
ABSTRACT
Introduction
Janus kinase (JAK) inhibitors are an emerging class of small-molecule drugs, providing targeted therapy for a variety of diseases, and have made their way into the treatment of armamentarium of ulcerative colitis (UC) in recent years.
Areas covered
This review focuses on the pharmacokinetics, safety, and efficacy of selective JAK1 inhibitors in the treatment of moderate-to-severe UC. The PubMed database and clinicaltrials.gov were consulted using keywords – further expanded in the methods section. The search was focused on full-text publications in English. No publication date restrictions were imposed.
Expert opinion
JAK1 inhibitors are small-molecule drugs used in the treatment of ulcerative colitis and other immune mediated inflammatory diseases. They are orally bioavailable and have a rapid mechanism of action and no immunogenicity. JAK inhibitors can be used for the management of both naïve patients and biological-experienced patients.
Particular attention should be paid to elderly patients or those with cardiovascular or oncological risk factors, in whom JAK inhibitors should be recommended only if no alternatives are available. In addition, JAK inhibitors have the potential to be combined with other biological drugs or small molecules for the management of difficult-to-treat cases.
1. Introduction
Ulcerative colitis (UC) is a chronic inflammatory bowel disease (IBD) characterized by relapsing and remitting mucosal inflammation [Citation1]. Mucosal inflammation typically begins in the rectum, and it extends to proximal segments of the colon in a continuous uninterrupted pattern involving part of, or the entire colon [Citation2]. It is estimated that 6,8 million people are living with IBD worldwide [Citation3,Citation4]. Many cytokines and pro-inflammatory pathways play a significant role in the development of inflammation in UC [Citation5].
Current treatments for UC consist of ‘conventional’ medications, biologics, JAK inhibitors, and a S1P receptor modulator – ozanimod, which is currently the only one of its class approved for the treatment of UC in Europe and the United States (US) [Citation6,Citation7]. However, many UC patients do not respond to currently available treatments. During anti-TNF induction therapy, primary failure occurs in 19–58% of patients in clinical trials, and rates of secondary failure (loss of response) are approximately 17–22% [Citation8]. Despite the recognized efficacy of biological agents, roughly more than 50% of patients with moderate-to-severe UC do not have sustained remission [Citation9]. UC treatment non-adherence rates were recently reported to range from 7% to 72%, with most studies reporting 30–45% of patients being non-adherent to treatment [Citation10]. Non-adherence is associated with an increased risk for relapse [Citation11]. Biologics are encumbered by their large molecular structure and must be administered parenterally (e.g. intravenous or subcutaneous) which poses a burden, since patients are required to visit the clinic frequently for maintenance therapy [Citation12]. The treatment possibilities of UC have been changing in recent years, one of them being the introduction of tofacitinib. Tofacitinib was first approved for the treatment of rheumatoid arthritis and psoriatic arthritis by the Food and Drug administration (FDA) [Citation13]. Following three successful phase III studies (OCTAVE), the indication for tofacitinib was further extended by the FDA to include adults with moderately to severely active ulcerative colitis [Citation14,Citation15]. Tofacitinib has been approved by EMA for the treatment of ulcerative colitis as well [Citation16,Citation17]. Tofacitinib is a small-molecule drug with a short half-life (3 h), and it is rapidly absorbed following oral administration (time to peak concentration 0,5 h) [Citation18]. Additionally, tofacitinib is not a monoclonal antibody, and therefore has the benefit of no immunogenicity risk [Citation19]. Tofacitinib is a pan-JAK inhibitor as it has moderate activity against JAK2 and TYK2, in addition to JAK1 and JAK3 [Citation20]. Cytopenias have been observed with JAK2 inhibition, as many hematopoietic growth factors (including erythropoietin, and granulocyte macrophage-colony stimulating factor) are dependent on JAK2 mediated signal transduction [Citation21]. Further, recent reports have raised safety concerns regarding the use of tofacitinib. RA patients being treated with tofacitinib 10 mg were at an increased risk for blood clots in the lungs and death [Citation22,Citation23]. Recent data from a noninferiority safety trial demonstrated that tofacitinib carried a higher risk of major adverse cardiovascular events (MACE) and cancer in comparison to patients receiving a TNF inhibitor [Citation24]. Due to the aforementioned points, nonselective pan-JAK inhibition could be associated with unwanted adverse effects and highlights the need for second-generation JAK inhibitors with selective inhibitory activity against specific JAKs [Citation21]. Filgotinib is a preferential JAK1 inhibitor primarily approved for the treatment of RA in Japan and Europe [Citation25,Citation26]. Filgotinib was subsequently evaluated for the treatment of UC patients [Citation27,Citation28], and approved for the treatment of UC in Europe and Japan [Citation25,Citation29]. Upadacitinib is another selective JAK1 inhibitor approved in the United States and Europe for the treatment of moderate-to-severe UC [Citation30,Citation31]. Another JAK1 inhibitor for the treatment of UC, being investigated is Ivarmacitinib (formerly SHR0302) [Citation32]. This review aims to report the advances in pharmacotherapy in ulcerative colitis with a focus in JAK1 inhibitors – Filgotinib, Upadacitinib, and Ivarmacitinib.
1.1. Methods
The PubMed database and clinicaltrials.gov were consulted using the following search terms: ‘JAK,’ ‘JAK inhibitor,’ ‘Janus Kinases,’ ‘Tofacitinib,’ ‘Filgotinib,’ ‘Upadacitinib,’ ‘Ivarmacitinib,’ ‘SHR0302’ individually or in combination with ‘IBD,’ ‘UC,’ ‘Ulcerative colitis,’ ‘inhibitors,’ ‘safety,’ ‘efficacy,’ ‘study,’ ‘trial.’ The search was focused on full-text papers published in English; no publication date restrictions were imposed.
1.2. JAK/STAT pathway
JAK inhibitors elicit their effect upon the JAK/STAT pathway (). JAKs belong to a large family of tyrosine kinases located intracellularly, and phosphorylate activated cytokine receptors leading to signal transduction [Citation33,Citation34]. So far four proteins have been identified in the JAK family: JAK1, JAK2, JAK3, and tyrosine-protein kinase 2 (TYK2) [Citation33,Citation34]. More than 50 cytokines and growth factors have been identified in this pathway, such as hormones, interferons, interleukins, and colony-stimulating factors [Citation35]. Cytokines bind to their specific receptors on the surface of target cells, leading to a conformational change of said receptor allowing JAK phosphorylation. Receptor activation via JAKs allows members of the signal transducer and activator of transcription (STAT) protein family to bind to the receptor. STATs become phosphorylated and form dimers, that then dissociate from the receptor and make their way into the nucleus to regulate gene transcription [Citation33,Citation35]. JAK/STAT mediated gene regulation leads to a variety of events, including hematopoiesis, immunoregulation, inflammation, tissue repair, apoptosis, and adipogenesis [Citation35,Citation36].
JAK1 is widely expressed in tissues and can phosphorylate all STATs [Citation35]. JAK1 is activated by multiple cytokine-receptor families. The type I cytokine receptor common γ-chain (γc) is associated with the JAK1 and JAK3 heterodimer, cytokines that bind to the γc receptor subunit are: IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 [Citation37]. Interferons bind to type II cytokine receptors, Interferon-γ (IFN-γ) needs JAK1 and JAK2, whereas interferon-α (IFN-α) and interferon-β (IFN-β) require JAK1 and TYK2 signaling [Citation37]. Cytokines from the IL-10 family are dependent on type II cytokine receptors and share common signaling pathways (JAK1, JAK2, TYK2) [Citation37]. Cytokines binding to the type I receptor common glycoprotein 130 (gp130) mediate their signal through JAK1, JAK2, TYK2 [Citation37]. Cytokines that bind to gp130 are IL-6, IL-11, IL-13, leukemia inhibitory factor (LIF), oncostatin M (OSM), and ciliary neurotrophic factor (CNTF) [Citation35,Citation37]. Inhibition of JAK-mediated inflammatory pathways can regulate the innate and adaptive immune responses, and thereby reduce chronic intestinal inflammation in UC [Citation38]. IL-6, IL-12, and IL-23 cytokines are important drivers of inflammation in IBD [Citation38]. JAK1 activation is involved in the signal transduction of IL-6, IFN, and γc cytokines [Citation33]. It is unclear if JAK1-inhibitor mediated suppression of IFN has therapeutic benefit. However, IFN plays a key role in antiviral immunity, which could explain the increased incidence of herpes zoster reactivation in tofacitinib studies [Citation24,Citation39,Citation40]. JAK2 inhibition is potentially related to undesirable adverse events, specifically suppression of hematopoiesis via erythropoietin, IL-3 and IL-5 inhibition [Citation33]. JAK1 selective molecules thereby aim to potentially ameliorate this issue by not inhibiting JAK2-dependent pathways.
2. Filgotinib
The safety and efficacy of filgotinib has been examined for multiple diseases. Studies have been conducted in patients, with active ankylosing spondylitis – TORTUGA, active psoriatic arthritis – EQUATOR, rheumatoid arthritis – DARWIN 1, DARWIN 2, DARWIN 3, Crohn’s disease – FITZROY and patients with ulcerative colitis – SELECTION [Citation41–46].
2.1. Filgotinib – pharmacokinetics
Filgotinib is orally bioavailable, and demonstrates rapid absorption, with maximum plasma concentration (Cmax) reached within 1–3-h postdose [Citation47]. Filgotinib exposure (both (Cmax) and area under the plasma concentration-time curve AUC) increases proportionally with dose (50–200 mg) increase [Citation47]. Inter-subject variability is low to moderate. Filgotinib’s primary metabolite is GS-829845, and is detected within 30 min in the patient's plasma, reaching a maximum plasma concentration in 3–8-h postdose [Citation47]. Primary metabolite exposure is on average 16 to 20-times higher than filgotinib exposure. It has a similar JAK1 selectivity; however, its potency is 10-fold lower in comparison to its parent compound. Filgotinib is available commercially in highly soluble maleate salt tablet formulation. Depending on a patient’s food intake, a slight delay in absorption can be seen (2–3 h) in comparison to fasting patients. Food intake, however, does not significantly affect Cmax and AUC of neither filgotinib or its primary metabolite. Both compounds have a low binding affinity to plasmatic proteins in humans (<60%). Steady-state concentration in plasma is reached for filgotinib by day 2 and for its primary metabolite by day 4. Filgotinib has a half-life of 4,9 to 10,7 h, GS-829845 has a half-life ranging between 19,6 and 27,3 h [Citation47]. Protein binding of filgotinib in humans is 55–59% [Citation47]. Carboxylesterases are the enzymes responsible for the formation of the primary metabolite. Following the administration of [14C]-filgotinib to healthy patients, the primary metabolite accounts for approximately 92% of the total radioactivity in plasma [Citation47]. The primary metabolite and its N-glucuronide derivative account for 68,6% of the total radioactivity in urine. Filgotinib and its metabolite are mainly eliminated via urine (86,9%). Filgotinib has low interaction potential with other drugs, without clinically significant interactions with commonly administered comedications [Citation47].
2.2. Efficacy in ulcerative colitis
The induction and maintenance efficacy of filgotinib was assessed in a phase IIb/III, double-blind, randomized, and placebo-controlled trial (SELECTION) [Citation45,Citation46]. The study design utilized two induction studies (biologic naïve – A and biologic experienced – B patients with UC) and one maintenance study [Citation45]. Patients aged 18–75 years with moderate-to-severe UC in accordance with Mayo Clinic Score (6–12) for at least 6 months were eligible for enrollment. Patients were stratified based on the use of corticosteroids and immunosuppressants in induction study A. Patients in induction study B were stratified by the same factors as the prior group as well as their previous exposure to 1 or more than 1 biologics. Patients in the aforementioned induction studies were randomized to receive 200 mg filgotinib, 100 mg filgotinib or placebo in a 2:2:1 ratio, respectively [Citation45]. Patients who had either clinical remission or Mayo Clinic Score (MCS) defined as response at week 10 were rerandomized to continue their filgotinib regimen or receive placebo in a 2:1 ratio, respectively, until week 58 [Citation45]. Placebo responders continued to receive placebo during the maintenance study. Patients who did not achieve clinical remission or MCS-defined response at week 10 had the option of joining the separate long-term extension study (SELECTION LTE) [Citation48]. Patients who encountered disease worsening criteria during the maintenance study were discontinued and offered open-label filgotinib in SELECTION LTE [Citation45]. Patients that completed week 58 were eligible for the long-term extension study as well [Citation45]. Clinical remission was set as the primary endpoint of week 10 and week 58 of the study. Clinical remission was defined as having a Mayo endoscopic subscore of 0 or 1, rectal bleeding subscore of 0, and at least a 1-point decrease in stool frequency from induction baseline for a subscore of 0 or 1. Secondary endpoints were MCS remission, endoscopic remission, histologic remission, and MCS remission (alternative definition) at week 10 and week 58 [Citation45]. MCS remission was defined as a total MCS of ≤2 with no single subscore higher than 1. The alternative definition of MCS was described as rectal bleeding, stool frequency, and physician’s global assessment subscores of 0 and an endoscopic subscore of 0 or 1; overall MCS of 1 or 0 [Citation45]. Endoscopic remission was defined as a Mayo endoscopic subscore of 0. Histologic remission was based on the Geboes scale [Citation49], defined as no or mild increase in chronic inflammatory infiltrate in the lamina propria, no neutrophils in the lamina propria or epithelium, and no erosion, ulceration, or granulation tissue [Citation45].
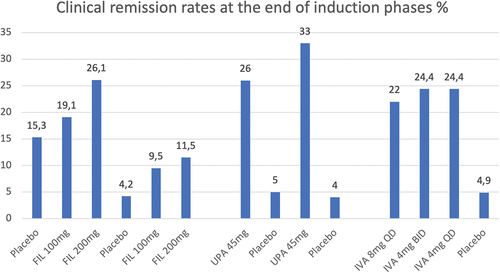
The efficacy of filgotinib during the induction phase was dose-dependent for both the biologic naïve and biologic experienced patient groups. In biologic-naïve group A (consisting of 659 participants), patients receiving filgotinib of 200 mg daily were significantly more likely to achieve clinical remission (absolute difference 10,8%, p = 0,0157). A greater proportion in endoscopic remission was seen in this group as well (absolute difference 8,6% p = 0,0047). Histologic remission was observed in over one-third of patients (35,1%) receiving daily filgotinib 200 mg (absolute difference 19,0%, p < 0,0001) [Citation32]. The MCS remission was also significant (absolute difference 12,1%, p = 0,0053). In biologic-experienced group B (consisting of 689 participants), patients receiving filgotinib 200 mg daily reached clinical remission in greater proportion in comparison to patients receiving placebo (absolute difference 7,2% p = 0,0103) as well as histologic remission (absolute difference 11,4%, p = 0,0019). MCS remission and endoscopic remission were not statistically significant in group B patients receiving filgotinib 200 mg during the induction phase. The differences in clinical remission or any of the secondary endpoints between patients on 100 mg filgotinib versus placebo were not statistically significant in either induction study.
The maintenance study enrolled 664 patients. During its course, 263 patients discontinued treatment, 401 patients completed the maintenance study to week 58. Filgotinib 200 mg group demonstrated superiority over placebo in achieving clinical remission (absolute difference 26.0%, p < 0,0001), corticosteroid-free remission (absolute difference 20,8%, p = 0,0055), sustained clinical remission (absolute difference 13,0%, p = 0,0024), MCS remission (absolute difference 25,5%, p < 0,0001), endoscopic remission (absolute difference 9,5%, p = 0,0157) and histological remission (absolute difference 24,9%, p < 0,0001) at week 58. Patients in the filgotinib 100 mg group had a greater rate of clinical remission in comparison to the placebo group at week 58 (absolute difference 10,4%, p = 0,0420) The treatment effect of filgotinib 200 mg in comparison to placebo in both the biologic naïve and experienced group was consistent in all key secondary endpoints [Citation45].
2.3. Safety in ulcerative colitis
In all three studies, most adverse events were mild to moderate in severity (). The distribution of treatment-emergent adverse events during the induction studies was similar in all groups (placebo − 56,3%, filgotinib 100 mg − 50,4%, and filgotinib 200 mg − 56,3%) [Citation45]. Adverse events were present in a similar proportion in all three groups during the maintenance study as well. The most frequent adverse events during the induction studies were nasopharyngitis, headache, and ulcerative colitis [Citation45]. The most frequent adverse events in the maintenance study were nasopharyngitis, arthralgia, headache, abdominal pain, and upper respiratory tract infections. Four patients during the induction studies and two patients in the maintenance study developed Herpes zoster infections, however they were not serious or complicated and did not result in discontinuation from the trial [Citation45]. One patient receiving filgotinib 200 mg during induction study A had an opportunistic infection – esophageal candidiasis, which resolved with treatment. One patient in the filgotinib 200 mg arm with a medical history of hypothyroidism, idiopathic pulmonary symptoms, and taking prednisone, had a pulmonary embolism during induction study B. Neither venous thrombosis or pulmonary embolism occurred in patients receiving filgotinib in the maintenance study. During induction studies, a small increase in total cholesterol, low-density lipoproteins and high-density lipoproteins was observed in patients receiving filgotinib. During the maintenance study lipid levels remained stable. Creatine kinase (CK) was elevated in patients receiving filgotinib in all three studies – mostly in patients of the maintenance study receiving filgotinib 200 mg (4%, n = 8). However, no rhabdomyolysis associated with increased CK was reported. Two patients died during the maintenance study, following investigator evaluation they were considered nonrelated to the treatment [Citation45]. Filgotinib 200 mg and 100 mg was well tolerated, and the incidence rate of adverse events was not increased in comparison to placebo [Citation45].
Table 1. Safety analysis data from Filgotinib studies. EAIR/100 PYE – Exposure-adjusted incidence rate per 100 patient-years of exposure, applies to DARWIN 3 only. AE, adverse events; SAE, serious adverse events; SI, serious infections; HZV, herpes zoster virus; VT, venous thrombosis; PE, pulmonary embolism; NMSC, non-melanoma skin cancer; Total, Total patients.
2.4. Safety in other diseases
The TORTUGA trial – a randomized, double-blind, placebo-controlled phase 2 study, demonstrated the efficacy and safety of filgotinib 200 mg in patients with active ankylosing spondylitis [Citation41]. Overall filgotinib was well tolerated, and adverse events were mostly mild-to-moderate. The proportion of treatment-emergent adverse events was equal for both the filgotinib and placebo group, the most common being nasopharyngitis. Among the serious treatment-emergent adverse events, grade 3 pneumonia was reported in a smoker in the filgotinib group, which led to treatment discontinuation. She recovered after antibiotic treatment. The other was a grade 2 deep vein thrombosis in a patient with a factor V Leiden heterozygous mutation three days following his last dose of filgotinib. No other potential class-related events were reported. Reports of infections did not differ between groups (both groups 12%) [Citation41].
The EQUATOR trial was a phase 2 study that showed 80% of patients in the filgotinib group met the primary endpoint of 20% improvement in ACR20 at week 16, compared to 33% in the placebo group (treatment difference 47%, p < 0,0001) [Citation42]. The distribution of treatment-emergent adverse events was similar across both groups (57% for filgotinib, 59% for placebo), most were mild to moderate in severity. One patient receiving filgotinib had pneumonia (onset day 106) with a fatal outcome at day 107 of the trial. Another patient in the filgotinib arm had a herpes zoster infection confined to one dermatome. The rest of treatment-emergent adverse events of interest in the filgotinib group was comprised of one urinary tract infection and respiratory tract infections. No other events of interest within the filgotinib arm were reported [Citation42].
The efficacy and safety of filgotinib for the treatment of rheumatoid arthritis was assessed in two randomized, dose-finding, phase IIb studies – DARWIN 1 and 2, and one ongoing long-term open-label extension study – DARWIN 3. Filgotinib was administered in combination with methotrexate (MTX) during the DARWIN 1 trial. In DARWIN 2 filgotinib was given as monotherapy. Treatment emergent adverse event (TEAE) frequency during DARWIN 1 was similar across all dose groups. TEAE considered related to study treatment occurred more frequently among the arms that continued to receive filgotinib (avg. 20,9%) than compared to placebo (10,7%) [Citation50]. Fifteen patients had more than 1 serious TEAE, of which one patient in the 100 mg twice daily group died as a result of pneumonia and septic shock. Herpes zoster infections were observed in one patient on placebo and four in the arm receiving filgotinib. During DARWIN 1, filgotinib as add-on to methotrexate was generally well tolerated and associated with rapid onset of action. In DARWIN 2 treatment, emergent adverse event frequency was similar across all dose groups ranging from 32,9% to 43,5% up to week 12 [Citation51]. Up to week 24 of the study 9 serious treatment emergent adverse events occurred of which 7 were in the treatment arms. Three occurred in the filgotinib 200 mg group – pneumonia, back pain, osteoarthritis, two in the filgotinib 100 mg group – cellulitis, vertigo, and two in the filgotinib 50 mg group – gastroenteritis, humeral fracture. In the placebo group, two serious TEAE were reported – worsening RA (prior to switching to filgotinib) and chronic pyelonephritis. There was one case of herpes zoster infection that occurred during week 12–24 in the filgotinib 50 mg group, which resolved after 10 days. Filgotinib as monotherapy was efficacious and generally well tolerated over the 24-week course of the study [Citation51].
The majority of patients (83%) from DARWIN1 and (85%) and DARWIN2 entered this open-label, long-term extension study [Citation43]. Over the 4-year period of the study, filgotinib demonstrated a safety profile comparable with the parent studies and a sustained efficacy when administered as 200 mg daily either as monotherapy or in combination with methotrexate [Citation43].
FITZROY [Citation44] was a randomized, double-blind, placebo-controlled phase 2 study to assess the efficacy and safety of filgotinib in patients with moderate-to-severe Crohn’s disease. In the pooled safety analysis at week 20, the proportion of patients experiencing at least 1 treatment emergent adverse event was similar in both the filgotinib receiving group (75%) and the placebo receiving group (67%) [Citation44]. Severe TEAE frequency was 9% for the filgotinib group and 4% for the placebo group [Citation44]. Serious infections were reported in 3% of the filgotinib group and none in the placebo arm. During the FITZROY trial, filgotinib had an acceptable safety profile and demonstrated significant efficacy in achieving clinical remission [Citation44].
Two safety studies examining the effect of filgotinib on sperm parameters were conducted, due to safety concerns regarding testicular toxicity. They were conducted in patients with IBD (MANTA, NCT03201445) or RA and other inflammatory arthropathies (MANTA-Ray, NCT03926195) [Citation52,Citation53]. Results of both studies concluded that filgotinib treatment is not associated with negative effects on testicular function [Citation54].
3. Upadacitinib
3.1. Upadacitinib pharmacokinetics
In terms of pharmacokinetics, upadacitinib immediate release (IR) formulation over a dose range of 1–48 mg demonstrated rapid absorption, with a median time to maximum plasma concentration of approximately 1 h under fasting conditions. After reaching maximum plasma concentration, upadacitinib plasma concentrations declined in a biphasic manner with a terminal elimination half-life of approximately 6–15 h for most doses [Citation55]. The upadacitinib maximum plasma concentration was dose proportional in the 1–48 mg dose range. The percentage of upadacitinib that was excreted via urine in its unchanged form ranged from 16% to 21% [Citation55]. A twice-daily (BID) IR formulation was tested as well and showed no accumulation at steady state BID dosing. Since once-daily (QD) is more ideal for patients an extended release (ER) formulation was developed. Following ER administration, the median time to maximum plasma concentration was 2–3 h under fasting conditions and 4 h under non-fasting conditions [Citation55]. Steady state was achieved by day 4 with ER formulation. The upadacitinib harmonic mean terminal half-life after ER administration ranged from 9 to 14 hours. Based on the analysis of phase 1 studies, the upadacitinib ER formulation had a bioavailability of 76% in comparison to the same dose ranges of IR formulation [Citation55].
3.2. Efficacy in ulcerative colitis
The induction and maintenance efficacy of upadacitinib was assessed in a phase 3, multicentre, randomized, double-blind, placebo-controlled clinical trial. Constructed of two induction studies – U-ACHIEVE induction (UC1) and U-ACCOMPLISH (UC2) and one maintenance study U-ACHIEVE maintenance (UC3) [Citation56]. Patients aged 16–75, with moderate-to-severe active ulcerative colitis (adapted Mayo score 5–9, endoscopic subscore 2 or 3) for at least 90 days met the inclusion criteria for the induction studies and were randomly assigned (2:1) to oral upadacitinib 45 mg or placebo once daily for a duration of 8 weeks. Patients had a history of inadequate response, loss of response or intolerance to conventional or biologic therapy (infliximab, adalimumab, golimumab, vedolizumab, or ustekinumab). Patients with at least 3 and more biologic treatment failures were limited to less than 30% of total population. Patients with prior biologic exposure who discontinued due to other reasons than inadequate response or intolerance were limited to 20% of the total population provided their exposure was less than 1 year. An 8 week and 12-week washout period were required for patients with previous use of anti-TNF or vedolizumab, and ustekinumab, respectively. Patients were stratified based on previous biologic treatment failure (yes vs. no), baseline corticosteroid use (yes vs. no), baseline adapted Mayo score (≤7 vs.>7), previous amount of biologic treatment received (1 vs.>1). Patients receiving upadacitinib 45 mg that achieved clinical response by week 8 were randomly assigned (1:1:1) to receive 15 mg or 30 mg upadacitinib or placebo in the UC3 maintenance study for 52 weeks [Citation56]. Placebo responders continued to receive placebo during the maintenance study. Concomitant UC-related medications were kept at a stable dose, biologics and immunosuppressants other than methotrexate were prohibited. At week 0 during the maintenance study, corticosteroid dose tapering was commenced in accordance with the predefined schedule [Citation56]. The primary endpoint was clinical remission defined as an adapted Mayo score ≤ 2, with a stool frequency score (SFS) ≤1 and no greater at baseline, rectal bleeding score (RBS) = 0 and endoscopic subscore ≤ 1 without friability. Secondary endpoints were endoscopic improvement, endoscopic remission, clinical response per adapted Mayo score, clinical response per partial adapted Mayo score, histological-endoscopic mucosal improvement (HEMI), no bowel urgency, no abdominal pain, histological improvement, mucosal healing, and change from baseline in two patient QoL questionnaires (IBDQ and FACIT-F) [Citation56]. In the UC1 induction study at week 8, clinical remission was achieved by 26% of patients receiving upadacitinib versus 5% of patients receiving placebo (adjusted treatment difference 21,6%, p < 0,0001). In the UC2 induction study at week 8, clinical remission was achieved by 33% of patients receiving upadacitinib versus 4% of patients receiving placebo (adjusted treatment difference 29,0%, p < 0,0001) [Citation56]. The rate of clinical remission at week 8 was consistent among all subgroups, including patients with prior biologic treatment failure. All secondary endpoints were met in both induction studies for patients receiving upadacitinib 45 mg vs patients receiving placebo. The proportion of clinical responders receiving upadacitinib was significantly greater than those receiving placebo in both UC1 (60% vs 27%, p < 0,0001) and UC2 (63% vs 26%, p < 0,0001) induction trials. Further, more upadacitinib treated patients demonstrated fecal calprotectin levels below 150 mg/kg at week 2 and 8 of induction, consistent with clinical and endoscopic status.
In the maintenance study, UC3 clinical remission was achieved by 42% of patients receiving 15 mg upadacitinib daily, 52% of patients receiving 30 mg upadacitinib daily and 12% of patients in the placebo arm [Citation56]. Adjusted treatment difference of 30,7% for 15 mg upadacitinib vs placebo and adjusted treatment difference of 39,0% for 30 mg upadacitinib vs placebo were found, both p < 0,0001. The rate of clinical remission at week 52 was consistent among all subgroups, including patients with prior biologic treatment failure. All secondary endpoints were achieved in both upadacitinib arms (15 mg and 30 mg) vs patients receiving placebo. Corticosteroid-free remission was reached in 57% of upadacitinib 15 mg group, 68% of upadacitinib 30 mg group and 22% of the placebo group p < 0,0001 [Citation56]. Consistent with clinical and endoscopic status fecal calprotectin levels below 150 mg/kg were more prominent in patients treated with upadacitinib.
3.3. Safety in ulcerative colitis
In the UC1 induction trial, the proportion of adverse effects reported was 56% in the upadacitinib 45 mg group and 60% in the placebo group (). In the UC2 induction trial, the proportions were higher in the upadacitinib 45 mg group − 53% in comparison to the group receiving placebo − 40% [Citation56]. The most common reported adverse event in UC1 was nasopharyngitis followed by creatine phosphokinase elevation and acne. The most common reported adverse event in UC2 was acne. The incidence of serious adverse events and adverse events leading to discontinuation of treatment was lower in the upadacitinib 45 mg group in comparison to the placebo group (serious adverse events in UC1–3% vs 6%, UC2–3% vs 5%, adverse events leading to discontinuation of treatment UC1–2% vs 9%, UC2–2% vs 5%) [Citation56]. Adverse events of special interest were scarce in both induction studies. No active tuberculosis, renal dysfunction, cancer, or major cardiovascular events were observed. One patient from the UC1 upadacitinib group had a herpes zoster reactivation, which led to treatment discontinuation, followed by post-herpes zoster virus neuralgia. Another in the UC1 upadacitinib group had an opportunistic oral fungal infection. One patient in the UC2 upadacitinib group had a herpes zoster reactivation involving one dermatome, another had an opportunistic cytomegalovirus infection leading to CMV colitis – both were considered non serious and did not lead to treatment discontinuation.
Table 2. Safety analysis data from upadacitinib studies. AE, adverse events; SAE, serious adverse events; SI, serious infections; HZV, herpes zoster virus; VT, venous thrombosis; PE, pulmonary embolism; NMSC, non-melanoma skin cancer; Total, Total patients.
In the maintenance study UC3 adverse events were reported in similar proportion across all groups, in the upadacitinib 15 mg group − 78%, in the upadacitinib 30 mg − 79% and in the placebo group − 76%. The incidence of serious adverse events and adverse events leading to discontinuation of treatment was lower in both upadacitinib groups in comparison to the placebo group. There were six events of herpes zoster reactivation each involving one dermatome, of which one led to treatment discontinuation. One non-serious opportunistic cytomegalovirus infection. Invasive breast cancer was reported in one patient of the placebo and one patient of the 15 mg upadacitinib group. Colon cancer and prostate cancer were both reported once in the upadacitinib 30 mg group. Other two patients in the 30 mg upadacitinib group had non-melanoma skin cancer. Hepatic disorders were more commonly reported in upadacitinib groups, however all were mild to moderate and the majority (86%) were transaminase elevations [Citation56]. Total cholesterol concentration was elevated in patients receiving upadacitinib across all three studies (UC1, UC2, UC3), although the LDL and HDL ratio remained stable. In a multivariate regression analysis of U-ACHIEVE and U-ACCOMPLISH studies patients receiving upadacitinib 45 mg were significantly more likely to achieve improvement of daily symptoms, demonstrated by SFS≤1 and RBS of 0. A greater proportion in this group also achieved abdominal pain = 0 and absence of bowel urgency within day 3 of treatment. The analysis further revealed that patients who reached SFS≤1 or lacking bowel urgency by day 7 were more likely to achieve clinical remission at week 8 [Citation57].
3.4. Safety in other diseases
The CELEST trial, a randomized, double-blind, phase 2 study, evaluated the efficacy and safety of upadacitinib in patients with Crohn’s disease (CD). Overall, nine patients receiving UPA during the induction phase developed serious infections, and six serious infections were observed during the maintenance phase. Three events of Herpes zoster in the active treatment arms were reported during the entire study, all resolved with antiviral therapy. One non-serious event of non-melanoma skin cancer (NMSC) and two malignancies occurred in the active treatment arms. During induction, two acute, serious intestinal perforations were reported, both in patients receiving upadacitinib. No events of deep vein thrombosis or pulmonary embolism occurred during the study [Citation58].
The safety and efficacy of upadacitinib was examined for atopic dermatitis as well in a completed trial (ref NCT02925117). The results are reported from the analysis from week 16. In the upadacitinib 7,5 mg group, two patients had serious adverse events – lower jaw pericoronitis and worsening atopic dermatitis following contact dermatitis. In the upadacitinib 15 mg group, one serious adverse event of appendicitis was reported. No serious adverse events were reported in the upadacitinib 30 mg group. One patient from the placebo group experienced atrial fibrillation, which was reported as a serious adverse advent. No deaths, opportunistic infections, herpes zoster infections, latent tuberculosis, or thromboembolic events occurred during this 16-week period [Citation59].
4. Ivarmacitinib
4.1. Efficacy in ulcerative colitis
The efficacy and safety of ivarmacitinib were evaluated in AMBER2 – a double-blind, placebo-controlled phase II trial, conducted across clinical centers in China, EU, and the US. Patients (n = 164) with moderate-to-severe UC aged 18–75, with inadequate response to conventional therapy were randomized 1:1:1:1 to receive ivarmacitinib 8 mg once-daily (QD), ivarmacitinib 4 mg twice-daily (BID), ivarmacitinib 4 mg once-daily (QD) or placebo for an 8-week treatment phase. Patients who completed the treatment phase had the opportunity to enter the blinded, active treatment, 8-week extension phase. The primary endpoint was the percentage of patients achieving clinical response at week 8, defined as a decrease from baseline in the 9-point modified Mayo Score of ≥ 2 points and at least 30%, along with a decrease in the rectal bleeding subscore of ≥ 1 point or rectal bleeding absolute subscore of 0 or 1. The clinical response rate at week 8 for the ivarmacitinib 8 mg QD was 46,3% (p = 0,066), for ivarmacitinib 4 mg BID was 46,3% (p = 0,059), for ivarmacitinib 4 mg QD was 43,9% (p = 0,095) and for placebo 26,8%. One of the secondary endpoints – clinical remission at week 8 was observed in 22%, 24,4%, 24,4% of the ivarmacitinib 8 mg QD, 4 mg BID, and 4 mg QD groups, respectively, as opposed to placebo where clinical remission rate was 4,9%. Another secondary endpoint – endoscopic improvement at week 8 was seen in higher rates in all active-dose groups in comparison to placebo, with the ivarmacitinib 4 mg QD achieving statistical significance over placebo (adjusted difference 22,7%, p = 0,018) [Citation60].
4.2. Safety in ulcerative colitis
The safety analysis set constituted 164 patients, including placebo patients that switched over ivarmacitinib during the extension phase (). The TEAE rate reported during the core 8-week treatment phase ranged from 43,9% to 48,8% for the ivarmacitinib groups and 39% for placebo. Severe adverse events were reported in five (3%) patients. TEAE considered by the investigator to be related to the study drug occurred in 49 (29,9%) patients. The study drug-related AE were predominantly mild-moderate in severity, and the most frequent being anemia. One SAE was considered drug-related in a patient with moderate synovitis. The other SAEs were UC worsening, cytomegalovirus infection, and liver or anal abscess. Safety findings from the entire 16-week period were comparable to the initial 8-week period. No major cardiovascular events, thromboembolic events, herpes zoster infections, or NMSC occurred during the duration of the study [Citation60].
Table 3. Safety analysis data from Ivarmacitinib studies. AE, adverse events; SAE, serious adverse events; SI, serious infections; HZV, herpes zoster virus; VT, venous thrombosis; PE, pulmonary embolism; Total, Total patients.
4.3. Safety in other diseases
The safety and efficacy of ivarmacitinib for the treatment of atopic dermatitis was evaluated in a Chinese study (ref NCT04162899). The study was a randomized, double-blind, placebo-controlled, multicentre, phase II trial. Patients (n = 105) aged 18–75 with moderate-to-severe atopic dermatitis who were nonresponsive or intolerant to topical or conventional systemic treatments were included. Patients were randomized in a 1:1:1 ratio, to receive ivarmacitinib 4 mg once daily, ivarmacitinib 8 mg once daily or placebo. The duration of the study was 12 weeks. Treatment of emergent adverse events were reported in 60%, 68,6%, and 51,4% in the ivarmacitinib 4 mg, 8 mg, and placebo groups, respectively. All serious adverse events were cases of worsening atopic dermatitis, a total of 3, of which all patients recovered.
No serious infections were reported. One patient in each active treatment arm had a herpes zoster infection, no cases of HZV were reported in the placebo group. Herpes simplex/herpes virus infection was reported in each group once. One case of Kaposi’s varicelliform eruption was reported in the 4 mg group. No cases of neoplasia, thrombosis, liver injury, or death were reported [Citation61]. The safety profile of ivarmacitinib was also examined in patients with RA, data from a study available in abstract form [Citation62,Citation63].
5. Efficacy comparison of filgotinib, upadacitinib, and ivarmacitinib
See .
Figure 1. Simplified JAK/STAT pathway. Cytokines and their associated receptors align with the downstream events depicted in the bottom.
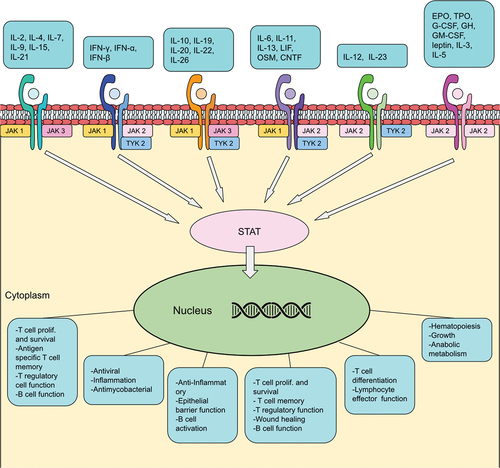
Figure 2. Clinical remission from the induction phases, of the three JAK1 inhibitors discussed prior.
6. Conclusion
The results of the phase II and phase III studies have shown the efficacy and safety of selective JAK1 inhibitors in the treatment of UC. However, further investigation is necessary regarding their long-term safety profile. Comparative research and head-to-head trials between selective JAK inhibitors and other advanced therapies are needed to determine which drug to use as first-line therapy for UC patients. To date, we can use indirect data from meta-analyses to guide the clinical use of JAK inhibitors. JAK selectivity seems promising, however, if this will translate into clinical benefit remains to be determined.
7. Expert opinion
While we have seen significant advances in treatment options for UC in recent years, there remains a high incidence of primary and secondary response failure rates. The UC treatment armamentarium has expanded over recent years with the addition of small-molecule drugs. JAK inhibitors have the advantage of simultaneously dampening the effect of multiple proinflammatory cytokines, oral administration, a rapid onset of action and no immunogenicity [Citation64]. Tofacitinib the first drug approved in this class, proved to be efficacious in UC, however there are still concerns regarding its safety profile.
A recent randomized, noninferiority, postauthorization trial examined patients with RA, 50 years or older with at least one cardiovascular risk factor to determine noninferiority of tofacitinib in comparison to TNF inhibition. The study demonstrated during a median follow-up time of 4 years that tofacitinib carried a higher risk of major adverse cardiovascular events (MACE) and cancer in comparison to patients receiving a TNF inhibitor. Serious infections, opportunistic infections, herpes zoster, and NMSC were higher with tofacitinib than with a TNF inhibitor. Thus, tofacitinib did not meet the noninferiority criteria [Citation24]. For that reason, EMA’s Pharmacovigilance Risk Assessment Committee (PRAC) has stated that JAK inhibitors only be used in the following patients if no other treatment alternatives are available: patients 65 years or older, those at increased risk of major cardiovascular events, smokers, or those who have done so for a long time, and patients with an increased risk of cancer. Furthermore, JAK inhibitors should be used with caution in patients with increased risk for blood clot formation in the lungs and deep veins, and dose reduction should be considered. The PRAC statement applies to abrocitinib, filgotinib, baricitinib, upadacitinib, and tofacitinib [Citation65]. Results from a recent study to identify subpopulations with different relative risk with tofacitinib versus TNFi were published. The study found that patients aged 65 years and older or ever smokers had an increased risk of malignancies (excluding NMSC), MACE, myocardial infarction, venous thromboembolism, and all-cause death with tofacitinib versus TNFi, and were defined as ‘high-risk’ [Citation66]. Patients under 65 years of age and never smokers were considered ‘low risk’ as there was no detectable risk increase with tofacitinib versus TNFi [Citation66].
Selective JAK1 inhibitors were developed to improve the risk-benefit profile of this class, however current data might indicate otherwise. A recent meta-analysis compared efficacy and safety data from randomized controlled trials of various biologics and small-molecule drugs for induction and maintenance treatment of moderate-to-severe UC [Citation67]. The results found that all treatments, except filgotinib 100 mg were significantly better than placebo in induction of clinical remission and endoscopic improvement. Upadacitinib ranked highest and was notably superior to all other interventions in the induction of clinical remission and endoscopic improvement, and infliximab was ranked second for both outcomes [Citation67]. Regarding safety however, upadacitinib was the worst performing agent, vedolizumab was ranked the safest drug for both adverse events and serious adverse events [Citation67]. Filgotinib ranked third highest for serious adverse events out of all interventions, however, was second lowest in terms of adverse events [Citation67]. Another meta-analysis similarly found that upadacitinib 45 mg ranked first for clinical remission in both biologic naïve and experienced patients [Citation68]. In terms of small molecules, upadacitinib ranked highest among all endpoints [Citation68]. However, in terms of safety, upadacitinib ranked last for the total number of adverse events [Citation68]. In both studies, upadacitinib was the most effective in inducing clinical remission, although that is polarized by the safety results, showing that remarkable efficacy might come at the cost of higher adverse event rates. We will need more evidence to justify the difference in effectiveness between these drugs. Further evaluation of drugs in this class is warranted to determine their impact on long term and corticosteroid-free remission in UC patients. It is crucial to understand how to position these drugs in the contemporary UC treatment algorithm. Therapeutic decisions are becoming increasingly personalized and should be guided by risk-benefit evaluation considering patients’ age, comorbidities, medical history, and disease severity. JAK inhibitors, thanks to their rapidity and efficacy, could potentially be used as an alternative treatment option in patients with acute severe UC who do not respond to intravenous steroids [Citation69]. Further, this drug class could also shine in advanced dual therapy. JAKi could potentially, be used in combination with biologics to enhance induction of remission, and once remission is achieved patients could be switched to a biologic monotherapy with a more reassuring safety profile [Citation70]. Other JAK inhibitors that do not target JAK1 are being investigated for the treatment of UC as well. One of them is OST-122, which inhibits JAK3/TYK2/ARK5 (ARK5 – AMPK- protein-related kinase 5) and has a gut-restricted pharmacokinetic profile [Citation71]. Deucravacitinib is a selective TYK2 inhibitor being investigated for the treatment of UC [Citation72]. Ritlecitinib, a JAK3/TEC (TEC, tyrosine kinase expressed in hepatocellular carcinoma), and brepocitinib, a TYK2/JAK1 inhibitor is being investigated as well for the treatment of moderate-to-severe UC [Citation73].
Article highlights
This drug class could potentially be used in advanced dual therapy, in combination with biologics to enhance induction of remission
JAK1 inhibitors show promise in treatment of UC but further evaluation is needed to determine their long-term safety and efficacy
JAK inhibitors dampen the effect of multiple proinflammatory cytokines
They are orally administered, have a rapid onset of action and no immunogenicity
Declaration of interest
F D’Amico served as a speaker for Sandoz, Jannsen, Omega Pharma, and Galapagos and served as an advisory board member for Galapagos. F Furfaro received consulting fees from Amgen, AbbVie and lecture fees from Janssen and Pfizer; M Allocca received consulting fees from Nikkiso Europe, Mundipharma, Janssen, AbbVie and Pfizer; G Fiorino received consultancy fees from Ferring, MSD, AbbVie, Takeda, Janssen, Amgen, Sandoz, Samsung Bioepis, Celltrion. L Peyrin-Biroulet declares personal fees from Galapagos, AbbVie, Janssen, Genentech, Ferring, Tillots, Celltrion, Takeda, Pfizer, Index Pharmaceuticals, Sandoz, Celgene, Biogen, Samsung Bioepis, Inotrem, Allergan, MSD, Roche, Arena, Gilead, Amgen, BMS, Vifor, Norgine, Mylan, Lilly, Fresenius Kabi, OSE Immunotherapeutics, Enthera, Theravance, Pandion Therapeutics, Gossamer Bio, Viatris, Thermo Fisher; grants from AbbVie, MSD, Takeda, Fresenius Kabi; stock options: CTMA; S Danese served as a speaker, consultant, and advisory board member for Schering-Plow, Abbott (AbbVie) Laboratories, Merck, UCB Pharma, Ferring, Cellerix, Millenium Takeda, Nycomed, Pharmacosmos, Actelion, Alfa Wasserman, Genentech, Grunenthal, Pfizer, AstraZeneca, Novo Nordisk, Cosmo Pharmaceuticals, Vifor, and Johnson and Johnson.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
S Danese conceived the study. A Goetsch and F D’Amico wrote the article and created tables and figures. M Allocca, G Fiorino, F Furfaro, A Zilli, T L Parigi, L Peyrin-Biroulet, and S Danese critically revised the manuscript. The manuscript was approved by all authors.
Additional information
Funding
References
- Ungaro R, Mehandru S, Allen PB, et al. Ulcerative colitis. Lancet. 2017;389:1756–1770.
- Ordás I, Eckmann L, Talamini M, et al. Ulcerative colitis. Lancet. 2012;380:1606–1619.
- Jairath V, Feagan BG. Global burden of inflammatory bowel disease. Lancet Gastroenterol Hepatol. 2020;5:2–3.
- Alatab S, Sepanlou SG, Ikuta K, et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet Gastroenterol Hepatol. 2020;5:17–30.
- Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14:329–342.
- EMA. Zeposia [Internet]. European Medicines Agency. 2020 [cited 2023 Jan 12]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/zeposia
- U.S. Food and Drug Administration Approves Bristol Myers Squibb’s Zeposia® (ozanimod), An oral treatment for adults with moderately to severely active ulcerative colitis1 [Internet]. [cited 2023 Jan 12]. Available from: https://news.bms.com/news/details/2021/U.S.-Food-and-Drug-Administration-Approves-Bristol-Myers-Squibbs-Zeposia-ozanimod-an-Oral-Treatment-for-Adults-with-Moderately-to-Severely-Active-Ulcerative-Colitis1/default.aspx
- Gordon JP, McEwan PC, Maguire A, et al. Characterizing unmet medical need and the potential role of new biologic treatment options in patients with ulcerative colitis and Crohn’s disease: a systematic review and clinician surveys. Eur J Gastroenterol Hepatol. 2015;27:804–812.
- McLornan DP, Pope JE, Gotlib J, et al. Current and future status of JAK inhibitors. Lancet. 2021;398:803–816.
- Casellas F, González-Lama Y, Ginard Vicens D, et al. Adherence improvement in patients with ulcerative colitis: a multidisciplinary consensus document. Rev Esp Enferm Dig. 2022;114:156–165.
- Danese S, Allez M, van Bodegraven AA, et al. Unmet medical needs in ulcerative colitis: an expert group consensus. DDI. 2019;37:266–283.
- Olivera P, Danese S, Peyrin-Biroulet L. Next generation of small molecules in inflammatory bowel disease. Gut. 2017;66:199–209.
- Padda IS, Bhatt R, Parmar M Tofacitinib [Internet]. PubMed. StatPearls Publishing; 2022 [cited 2022 Jan 1]. Available from: https://pubmed.ncbi.nlm.nih.gov/34283514/
- Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376:1723–1736.
- Commissioner O of the. FDA approves new treatment for moderately to severely active ulcerative colitis [Internet]. FDA. FDA; 2020 [cited 2023 Jan 12]. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-moderately-severely-active-ulcerative-colitis
- XELJANZ® (tofacitinib citrate) receives marketing authorization in the European Union for moderately to severely active ulcerative colitis | Pfizer [Internet]. [cited 2023 Jan 12]. Pfizer [Internet] https://www.pfizer.com/news/press-release/press-release-detail/xeljanz_tofacitinib_citrate_receives_marketing_authorization_in_the_european_union_for_moderately_to_severely_active_ulcerative_colitis-0
- EMA. Xeljanz [Internet]. European Medicines Agency. 2018 [cited 2023 Jan 12]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/xeljanz
- Danese S, Grisham M, Hodge J, et al. JAK inhibition using tofacitinib for inflammatory bowel disease treatment: a hub for multiple inflammatory cytokines. Am J Physiol Gastrointest Liver Physiol. 2016;310:G155–162.
- Lefevre PLC, Vande Casteele N. Clinical pharmacology of janus kinase inhibitors in inflammatory bowel disease. J Crohn’s Colitis. 2020;14:S725–736.
- McInnes IB, Byers NL, Higgs RE, et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res Ther. 2019;21:183.
- JAK–STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects | SpringerLink [Internet]. [cited 2023 Jan 18]. [Internet]: https://link.springer.com/article/10.1007/s40265-017-0701-9
- EMA. Increased risk of blood clots in lungs and death with higher dose Xeljanz (tofacitinib) for rheumatoid arthritis [Internet]. European Medicines Agency. 2019 [cited 2023 Jan 18]. Available from: https://www.ema.europa.eu/en/news/increased-risk-blood-clots-lungs-death-higher-dose-xeljanz-tofacitinib-rheumatoid-arthritis
- Research C for DE and. Safety trial finds risk of blood clots in the lungs and death with higher dose of tofacitinib (Xeljanz, Xeljanz XR) in rheumatoid arthritis patients; FDA to investigate. FDA [Internet]. 2019 [cited 2023 Jan 18]; Available from: https://www.fda.gov/drugs/drug-safety-and-availability/safety-trial-finds-risk-blood-clots-lungs-and-death-higher-dose-tofacitinib-xeljanz-xeljanz-xr
- Ytterberg SR, Bhatt DL, Mikuls TR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386:316–326.
- NV G. Jyseleca® approved in Japan for ulcerative colitis [Internet]. GlobeNewswire News Room. 2022 [cited 2022 Jan 1]. Available from: https://www.globenewswire.com/en/news-release/2022/03/28/2410628/0/en/Jyseleca-approved-in-Japan-for-ulcerative-colitis.html
- EMA. Jyseleca - European medicines agency [internet]. European Medicines Agency. 2020 [cited 2022 Jan 1]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/jyseleca
- D’Amico F, Magro F, Peyrin-Biroulet L, et al. Positioning filgotinib in the treatment algorithm of moderate to severe ulcerative colitis. J Crohn’s Colitis. 2021;16:835–844.
- Genovese MC, Kalunian K, Gottenberg J-E, et al. Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease-modifying antirheumatic drug therapy. JAMA. 2019;322:315.
- Hanzel J, Hulshoff MS, Grootjans J, et al. Emerging therapies for ulcerative colitis. Expert Rev Clin Immunol. 2022;18:513–524.
- RINVOQ® (Upadacitinib) Receives its sixth U.S. FDA approval | AbbVie news center [Internet]. news.abbvie.com. 2022 [cited 2022 Jan 1]. Available from: https://news.abbvie.com/news/press-releases/rinvoq-upadacitinib-receives-its-sixth-us-fda-approval.htm
- EMA. Rinvoq - European medicines agency [internet]. European Medicines Agency. 2019. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/rinvoq
- Reistone Biopharma Company Limited. A phase 3 study to evaluate the efficacy and long-term safety of SHR0302 therapy in subjects with moderately to severely active ulcerative colitis [Internet]. clinicaltrials.gov. 2022 [cited 2022 Jan 1]. Available from: https://clinicaltrials.gov/ct2/show/NCT05181137?term=SHR0302&draw=3&rank=19
- Danese S, Argollo M, Le Berre C, et al. JAK selectivity for inflammatory bowel disease treatment: does it clinically matter? Gut. 2019;68:1893–1899.
- Babon JJ, Lucet IS, Murphy JM, et al. The molecular regulation of janus kinase (JAK) activation. Biochem J. 2014;462:1–13.
- Hu X, Li J, Fu M, et al. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6:402.
- Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers (Basel). 2019;11:2002.
- Salas A, Hernandez-Rocha C, Duijvestein M, et al. JAK–STAT pathway targeting for the treatment of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17:323–337.
- Harris C, Cummings JRF. JAK1 inhibition and inflammatory bowel disease. Rheumatology (Oxford). 2021;60:ii45–51.
- Kremer JM, Bingham CO, Cappelli LC, et al. Postapproval comparative safety study of tofacitinib and biological disease-modifying antirheumatic drugs: 5-year results from a United States-based rheumatoid arthritis registry. ACR Open Rheumatol. 2021;3:173–184.
- Burmester GR, Nash P, Sands BE, et al. Adverse events of special interest in clinical trials of rheumatoid arthritis, psoriatic arthritis, ulcerative colitis and psoriasis with 37 066 patient-years of tofacitinib exposure. RMD Open. 2021;7:e001595.
- van der Heijde D, Baraliakos X, Gensler LS, et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): results from a randomised, placebo-controlled, phase 2 trial. Lancet (London, England). 2018;392:2378–2387.
- Mease P, Coates LC, Helliwell PS, et al. Efficacy and safety of filgotinib, a selective janus kinase 1 inhibitor, in patients with active psoriatic arthritis (EQUATOR): results from a randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392:2367–2377.
- Kavanaugh A, Westhovens RR, Winthrop KL, et al. Safety and efficacy of filgotinib: up to 4-year results from an open-label extension study of phase ii rheumatoid arthritis programs. J Rheumatol. 2021;48:1230–1238.
- Vermeire S, Schreiber S, Petryka R, et al. Clinical remission in patients with moderate-to-severe Crohn’s disease treated with filgotinib (the FITZROY study): results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet. 2017;389:266–275.
- Feagan BG, Danese S, Loftus EV, et al. Filgotinib as induction and maintenance therapy for ulcerative colitis (SELECTION): a phase 2b/3 double-blind, randomised, placebo-controlled trial. Lancet. 2021;397:2372–2384.
- Hibi T, Motoya S, Hisamatsu T, et al. Efficacy and safety of filgotinib as induction and maintenance therapy for Japanese patients with moderately to severely active ulcerative colitis: a post-hoc analysis of the phase 2b/3 SELECTION trial. Intest Res. 2022;21:110–125.
- Namour F, Anderson K, Nelson C, et al. Filgotinib: a clinical pharmacology review. Clin Pharmacokinet. 2022;61:819–832.
- Galapagos NV, Gilead Sciences. A long-term extension study to evaluate the safety of filgotinib in subjects with ulcerative colitis [Internet]. clinicaltrials.gov. 2021 [cited 2022 Jan 1]. Available from: https://clinicaltrials.gov/ct2/show/NCT02914535
- Geboes K. A reproducible grading scale for histological assessment of inflammation in ulcerative colitis. Gut. 2000;47:404–409.
- Westhovens R, Taylor PC, Alten R, et al. Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (mtx) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose-finding study (DARWIN 1). Ann Rheumatic Dis. 2017;76:998–1008.
- Kavanaugh A, Kremer J, Ponce L, et al. Filgotinib (GLPG0634/GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose-finding study (DARWIN 2). Ann Rheumatic Dis. 2017;76:1009–1019.
- Galapagos NV, A randomized double-blind, placebo-controlled phase 2 study to evaluate the testicular safety of filgotinib in adult males with moderately to severely active inflammatory bowel disease [Internet]. 2022 [cited 2023 Jan 18]. Report No.: study/NCT03201445. [Internet]: https://clinicaltrials.gov/ct2/show/study/NCT03201445
- Galapagos NV, A randomized double-blind, placebo-controlled phase 2 study to evaluate the effect of filgotinib on semen parameters in adult males with active rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis or non-radiographic axial spondyloarthritis [Internet]. 2022 [cited 2023 Jan 18]. Report No.: NCT03926195. [Internet]: https://clinicaltrials.gov/ct2/show/NCT03926195
- Galapagos receives positive CHMP opinion for Jyseleca® European label update based on testicular function safety data from MANTA/RAy studies [Internet]. [cited 2023 Feb 27]. Available from: https://www.glpg.com/press-release/3687/galapagos-receives-positive-chmp-opinion-for-jyseleca-european-label-update-based-on-testicular-function-safety-data-from-manta-ray-studies
- Mohamed ME, Klünder B, Othman AA. Clinical pharmacokinetics of upadacitinib: review of data relevant to the rheumatoid arthritis indication. Clin Pharmacokinet. 2019;59:531–544.
- Danese S, Vermeire S, Zhou W, et al. Upadacitinib as induction and maintenance therapy for moderately to severely active ulcerative colitis: results from three phase 3, multicentre, double-blind, randomised trials. Lancet. 2022;399:2113–2128.
- Vermeire S European Crohn´s and colitis organisation - ECCO - DOP38 upadacitinib therapy reduces ulcerative colitis symptoms as early as Day 1 [Internet]. [cited 2022 Jan 1]. [Internet] https://www.ecco-ibd.eu/publications/congress-abstracts/item/dop38-upadacitinib-therapy-reduces-ulcerative-colitis-symptoms-as-early-as-day-1.html
- Sandborn WJ, Feagan BG, Loftus EV, et al. Efficacy and safety of upadacitinib in a randomized trial of patients with crohn’s disease. Gastroenterology. 2020;158:2123–2138.e8.
- Guttman-Yassky E, Thaçi D, Pangan AL, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol. 2020;145:877–884.
- Chen B, Zhong J, Li X, et al. Efficacy and safety of ivarmacitinib in patients with moderate-to-severe, active, ulcerative colitis: a phase II study. Gastroenterology. 2022;163:1555–1568.
- Zhao Y, Zhang L, Ding Y, et al. Efficacy and safety of SHR0302, a highly selective janus kinase 1 inhibitor, in patients with moderate to severe atopic dermatitis: a phase ii randomized clinical trial. Am J Clin Dermatol. 2021;22:877–889.
- A multicenter, randomized, placebo-controlled, double-blind phase 2 study of SHR0302 versus placebo in Chinese subjects with moderate to severe active rheumatoid arthritis (RA) [Internet]. ACR Meeting Abstracts. [cited 2023 Jan 12]. Available from: https://acrabstracts.org/abstract/a-multicenter-randomized-placebo-controlled-double-blind-phase-2-study-of-shr0302-versus-placebo-in-chinese-subjects-with-moderate-to-severe-active-rheumatoid-arthritis-ra/
- Jiangsu HengRui Medicine Co., Ltd. Randomized, double-blind, placebo-controlled, multicenter, phase ii study to assess the safety and efficacy of SHR0302 in patients with moderate to severe active rheumatoid arthritis [Internet]. clinicaltrials.gov; 2021 [cited 2023 Jan 10]. Report No.: study/NCT03254966. [Internet]. clinicaltrials.gov: https://clinicaltrials.gov/ct2/show/study/NCT03254966
- D’Amico F, Peyrin-Biroulet L, Danese S. Is selectivity the JAKpot winner for inflammatory bowel disease treatment? Gastroenterology. 2022;163:1482–1484.
- EMA. Meeting highlights from the pharmacovigilance risk assessment committee (PRAC) 24 - 27 October 2022 European medicines agency [Internet]. European Medicines Agency. 2022 [cited 2023 Jan 1]. Available from: https://www.ema.europa.eu/en/news/meeting-highlights-pharmacovigilance-risk-assessment-committee-prac-24-27-october-2022
- Kristensen LE, Danese S, Yndestad A, et al. Identification of two tofacitinib subpopulations with different relative risk versus TNF inhibitors: an analysis of the open label, randomised controlled study ORAL Surveillance. Annals of the Rheumatic Diseases [Internet]. 2023 [cited 2023 Mar 26]; Available from: https://ard.bmj.com/content/early/2023/03/16/ard-2022-223715
- Lasa JS, Olivera PA, Danese S, et al. Efficacy and safety of biologics and small molecule drugs for patients with moderate-to-severe ulcerative colitis: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. 2022;7:161–170.
- Burr NE, Gracie DJ, Black CJ, et al. Efficacy of biological therapies and small molecules in moderate to severe ulcerative colitis: systematic review and network meta-analysis. Gut. 2021;71: gutjnl-2021-326390.
- D’Amico F, Peyrin-Biroulet L, Danese S. Tofacitinib for acute severe colitis: when the going gets tough, the tough get going. J Crohn’s Colitis. 2020;14:883–885.
- Danese S, Solitano V, Jairath V, et al. The future of drug development for inflammatory bowel disease: the need to ACT (advanced combination treatment). Gut. 2022;71:2380–2387.
- Oncostellae SL A phase Ib/IIa, randomized, double blind, placebo controlled, multicenter clinical trial to evaluate the safety, pharmacokinetics and efficacy of oral treatment with OST-122 in patients with moderate to severe ulcerative colitis [Internet]. clinicaltrials.gov; 2023 [cited 2023 Mar 28]. Report No.: NCT04353791. [Internet]. clinicaltrials.gov: https://clinicaltrials.gov/ct2/show/NCT04353791
- Bristol-Myers Squibb. A phase 2 randomized, double-blind, placebo-controlled study of the safety and efficacy of BMS-986165 in subjects with moderate to severe ulcerative colitis [Internet]. clinicaltrials.gov;2023 [cited 2023 Mar 28]. Report No.: NCT03934216. [Internet]. clinicaltrials.gov: https://clinicaltrials.gov/ct2/show/NCT03934216
- Sandborn WJ, Danese S, Leszczyszyn J, et al. Oral Ritlecitinib And Brepocitinib For Moderate-to-Severe Ulcerative Colitis: Results From A Randomized, Phase 2b study. Clinical Gastroenterology and Hepatology [Internet]. 2023 [cited 2023 Mar 31]. Available from: https://www.cghjournal.org/article/S1542-35652300007-1/fulltext