
1. Introduction
Drug-induced immune hemolytic anemia (DIHA) is a rare phenomenon with an estimated incidence of 1 per million/year [Citation1], difficult to quantify, and probably underestimated due the challenging diagnosis. Since the first reports in the early 1950s, the number of drugs thought to cause DIHA has been progressively increasing and nowadays more than a hundred case reports have been published [Citation2]. The epidemiology is further complicated by the various usages of dugs over time and in different countries. For instance, intravenous penicillin, commonly used about 30–40 years ago, was progressively replaced by cefotetan, and more recently ceftriaxone, beta-lactamase inhibitors, and piperacillin, all drugs associated with DIHA. Additionally, various drugs have been reported to induce DIHA, such as methyl-dopa, procainamide, rifampin, nafcillin, erythromycin, ticarcillin, trimethoprim, sulfamethoxazole, probenecid, quinine, phenacetin, diclofenac, cimetidine, hydrochlorothiazide, chloropropamide, fludarabine, tolectin, mefloquine, oxaliplatin, and carboplatin.
The main issue for these reports is the certainty of the diagnosis, as it is mostly based on temporal association and resolution of the hemolytic process after stopping the suspected drug, while the confirmatory serological testing is complex and not always performed [Citation3,Citation4]. Moreover, the temporal association is a weak evidence, as hemolysis may occur from days to weeks or even months from the introduction of a suspected drug, and the role of concomitant medications is hard to assess. To further complicate the picture, recent biological drugs, such as immune check-point inhibitors (nivolumab, pembrolizumab, ipilimumab, and atezolizumab) in attempt to enhance host immune response against tumor cells and to inhibit programmed death, may evoke severe autoimmune reactions, including fulminant autoimmune hemolytic anemia (AIHA) [Citation5]. Furthermore, although not strictly drugs, hematopoietic stem cell transplant (HSCT) or solid organ transplantation and CAR-T therapy have been associated with severe and refractory AIHAs [Citation6,Citation7]. In these complex clinical scenarios, several drugs are administered at the same time, making it impossible to attribute the autoimmune disease to a dysregulation of immunosurveillance or to specific anti-drug antibodies. Finally, it is worth citing the false-Coombs positivity in patients treated with anti‐CD38 monoclonal antibodies (daratumumab and isatuximab) due to the low-level expression of CD38 on erythrocytes; the strategies for overcoming this interference have been recently reviewed [Citation8].
Historically, anti-drug antibodies have been distinguished in drug-independent and drug-dependent antibodies [Citation1–3]. The former do not require the addition of the drug into in vitro test systems (the Coombs test or direct antiglobulin test, DAT) for their detection, are authentic anti-erythrocyte antibodies, and thus indistinguishable from those causing AIHA. Drug-dependent antibodies are detectable by DAT when the drug (or its metabolites) is added in vitro and can be distinguished in those directed against the drug covalently bound to the erythrocyte membrane and those reacting with the drug not firmly bound. In the first case, the drug–erythrocyte complex will be preserved after the several washes of the assay (resulting in a positive test), while in the second case, anti-drug antibodies can only be demonstrated in patient’s serum by the indirect antiglobulin test [Citation1–3]. Usually, antibodies are of the IgG class (DAT-positive with IgG antisera) that weakly activate complement and induce a subacute clinical picture. More rarely, they are of the IgM class that strongly activate complement (DAT-positive with anti-C reagents) and can cause acute and severe intravascular hemolysis, possibly fatal. Finally, DIHA can be confused with a hemolytic transfusion reaction, possibly delaying appropriate management [Citation1,Citation2,Citation9]. A simple algorithm for investigating DIHA is shown in . The first step is demonstrating the hemolytic nature of anemia, followed by a positive DAT and a clinical suspect based on a pharmacological anamnesis and temporal association with a drug. Further actions include stopping the suspected drug (if possible), treat the patient on a clinical basis (transfusions, steroids, etc.), and refer to a reference laboratory for diagnostic confirmation. A definite diagnosis of DIHA is achieved by the demonstration of drug-dependent antibodies in patient’s serum (and/or eluate) using erythrocytes pretreated with the suspected drug or in the presence of the suspected drug or its metabolites.
Figure 1. Diagnostic algorithm for drug-induced hemolytic anemia.
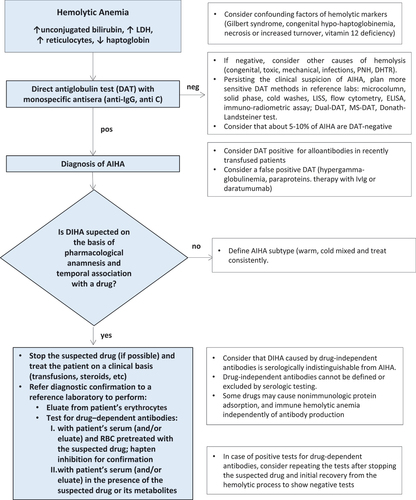
Therapy of DIHA is mainly based on drug discontinuation, usually determining a hematological improvement/recovery in 1–2 weeks [Citation4]. In acute and severe cases, blood transfusions may be required as well as intensive care admission and temporary dialysis. Steroids are usually administered in most patients, as in classic AIHAs, although their benefit is unclear and indistinguishable from that of drug discontinuation. In the Berlin Case–Control Surveillance Study, approximately 55% of patients required blood transfusions and 105/124 (85%) patients received corticosteroids [Citation10]. Second-line treatment with rituximab, and plasma exchange have been adopted in few refractory cases associated with check-point inhibitors and biologic therapies. An important and still unsolved question is whether to re-introduce the suspected drug, being the treatment lifesaving [Citation5–7,Citation11].
Several new drugs are populating the therapeutic armamentarium of AIHA, and some of them are already available in the clinical practice while most are in clinical trials (). However, none of them has been investigated in DIHA, and will unlikely be indicated/reimbursed in real-world treatment of DIHA. Therefore, these new drugs will probably be administered in drug-independent DIHA that is indistinguishable from idiopathic AIHA, and/or off-label in urgent or severe/refractory cases. Steroids remain the standard first-line treatment of wAIHA while they are acceptable in acute CAD but discouraged in the chronic setting, as effective at high/unacceptable doses. Rituximab is the preferred second-line in wAIHA and is recommended as first-line in CAD. Further lines (splenectomy or immunosuppressants for wAIHA, complement inhibitors or rituximab plus bendamustine or fludarabine for CAD, bortezomib for both) depend on medical experience, patient’s preferences, and availability of drugs or clinical trials with new drugs (). Curiously, cases of AIHA have been described following therapy with fludarabine or bendamustine, adding intricacy to the diagnosis and management of DIHA. Besides B-cell and plasma cell-target therapies (rituximab, ibrutinib, rilzabrutinib, parsaclisib, ianalumab, obexelimab, bortezomib, daratumumab, isatuximab) aimed at inhibiting the source of autoantibody production, complement inhibitors and neonatal Fc receptor (FcRn) inhibitors deserve special attention for their novel mechanism of action and prompt effect, which may be pivotal in acute/severe DIHA.
Table 1. Standard and new drugs for warm AIHA (wAIHA) and cold agglutinin disease (CAD).
The FcRn was described about 50 years ago, as the receptor that transports maternal IgG from mother to child via mother’s milk. It is structurally homologous to the MHC Class I heterodimeric receptor family, and is expressed by several cells including macrophages, monocytes, B cells, and dendritic cells. The main ‘task’ of the FcRn is the salvage of IgG from catabolism, resulting in increased half-life of these antibodies. The engineering of the Fc portion of several therapeutic monoclonal antibodies, and thus their binding to the FcRn, has implemented the development of several molecules with longer half-life than original ones. Conversely, blocking the FcRn may increase IgG clearance, resulting in reduced IgG (including pathogenic autoantibodies), an outcome assimilable to a pharmacological plasma exchange. Several FcRn blocking strategies are under investigation in various autoimmune diseases: anti-FcRn heavy and light chain antibodies, Fc-engineered IgGs that have increased affinity for FcRn, peptides and small molecules that compete with IgG for binding to FcRn. Interestingly, studies on the FcRn have provided evidence that high-dose IV immunoglobulins, a therapeutic option in primary immune thrombocytopenia (ITP) and AIHA, can saturate FcRn and accelerate the clearance of endogenous IgG. This mechanism adds to the known therapeutic effect of masking Fc receptors and thus phagocytosis by macrophages and antibody-mediated cellular cytotoxicity. The efficacy and safety of the FcRn inhibitor efgartigimod has been proved in adults with ITP and myasthenia gravis, two autoimmune diseases where the autoantibody plays a primary role [Citation12–14]. Overall, the drug may act as a ‘plasma exchange in a bottle,’ a result that may be of great importance in DIHA.
Complement inhibition is another emerging therapeutic strategy for diseases where complement activation pays a pivotal role [Citation15]. The paradigmatic disease is paroxysmal nocturnal hemoglobinuria, characterized by an abnormal sensitivity of erythrocytes to the lytic activity of several complement fractions. The MoAb eculizumab and, more recently, the FcRn-engineered anti C5 ravulizumab have dramatically changed the prognosis and the natural history of the disease. Additional complement inhibitors, such as pegcetacoplan (anti-C3), Iptacopan (anti-Factor B), and danicopan (anti-Factor D), are at different stages of clinical development and availability on the market. Some of the later are in clinical trials in wAIHA and CAD with promising results. Finally, sutimlimab, an anti-C1s inhibitor, has demonstrated clinical efficacy in pilot studies and in a double-blind, placebo-controlled trial in CAD and is now approved by FDA and EMA in this disease. Importantly, the drug has a very rapid effect with meaningful hemoglobin increase and blocking of the classical complement cascade [Citation15]. These characteristics may be of great utility in DIHA with complement involvement, which are known to be particularly severe and possibly fatal.
2. Expert opinion
DIHA are rare and difficult to diagnose. Knowledge and clinical suspicion are fundamental to identify the phenomenon, and close interaction with an experienced immune-hematology laboratory is essential to confirm the diagnosis. With the increasing use of biological therapies in complex clinical setting, the diagnosis is even more challenging. Therapy is based on discontinuation of the suspected drug, but this is not always possible when alternative therapies are not available. In these cases, re-introducing the suspected drug is even more doubtful. Steroids are commonly used in DIHA, but their efficacy is questionable, being biased by the simultaneous discontinuation of the offending drug. Other therapies such as rituximab and plasma exchange have been used in few severe cases and are not available worldwide. On the other hand, there is increasing knowledge on the pathogenesis of wAIHA and CAD, as well as growing availability of new drugs directed at the several pathogenic mechanisms of these diseases. They include B-cell-directed therapies (rituximab alone or in combination with bendamustine or fludarabine, inhibitors of Bruton tyrosine kinase, phosphatidylInositol 3-kinase, spleen tyrosine kinase, B-cell activating factor, and proteasome), as well as ‘smart’ drugs such as the neonatal Fc receptor and complement inhibitors. The latter act quickly and have novel mechanisms of action, i.e. a ‘plasma exchange in a bottle’ and a neutralizing effect of complement activation, respectively. Thus, they have a potential utility in emergency situations of DIHA. However, most of the trials with these drugs are being conducted in wAIHA or CAD, leaving an unmet need even in some rare AIHAs (mixed forms, wAIHA with complement involvement, DAT-negative AIHAs, paroxysmal cold hemoglobinuria). The feasibility of clinical trials in DIHA is even more challenging, given the difficult diagnosis and the heterogeneity of drugs and mechanisms involved. While stopping the suspected drug and supportive therapy are the mainstay of management, new target therapies are highly advisable, particularly in severe and acute forms. However, their actual application in DIHA will be possible off-label when the drugs will be in the market and clinicians have gained enough experience to use them confidently.
Declaration of interest
W Barcellini has received research support from Alexion, participated in advisory boards for Agios, Alexion, Bioverativ, Incyte, Novartis, Sobi, and Sanofi, and has been an invited speaker for Alexion, Novartis, and Sanofi.
B Fattizzo received consultancy honoraria and speaker’s bureau from Alexion, Janssen, Novartis, Roche, Sanofi, and Sobi.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24(4–5):143–150. doi: 10.1016/j.blre.2010.06.004
- Garratty G. Immune hemolytic anemia caused by drugs. Expert Opin Drug Saf. 2012;11(4):635–642. doi: 10.1517/14740338.2012.678832
- Leger RM, Arndt PA, Garratty G. How we investigate drug-induced immune hemolytic anemia. Immunohematology. 2014;30(2):85–94. PMID: 25247618 doi: 10.21307/immunohematology-2019-102
- Hill QA, Stamps R, Massey E, et al. British Society for Haematology Guidelines. Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br J Haematol. 2017 Apr;177(2):208–220. doi: 10.1111/bjh.14654
- Tanios GE, Doley PB, Munker R. Autoimmune hemolytic anemia associated with the use of immune checkpoint inhibitors for cancer: 68 cases from the food and drug administration database and review. Eur J Haematol. 2019;102(2):157–162. doi: 10.1111/ejh.13187
- Fattizzo B, Barcellini W. Autoimmune hemolytic anemia: causes and consequences. Expert Rev Clin Immunol. 2022;18(7):731–745. doi: 10.1080/1744666X.2022.2089115
- Schubert ML, Schmitt M, Wang L, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol. 2021;32(1):34–48.
- Feng CC, Chang CW, Lien ZY, et al. Better resolving of anti-CD38 antibody interference with blood compatibility testing by using manual polybrene method compared with dithiothreitol-pretreatment indirect antiglobulin test. J Clin Lab Anal. 2023;37(8):e24891.
- Ahrens N, Genth R, Kiesewetter H, et al. Misdiagnosis in patients with diclofenac-induced hemolysis: new cases and a concise review. Am J Hematol. 2006;81:128–131. doi: 10.1002/ajh.20494
- Garbe E, Andersohn F, Bronder E, et al. Drug induced immune haemolytic anaemia in the Berlin case–control surveillance study. Br J Haematol. 2011;154:644–653. doi: 10.1111/j.1365-2141.2011.08784.x
- Leaf RK, Ferreri C, Rangachari D, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94(5):563–574.
- Yunce M, Katyal N, Monis GF, et al. Neonatal Fc receptor blockade as emerging therapy in diseases with plasma exchange indications. J Clin Apheresis. 2023;38(5):632–640.
- Bhandari V, Bril V. FcRN receptor antagonists in the management of myasthenia gravis. Front Neurol. 2023;14:1229112. doi: 10.3389/fneur.2023.1229112
- Broome CM, McDonald V, Miyakawa Y, et al. Efficacy and safety of the neonatal Fc receptor inhibitor efgartigimod in adults with primary immune thrombocytopenia (ADVANCE IV): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2023;S0140-6736(23):01460–5.
- Bortolotti M, Barcellini W, Fattizzo B. Molecular pharmacology in complement-mediated hemolytic disorders. Eur J Haematol. 2023;111(3):326–336. doi: 10.1111/ejh.14026