed therapy for MF. The population prevalence of MF is 4–6 per 100 000, with 1 in 5 prescribed ruxolitinib [Citation10,Citation11]. In specialized centers, up to 35–40% of MF patients will receive JAKi during their disease course (unpublished data from prospective MPN registry at the Princess Margaret Cancer Center; NCT02760238). The COMFORT-I and COMFORT-II trials demonstrated that ruxolitinib improves splenomegaly, MF-related symptom burden, and health-related quality of life in patients with Int-2/High DIPSS MF; with the JUMP study observing benefit in low-/Int-1 risk MF [Citation6,Citation7,Citation12]. Ruxolitinib does not exclusively inhibit JAK2 (50% inhibitory concentration [IC50]2.8 nanomolar [nM]) but also has activity against JAK1 (IC50 3.3 nM) and TYK2 (IC50 19 nM) [Citation13]. The degree of improvement in splenomegaly and development of treatment-related cytopenias are both dose dependent and development of therapies that ameliorate dose-limiting cytopenias has been a priority [Citation4,Citation14]. Ruxolitinib is also limited by duration of benefit with 50–71% of patients experiencing failure or discontinuation at 3 years; and poor subsequent overall survival of 11–14 months in retrospective cohorts [Citation15–19].
Figure 1. Clinical development of JAK-inhibitors for use in myelofibrosis.
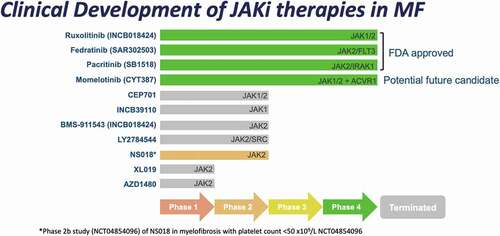
Momelotinib is a JAK1/JAK2 inhibitor with additional activity against ACVR1 leading to reduced activity of hepcidin and improvement of inflammation-driven anemia [Citation14,Citation20,Citation21]. The SIMPLIFY clinical trials demonstrated clinical efficacy in spleen reduction with lower risk of development of significant new or worsening anemia, and overall improvement in hemoglobin level [Citation22,Citation23]. The MOMENTUM phase 3 study of MF patients previously treated with JAKi and hemoglobin <100 g/L compared momelotinib to danazol; with initial results reporting significant efficacy in total symptom score (TSS) improvement, spleen response, and higher rates of transfusion independence [Citation24].
Pacritinib is an inhibitor of JAK2 (IC50 23 nM) and TYK2 (IC50 50 nM) without effect on JAK1 (IC50 1280 nM) which has demonstrated symptom and spleen benefit with less impact on platelet count [Citation25,Citation26]. The PERSIST-1 study demonstrated clinical efficacy of pacritinib compared to best-available non-JAKi therapy with particular benefit amongst patients with baseline thrombocytopenia. The PERSIST-2 study observed similar efficacy in patients with platelet count <100 x 109/L, of whom, half were previously treated with ruxolitinib. PERSIST-2 was terminated early in 2016 after an FDA clinical hold was placed on pacritinib due to concerns of bleeding, cardiovascular events, and death [Citation26–28]. With longer follow-up and further investigation into optimal dosing the hold was lifted in 2017. The ongoing PACIFICA study is enrolling patients with MF with platelets <50 x 109/L to receive pacritinib or physician-selected best available therapy [Citation28]. Initial FDA approval in 2022 for use of pacritinib in intermediate- and high-risk MF patients with platelet count <50 x 109/L highlights the niche that this agent has in a difficult-to-treat subset of patients.
A number of novel agents in late stage development are being studied in combination with JAKi therapy with the goals of synergistic improvement in clinical symptoms, potential disease-modifying effects, or ameliorating cytopenias [Citation4,Citation14]. Combinations with JAKi include pelabresib (MANIFEST-2), navitoclax (TRANSFORM-1&2), parsaclisib (LIMBER-313), and luspatercept (INDEPENDENCE).
3. Introduction to the compound
3.1. Chemistry
Fedratinib (SAR-302503; TG-101348; INREBIC®) is a kinase inhibitor with molecular weight 615.62 g/mol. The empirical formula is C27H36N6O3S·2HCl·H2O, while the chemical name is N-tert-butyl-3-[(5-methyl-2-{[4-(2pyrrolidin-1-ylethoxy)phenyl]amino}pyrimidin-4-yl)amino]benzenesulfonamide dihydrochloride monohydrate [Citation29]. It is available in gelatin capsules for oral administration (Box 1).
Box 1. Drug summary box
3.2. Pharmacodynamics
Initial development of fedratinib used rational structure-based techniques, combining kinase-screening and molecular modeling of crystal structures [Citation13]. Fedratinib has a high selectivity for JAK2 with IC50 of 3 nM and less effect on JAK1, JAK3, and TYK2; which is hypothesized to reduce immunosuppression and the risk of infection [Citation13,Citation30–37]. Fedratinib has also been shown to inhibit FLT3, a tyrosine kinase regulating hematopoiesis, and BRD4, a bromo and extra terminal (BET) protein, at therapeutic concentrations of IC50 15 nM and 164 nM, respectively [Citation13,Citation38–40]. Reduction of NF-kB-mediated inflammation through concurrent inhibition of JAK/STAT and BET pathways may be of particular interest given the promising results and potential disease-modifying effects with the combination of ruxolitinib with pelabresib though clinical significance with fedratinib is not known [Citation41]. Consistent with prior preclinical models, treatment of MF patients with fedratinib had a demonstrated reduction in downstream STAT3 phosphorylation [Citation13,Citation30]. In contrast to earlier mouse models, the use of fedratinib had no demonstrated reduction in mutant JAK2 V617F allele frequency [Citation30].
3.3. Pharmacokinetics and metabolism
In doses of 10–680 mg, fedratinib is rapidly absorbed without food; though low- and high-fat meals increased AUC and Cmax by 24% and 14%, respectively [Citation42,Citation43]. Time to maximum concentration is 2–4 hours following administration. Fedratinib is ≥90% plasma protein bound with steady state concentrations achieved after 15 days and apparent volume of 1770 L [Citation42,Citation43]. Fedratinib is metabolized by CYP3A4, CYP2C19, and flavin-containing monooxygenase-3; though 80% of circulating drug is unmodified fedratinib [Citation42–45]. Elimination of radiolabeled fedratinib is 77% through fecal excretion with only 5% expelled in urine [Citation42,Citation43,Citation46]. The effective half-life of fedratinib is 41 h with apparent clearance of 13 L/h in MF patients [Citation29]. There was no observed impact of sex, age, body weight, or mild-moderate hepatic impairment on the pharmacokinetics of fedratinib [Citation42,Citation43,Citation45,Citation46]. Fedratinib levels increased 1.5- and 1.9-fold in the setting of moderate (creatinine clearance [CrCl], 30–59 cc/min) and severe (CrCl 15–29 cc/min) renal dysfunction, respectively, and dose reduction for to 200 mg is recommended in patients with CrCl 15–29 cc/min [Citation42–46].
Co-administration of fedratinib with strong CYP3A4-inhibitors has been shown to increase fedratinib exposure, and dose reduction to 200 mg is recommended [Citation42,Citation44,Citation45]. Avoidance of moderate/strong CYP3A4 inducers or dual CYP3A4/CYP2C19 inhibitors is recommended. Administration of fedratinib alongside substrates of CYP3A4 or CYP2C19 may increase levels of the other drugs and dose modification/monitoring for adverse events is suggested [Citation42,Citation44,Citation45].
4. Clinical efficacy
The first phase I study, NCT00631462, () of fedratinib consisted of 59 patients with intermediate/high-risk MF and evaluated 4-week cycles of 30–800 mg/day in a 3 + 3 cohort design [Citation47,Citation48]. The maximum tolerated dose was 680 mg/day of fedratinib; while the lowest dose with clinical activity was 240 mg/day. Adverse events (AEs) were more common amongst patients treated at doses exceeding 680 mg/day with the most common dose-limiting toxicity being asymptomatic rise in amylase. In the extended follow-up (NCT00724334) of participating patients, 39% remained on study with median duration of therapy 30 (range 13–44) months [Citation47].
Table 1. Completed and ongoing clinical trials of fedratinib monotherapy in myelofibrosis.
A phase II trial (NCT01420770) of treatment-naïve patients enrolled 31 patients to receive 300 mg, 400 mg, or 500 mg per day of fedratinib [Citation49]. At 24 weeks of the proportion of patients achieving spleen volume reduction of 35% (SVR35) was 30%, 60%, and 55%; and at 48 weeks was 30%, 80%, and 45%, respectively. Toxicities were similar to what was observed in the phase I study with grade 3–4 AEs, predominately hematological, observed in 27/31 patients. Anemia in the 300 mg and 400 mg groups was observed to reach nadir in weeks 12–16 weeks and then increased to near-baseline levels by week 48; while patients treated with 500 mg/day did not recover hemoglobin levels.
The JAKARTA (NCT01437787) trial was a phase III multicentre, double-blind study with JAKi-naïve patients with int-2/high risk MF randomized to receive fedratinib 400 mg, 500 mg, or placebo [Citation50]. The primary endpoint of SVR35% after 24 weeks and was achieved in 36%, 40%, and 1% of patients, respectively. TSS reduction ≥50% was observed in 36%, 34%, and 7% of the cohorts. There was no difference in outcomes based on baseline platelet counts, DIPSS risk, JAK2 mutation status, or diagnosis (primary vs. secondary MF). New or worsening grade 3 anemia was observed in 34% of patients treated with fedratinib 400 mg with nadir hemoglobin occurring between 12 and 16 weeks. Grade ≥3 thrombocytopenia was observed in 12% of patients with all cases developing within the first 16 weeks.
The phase II JAKARTA2 trial (NCT01523171) enrolled patients that discontinued ruxolitinib due to intolerance, resistance, or loss of response [Citation51]. A total of 97 patients were treated with a starting dose of 400 mg of fedratinib. The primary endpoint was SVR35 following 24 weeks of therapy; while key secondary endpoints were TSS reduction of ≥50%. On initial analysis of per-protocol population with the last observation carried forward (LOCF), the primary endpoint was met in 55% of evaluable patients (n = 83) and TSS≥50% was achieved in 25% of patients following 6 cycles [Citation51]. In patients resistant to ruxolitinib the SVR35 was 53% with TSS≥50% in 21%; while patients who were intolerant of ruxolitinib had spleen responses in 63% and symptom responses in 32%. Due to improved understanding and evolving definitions of ruxolitinib failure, reanalysis of the JAKARTA2 data was conducted with an intention-to-treat (ITT) population without LOCF, along with a sensitivity analysis (‘Stringent Criteria Cohort’) using more refined criteria for ruxolitinib intolerance, refractoriness, or loss of response [Citation52]. In the stringent cohort, after excluding patients with insufficient data or ongoing response at the time of trial enrollment, 23% of the cohort had loss of clinical response, 59% were refractory, and 18% were intolerant to ruxolitinib [Citation52]. The median time on prior ruxolitinib was 10.7 months in the ITT population and 11.5 months in the Stringent Criteria Cohort [Citation51,Citation52]. This reanalysis demonstrated SVR35% of 31% and TSS≥50% in 27% in the ITT population and no overall differences in outcomes when restricted to the ‘Stringent Criteria Cohort.’
Post-hoc subgroup analyses of the JAKARTA/JAKARTA2 trials have observed comparable clinical responses in fedratinib treated patients despite baseline thrombocytopenia (platelet count 50–100 x 109/L), or significant splenomegaly [Citation53,Citation54]. The long-term survival analysis, though limited by the premature study termination, demonstrated improved progression-free survival compared to placebo in treatment-naïve patients; and 18-month overall survival rate of 67% in second-line treated patients [Citation55].
The FREEDOM (NCT03755518) and FREEDOM2 (NCT03952039) clinical trials have been opened to further evaluate the efficacy and safety of fedratinib 400 mg in patients experiencing ruxolitinib failure [Citation56,Citation57]. FREEDOM is an open label, single-arm Phase IIIb study of 400 mg of fedratinib; while FREEDOM2 is a randomized, open-label study of fedratinib 400 mg compared to best available therapy (BAT).The primary outcomes remains SVR35 at 24 weeks, with secondary outcomes of TSS≥50% reduction, and key safety outcomes of gastrointestinal adverse events, WE, and thiamine level monitoring.
Following the approval of fedratinib, studies of real-world use and outcomes have confirmed similar short-term benefit as observed in clinical trials; though ongoing monitoring of key safety and long-term outcomes are still required [Citation58–60].
5. Safety and tolerability
5.1. Gastrointestinal toxicity
Gastrointestinal (GI) symptoms, including diarrhea, nausea, and vomiting, are the most common non-hematological AEs observed with fedratinib therapy (). Efforts to prevent and mitigate GI AEs were implemented in the design of FREEDOM and FREEDOM2; with initial reports of lower rates observed in the FREEDOM trial (diarrhea 35%, vomiting 18%, nausea 26%) that were highest during the first cycle and less prevalent as treatment continued [Citation61].
Strategies and tips to prevent and manage GI toxicities are summarized in and . Prophylactic antiemetics (i.e. ondansetron) are recommended for the first 2 cycles of therapy and continued as necessary. Diarrhea can be managed with antidiarrheal medication (i.e. loperamide) in addition to dietary and lifestyle modifications such as reduction of insoluble fiber, and caffeine avoidance. In patients experiencing significant GI toxicity thiamine levels should be more closely monitored and supplemented as necessary due to the potential for Wernicke’s encephalopathy (WE).
Figure 2. How we initiate fedratinib following failure of first-line ruxolitinib in myelofibrosis.
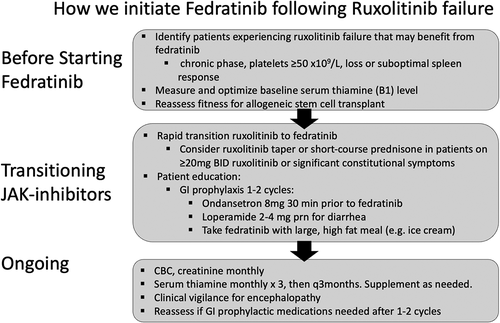
Table 2. Management of fedratinib-associated non-hematological toxicities.
5.2. Wernicke’s encephalopathy
The FDA approval for fedratinib contains a black box warning for the risk of encephalopathy including WE, and highlights the need to identify and manage thiamine deficiency prior to and during therapy. WE is a rare neurologic syndrome that develops due to deficiency of thiamine (vitamin B1) a cofactor necessary for multiple reactions in energy metabolism, in-particular for metabolically active cells of the central nervous system [Citation62,Citation63]. A classic triad of encephalopathy, oculomotor dysfunction, and gait ataxia is described, with hypotension, hypothermia, vestibular dysfunction, peripheral neuropathy, and decreased level of consciousness also observed [Citation62,Citation63].
Risk factors for thiamine deficiency include poor nutrition and patients with MF may be at risk due to splenomegaly associated early satiety, and cytokine-mediated cachexia. Prospective and retrospective studies, however, have found baseline thiamine deficiency to be rare in myelofibrosis [Citation64–66]. Fedratinib may contribute to thiamine deficiency indirectly through GI toxicity exacerbating malnutrition, or directly through inhibition of the human thiamine transporter-2 (THTR2/SLC19A3); though this finding has not been confirmed in rodent models [Citation67,Citation68].
Clinical events consistent with WE were observed in 8 of 670 patients participating in fedratinib clinical trials leading to clinical hold from 2013 to 2017 [Citation63]. Seven of the patients were receiving 500 mg of fedratinib, and 7 also had risk factors for malnutrition or GI AEs. In central review of all cases of suspected WE one definitive case (confirmed with clinical assessment and consistent MRI evaluation) was reported in a patient with baseline weight loss >10%, poor performance status, and preexisting ataxia; who later developed GI toxicity. An additional two cases of suspected WE were described, with suggestive symptoms and MRI findings, but confounded by cerebral infarct, and GI dysfunction. Most events resolved with some residual neurological symptoms including memory loss, cognitive impairment, and dizziness. One patient with metastatic head and neck cancer and severe malnutrition died, though his symptoms could not be confirmed to represent WE [Citation63]. The conclusion was that there was no definitive evidence for fedratinib causing WE, though baseline nutritional assessment and thiamine levels are recommended and suspicion of any encephalopathy should prompt fedratinib discontinuation and parental thiamine supplementation ().
5.3. Increased creatinine
Patients who initiate fedratinib are commonly observed to have an elevation in creatinine, typically developing in the first cycle of therapy. In our experience this initial rise is followed by a plateau that continues while patients remain on the medication and resolves shortly after discontinuation. Initial hypotheses suggested pre-renal injury from hypovolemia secondary to increased losses and reduced fluid intake from GI toxicity. In our experience, this phenomenon also develops in patients with adequate GI prophylaxis and minimal or no GI side effects. Pharmacodynamic studies have demonstrated that fedratinib can inhibit renal transporters MATE2-K and OCT2, for which creatinine is also a substrate [Citation42,Citation44,Citation45]. This observation offers a potential explanation for the increased serum creatinine levels that resolves with discontinuation of drug [Citation42,Citation44,Citation45]. We recommend careful monitoring of renal function when initiating fedratinib, especially in patients with preexisting renal dysfunction. We tolerate a mild rise in serum creatinine if there is no clinical sequelae or potential toxicity from co-administered medications that are renally cleared; in particular medications that are eliminated through OCT2 and MATE2-K such as metformin [Citation42,Citation44,Citation45].
6. Regulatory affairs
Fedratinib has been approved for the use in symptomatic int-2, and high-risk myelofibrosis by the FDA, European Medicines Agency, and Health Canada. The FDA maintains a black box warning for the risk of encephalopathy, including WE.
7. Expert opinion
As front-line therapy fedratinib has been shown to be efficacious for management of symptomatic splenomegaly and MF-related symptoms in comparison to placebo. There has been no direct comparison to the current standard of care, ruxolitinib, in the first-line setting. Industry-sponsored Indirect Treatment Comparison (ITC) using Matching-Adjusted Indirect Comparisons (MAICs) analysis compared patient level data from the JAKARTA, COMFORT-I, and COMFORT-II trials using the subset of patients with baseline platelet count ≥100 x 109/L, and demonstrated comparable efficacy for spleen volume response [Citation69]. The increased cost compared to ruxolitinib and need for GI prophylaxis, thiamine monitoring, and clinical vigilance for encephalopathy are added complexities that will likely dissuade front-line use of fedratinib without evidence of clear superiority ()[Citation70]. Clinical factors including transfusion requirement, peripheral blasts, clinical response at 6 months, in addition to mutational risk factors have been shown to predict outcomes with ruxolitinib therapy [Citation15,Citation71–74]. Future studies may identify patients that would benefit from front-line fedratinib due to these, or other identified factors.
Table 3. Potential uses of fedratinib in clinical practice.
Fedratinib and pacritinib (for patients with platelets <50 x109/L) are currently the only approved MF therapies for which there is clinical trial evidence of efficacy following first-line ruxolitinib. While outcomes following ruxolitinib failure are poor, many patients are not candidates for HCT and fedratinib is an excellent option for managing splenomegaly or cytokine-mediated symptoms in patients with platelets ≥50 x 109. In patients requiring optimization of splenomegaly prior to HCT who are refractory to or have lost response to ruxolitinib, fedratinib is an additional option to avoid splenectomy and its associated postoperative and infectious complications.
Care and clinical vigilance is required when transitioning from ruxolitinib as a discontinuation/withdrawal syndrome is associated with acute worsening symptoms in up to 15% of patients with potential for significant complications and death [Citation75]. The half-life of ruxolitinib is short (~3 hours) and while fedratinib is rapidly absorbed, it may take up to 2 weeks to achieve steady-state concentrations [Citation42,Citation45]. Several strategies have been suggested to transition from ruxolitinib to fedratinib. Various clinical trials recommended taper of ruxolitinib followed by 2 week gap prior to starting fedratinib. The gap period can cause significant symptom exacerbation in some patients and we instead start fedratinib the day after stopping ruxolitinib. Patients on high-doses (≥20 mg BID) of ruxolitinib or with breakthrough constitutional symptoms should be monitored closely and short-courses of steroid can be considered while waiting for fedratinib to achieve steady-state concentrations.
As more options become available for patients with myelofibrosis understanding of unique strengths and toxicities is needed to define a role for fedratinib. Atypical and opportunistic infections are common in MF and risk is increased with ruxolitinib therapy; while an increased risk of cutaneous malignancies has also been suggested [Citation33]. Though fedratinib was initially chosen for development due to JAK2 specificity with the hope this would translate to less nonspecific immunosuppression, the long-term rates of infectious complications and secondary malignancies are not known. With the clinical hold of the JAKARTA and JAKARTA2 trials long-term AEs and durability of responses are not well evaluated. Data from the FREEDOM and FREEDOM2 clinical trials will improve our understanding of extended complications, and help optimize mitigation strategies. The incidence of WE and utility and effectiveness of thiamine monitoring and supplementation will be key long-term safety observations.
The use of JAKi prior to HCT has been investigated in previous studies of ruxolitinib and may be beneficial for pretransplant disease and splenomegaly optimization. The safety of fedratinib use and discontinuation leading into transplant conditioning therapy has not been studied. With a longer half-life (41 hrs vs. 4 hrs for ruxolitinib) the risk of acute withdrawal complications is predicted to be less; though data are currently lacking to establish optimal use of fedratinib in the pre-transplant period.
8. Conclusion
The role of fedratinib will evolve as additional agents in advanced clinical study are approved for patients following ruxolitinib failure (). Agents such as luspatercept or momelotinib may become favored in patients with transfusion-requiring anemia, while pacritinib is an option for patients with severe thrombocytopenia. Drugs with evidence of disease-modifying activity may have preference in the first-line either alone or in combination with ruxolitinib. Fedratinib is a useful option for patients with constitutional- and splenomegaly-related symptoms and less severe cytopenias.
Article highlights
Fedratinib is an inhibitor of the tyrosine kinases JAK2, FLT3, and BRD4
Fedratinib has been demonstrated to improve splenomegaly and symptoms in JAK-inhibitor naïve (JAKARTA) and previously ruxolitinib-treated (JAKARTA2) patients with myelofibrosis
Fedratinib was approved by the FDA in 2019 for use in adult patients with intermediate-2 or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) myelofibrosis
Initial reports for Wernicke’s encephalopathy have lead to a black box warning and recommendation for evaluation and monitoring of thiamine levels
Common side effects include GI intolerance and elevation in creatinine; and can be effectively mitigated with prophylactic strategies
Declaration of interest
V Gupta has received research funding through his institution and honoraria from Novartis and Incyte and has served on the advisory board of Novartis, Incyte, Bristol Myers Squibb-Celgene, Abb Vie, Sierra Oncology, Pfizer, Takeda, and Constellation Biopharma.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
A reviewer on this manuscript has received honoraria for advisory board participation and research support from Bristol Myers Squibb, the manufacturer of fedratinib.
Company review
Bristol Myers Squibb provided a scientific accuracy review at the request of the journal editor.
Additional information
Funding
References
- Kröger N, Deeg J, Olavarria E, et al. Indication and management of allogeneic stem cell transplantation in primary myelofibrosis: a consensus process by an EBMT/ELN international working group. Leukemia. 2015;29(11):2126–2133.
- Mesa RA, Jamieson C, Bhatia R, et al. NCCN guidelines insights: myeloproliferative neoplasms, version 2.2018. J National Compr Cancer Network. 2017;15(10):1193–1207.
- Gupta V, Hari P, Hoffman R. Allogeneic hematopoietic cell transplantation for myelofibrosis in the era of JAK inhibitors. Blood J Am Soc Hematol. 2012;120(7):1367–1379.
- England J, Gupta V. Novel therapies vs hematopoietic cell transplantation in myelofibrosis: who, when, how? Hematology. 2021;2021(1):453–462.
- Koschmieder S, Chatain N. Role of inflammation in the biology of myeloproliferative neoplasms. Blood Rev. 2020;42:100711.
- Harrison CN, Vannucchi AM, Kiladjian -J-J, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30(8):1701–1707.
- Verstovsek S, Gotlib J, Mesa RA, et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and-II pooled analyses. J Hematol Oncol. 2017;10(1):1–6.
- Barosi G, Gale RP. Does ruxolitinib really prolong survival in individuals with myelofibrosis? The never-ending story. Blood Adv. 2022;6(7):2331–2333.
- Passamonti F, Vannucchi A, Cervantes F, et al. Ruxolitinib and survival improvement in patients with myelofibrosis. Leukemia. 2015;29(3):739–740.
- Mehta J, Wang H, Iqbal SU, et al. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2014;55(3):595–600.
- Bankar A, Zhao H, Iqbal J, et al. Healthcare resource utilization in myeloproliferative neoplasms: a population-based study from Ontario, Canada. Leuk Lymphoma. 2020;61(8):1908–1919.
- Gupta V, Griesshammer M, Martino B, et al. Analysis of predictors of response to ruxolitinib in patients with myelofibrosis in the phase 3b expanded-access JUMP study. Leuk Lymphoma. 2021;62(4):918–926.
- Quintás-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood J Am Soc Hematol. 2010;115(15):3109–3117.
- Bankar A, Gupta V. Investigational non-JAK inhibitors for chronic phase myelofibrosis. Expert Opin Investig Drugs. 2020;29(5):461–474.
- England JT, McNamara CJ, Kennedy JA, et al. Clinical and molecular correlates of JAK-inhibitor therapy failure in myelofibrosis: long-term data from a molecularly annotated cohort. Leukemia. 2022;36(1):1–4.
- Kuykendall AT, Shah S, Talati C, et al. Between a rux and a hard place: evaluating salvage treatment and outcomes in myelofibrosis after ruxolitinib discontinuation. Ann Hematol. 2018;97(3):435–441.
- Mascarenhas J, Mehra M, He J, et al. Patient characteristics and outcomes after ruxolitinib discontinuation in patients with myelofibrosis. J Med Econ. 2020;23(7):721–727.
- Newberry KJ, Patel K, Masarova L, et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood J Am Soc Hematol. 2017;130(9):1125–1131.
- Palandri F, Breccia M, Bonifacio M, et al. Life after ruxolitinib: reasons for discontinuation, impact of disease phase, and outcomes in 218 patients with myelofibrosis. Cancer. 2020;126(6):1243–1252.
- Mesa RA, Catalano J, Cervantes F, et al. Dynamic and time-to-event analyses demonstrate marked reduction in transfusion requirements for janus kinase inhibitor-naïve myelofibrosis patients treated with momelotinib compared head to head with ruxolitinib. Blood. 2019;134(Supplement_1):1663.
- Chifotides HT, Bose P, Verstovsek S. Momelotinib: an emerging treatment for myelofibrosis patients with anemia. J Hematol Oncol. 2022;15(1):1–17.
- Harrison CN, Vannucchi AM, Platzbecker U, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol. 2018;5(2):e73–e81.
- Mesa RA, Kiladjian -J-J, Catalano JV, et al. Simplify-1: a phase III randomized trial of momelotinib versus ruxolitinib in janus kinase inhibitor–naïve patients with myelofibrosis. J clin oncol. 2017;35(34):3844.
- Mesa RA, Gerds AT, Vannucchi A, et al. MOMENTUM: phase 3 randomized study of momelotinib (MMB) versus danazol (DAN) in symptomatic and anemic myelofibrosis (MF) patients previously treated with a JAK inhibitor. J clin oncol. 2022;40(16_suppl):7002.
- Hart S, Goh K, Novotny-Diermayr V, et al. Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer J. 2011;1(11):e44–e.
- Mesa RA, Vannucchi AM, Mead A, et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol. 2017;4(5):e225–e36.
- Gerds AT, Savona MR, Scott BL, et al. Determining the recommended dose of pacritinib: results from the PAC203 dose-finding trial in advanced myelofibrosis. Blood Adv. 2020;4(22):5825–5835.
- Mascarenhas J, Hoffman R, Talpaz M, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol. 2018;4(5):652–659.
- Blair HA. Fedratinib: first approval. Drugs. 2019;79(15):1719–1725.
- Wernig G, Kharas MG, Okabe R, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13(4):311–320.
- Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat Immunol. 2017;18(4):374–384.
- Tremblay D, King A, Li L, et al. Risk factors for infections and secondary malignancies in patients with a myeloproliferative neoplasm treated with ruxolitinib: a dual-center, propensity score-matched analysis. Leuk Lymphoma. 2020;61(3):660–667.
- Landtblom AR, Andersson TM, Dickman PW, et al. Risk of infections in patients with myeloproliferative neoplasms—a population-based cohort study of 8363 patients. Leukemia. 2021;35(2):476–484.
- Caldarola G, FoSSati B, de Simone C, et al. New JaK inhibitors for the treatment of psoriasis and psoriatic arthritis. G Ital Dermatol Venereol. 2020;155:411–20.
- Yarilina A, Xu K, Chan C, et al. Regulation of inflammatory responses in tumor necrosis factor–activated and rheumatoid arthritis synovial macrophages by JAK inhibitors. Arthritis Rheumatism. 2012;64(12):3856–3866.
- T Virtanen A, Haikarainen T, Raivola J, et al. Selective JAKinibs: prospects in inflammatory and autoimmune diseases. BioDrugs. 2019;33(1):15–32.
- Zhou T, Georgeon S, Moser R, et al. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348). Leukemia. 2014;28(2):404–407.
- Ciceri P, Müller S, O’mahony A, et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat Chem Biol. 2014;10(4):305–12.
- Davis RR, Li B, Yun SY, et al. Structural insights into JAK2 inhibition by ruxolitinib, fedratinib, and derivatives thereof. J Med Chem. 2021;64(4):2228–2241.
- Hantschel O. Unexpected off-targets and paradoxical pathway activation by kinase inhibitors. ACS Chem Biol. 2015;10(1):234–245.
- Mascarenhas J, Kremyanskaya M, Hoffman R, et al. MANIFEST, a phase 2 study of CPI-0610, a Bromodomain and Extraterminal Domain Inhibitor (BETi), as monotherapy or “add-on” to ruxolitinib, in patients with refractory or intolerant advanced myelofibrosis. Blood. 2019;134(Supplement_1):670.
- Zhang M, Xu CR, Shamiyeh E, et al. A randomized, placebo‐controlled study of the pharmacokinetics, pharmacodynamics, and tolerability of the oral JAK 2 inhibitor fedratinib (SAR 302503) in healthy volunteers. J Clin Pharmacol. 2014;54(4):415–421.
- Zhang M, Xu C, Ma L, et al. Effect of food on the bioavailability and tolerability of the JAK2‐selective inhibitor fedratinib (SAR302503): results from two phase I studies in healthy volunteers. Clin Pharmacol Drug Dev. 2015;4(4):315–321.
- Ogasawara K, Wood-Horrall RN, Thomas M, et al. Impact of fedratinib on the pharmacokinetics of transporter probe substrates using a cocktail approach. Cancer Chemother Pharmacol. 2021;88(6):941–952.
- Ogasawara K, Zhou S, Krishna G, et al. Population pharmacokinetics of fedratinib in patients with myelofibrosis, polycythemia vera, and essential thrombocythemia. Cancer Chemother Pharmacol. 2019;84(4):891–898.
- Ogasawara K, Xu C, Kanamaluru V, et al. Excretion balance and pharmacokinetics following a single oral dose of [14C]-fedratinib in healthy subjects. Cancer Chemother Pharmacol. 2020;86(2):307–314.
- Pardanani A, Gotlib JR, Jamieson C, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J clin oncol. 2011;29(7):789.
- Gotlib J, Pardanani A, Jamieson C, et al. Long-term follow up of a phase 1/2 study of SAR302503, an oral JAK2 selective inhibitor, in patients with myelofibrosis (MF), Haematologica. 2012, Ferrata Storti Foundation Via Giuseppe Belli; Vol. 4:27100. Pavia Italy
- Pardanani A, Tefferi A, Jamieson C, et al. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J. 2015;5(8):e335–e.
- Pardanani A, Harrison C, Cortes JE, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1(5):643–51.
- Harrison CN, Schaap N, Vannucchi AM, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (Jakarta-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017;4(7):e317–e24.
- Harrison CN, Schaap N, Vannucchi AM, et al. Fedratinib in patients with myelofibrosis previously treated with ruxolitinib: an updated analysis of the JAKARTA2 study using stringent criteria for ruxolitinib failure. Am J Hematol. 2020;95(6):594–603.
- Harrison CN, Schaap N, Vannucchi AM, et al. Fedratinib induces spleen responses and reduces symptom burden in patients with myeloproliferative neoplasm (MPN)-associated myelofibrosis (MF) and low platelet counts, who were either ruxolitinib-naïve or were previously treated with ruxolitinib. Blood. 2019;134(Supplement_1):668.
- Kiladjian -J-J, Tefferi A, Passamonti F, et al. P1041: impact of fedratinib on spleen volume and myelofibrosis symptoms in patients with substantial splenomegaly: post hoc analyses from the Jakarta and jakarta2 trials. HemaSphere. 2022;6:931–932.
- Harrison C, Kiladjian J, Verstovsek S, et al. Overall and progression-free survival in patients treated with fedratinib as first-line myelofibrosis (MF) therapy and after prior ruxolitinib (RUX): results from the Jakarta and JAKARTA2 trials. Hemasphere. 2021;5:S203.
- Verstovsek S, Harrison CN, Barosi G. FREEDOM: aphase 3b efficacy and safety study of fedratinib in intermediate-or high-risk myelofibrosis patients previously treated with ruxolitinib. Journal of Clinical Oncology. 2019;37:15. DOI:10.1200/JCO.2019.37.15_suppl.TPS7072
- Vianelli N, Benevolo G, Vannucchi A, et al. A randomized, Phase 3 trial of fedratinib versus best available therapy in patients with Intermediate-2 or High-Risk myelofibrosis previously treated with ruxolitinib (FREEDOM2). Blood. 2021;138(Supplement 1):3643.
- Harrison CN, Mascarenhas J, Abraham P, et al. Real-World utilization of fedratinib for myelofibrosis Post-Ruxolitinib: patient characteristics, treatment patterns, and characterization of ruxolitinib failure. Blood. 2021;138(Supplement 1):3059.
- Mascarenhas J, Gerds A, Kish J, et al. P1060: Real-World clinical outcomes after 3 and 6 months of treatment with fedratinib following ruxolitinib failure. HemaSphere. 2022;6:950–951.
- Passamonti F, Lou Y, Chevli M, et al. P1058: REAL-WORLD OUTCOMES WITH FEDRATINIB THERAPY IN PATIENTS WITH PRIMARY MYELOFIBROSIS POST-RUXOLITINIB DISCONTINUATION. HemaSphere. 2022;6:948–949.
- Gupta V, Yacoub A, Verstovsek S, et al. Safety and Tolerability of Fedratinib (FEDR), an Oral Inhibitor of Janus Kinase 2 (JAK2), in Patients with Intermediate-or High-Risk Myelofibrosis (MF) Previously Treated with Ruxolitinib (RUX): results from the Phase 3b FREEDOM Trial. Blood. 2021;138(Supplement 1):389.
- Sinha S, Kataria A, Kolla BP, Thusius N, Loukianova LL. Wernicke encephalopathy—clinical pearls. Mayo Clinic ProceedingsElsevier. 2019; Vol. 94, No. 6:1065–1072. DOI:10.1016/j.mayocp.2019.02.018
- Harrison CN, Mesa RA, Jamieson C, et al. Case series of potential Wernicke’s encephalopathy in patients treated with fedratinib. Washington DC: American Society of Hematology; 2017.
- Yin H, Xu Q, Cao Y, et al. Nonalcoholic Wernicke’s encephalopathy: a retrospective study of 17 cases. J Int Med Res. 2019;47(10):4886–4894.
- Curto‐García N, Harrison CN, McLornan DP, et al. Thiamine deficiency appears uncommon in patients with myeloproliferative neoplasms. Br J Haematol. 2017;178(2):338–340.
- Gangat N, Phelps A, Lasho TL, et al. A prospective evaluation of vitamin B1 (thiamine) level in myeloproliferative neoplasms: clinical correlations and impact of JAK2 inhibitor therapy. Blood Cancer J. 2019;9(2):1–4.
- Hood J, Hazell A. Fedratinib does not inhibit thiamine uptake or induce experimental Wernicke’s encephalopathy in nonclinical studies. Blood. 2017;130:4993.
- Zhang Q, Zhang Y, Diamond S, et al. The Janus kinase 2 inhibitor fedratinib inhibits thiamine uptake: a putative mechanism for the onset of Wernicke’s encephalopathy. Drug Metab Dispos. 2014;42(10):1656–1662.
- Tang D, Taneja A, Smith S, et al. MPN-115: indirect treatment comparisons (ITCs) of the effect of fedratinib versus ruxolitinib (RUX) on spleen volume for patients with myelofibrosis (MF) and no prior RUX treatment. Clin Lymphoma Myeloma Leukemia. 2020;20:S329.
- Team R. Fedratinib (Inrebic). Canadian Journal of Health Technologies. 2021;1(6). DOI: 10.51731/cjht.2021.86.
- Coltro G, Rotunno G, Mannelli L, et al. RAS/CBL mutations predict resistance to JAK inhibitors in myelofibrosis and are associated with poor prognostic features. Blood Adv. 2020;4(15):3677–3687.
- Maffioli M, Mora B, Ball S, et al. A prognostic model to predict survival after 6 months of ruxolitinib in patients with myelofibrosis. Blood Adv. 2022;6(6):1855–1864.
- Palandri F, Bartoletti D, Iurlo A, et al. Peripheral blasts are associated with responses to ruxolitinib and outcomes in patients with chronic‐phase myelofibrosis. Cancer. 2022;128(13):2449–2454.
- Spiegel JY, McNamara C, Kennedy JA, et al. Impact of genomic alterations on outcomes in myelofibrosis patients undergoing JAK1/2 inhibitor therapy. Blood Adv. 2017;1(20):1729–1738.
- Palandri F, Palumbo GA, Elli EM, et al. Ruxolitinib discontinuation syndrome: incidence, risk factors, and management in 251 patients with myelofibrosis. Blood Cancer J. 2021;11(1):1–4.