
ABSTRACT
Introduction
Juxtaglomerular apparatus (JGA)-mediated homeostatic mechanism links to how sodium-glucose cotransporter 2 inhibitors (SGLT2is) slow progression of chronic kidney disease (CKD) and may link to how tolvaptan slows renal function decline in autosomal dominant polycystic kidney disease (ADPKD).
Area covered
JGA-mediated homeostatic mechanism has been hypothesized based on investigations of tubuloglomerular feedback and renin-angiotensin system. We reviewed clinical trials of SGLT2is and tolvaptan to assess the relationship between this mechanism and these drugs.
Expert opinion
When sodium load to macula densa (MD) increases, MD increases adenosine production, constricting afferent arteriole (Af-art) and protecting glomeruli. Concurrently, MD signaling suppresses renin secretion, increases urinary sodium excretion, and counterbalances reduced sodium filtration. However, when there is marked increase in sodium load per-nephron, as in advanced CKD, MD adenosine production increases, relaxing Af-art and maintaining sodium homeostasis at the expense of glomeruli. The beneficial effects of tolvaptan on renal function in ADPKD may also depend on the JGA-mediated homeostatic mechanisms since tolvaptan inhibits sodium reabsorption in the thick ascending limb.
The JGA-mediated homeostatic mechanism regulates Af-arts, constricting to relaxing according to homeostatic needs. Understanding this mechanism may contribute to the development of pharmacotherapeutic compounds and better care for patients with CKD.
1. Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited renal disorder characterized by the development of renal cysts and renal function decline [Citation1]. The natural history observation studies in ADPKD proved that the increase in total kidney volume (TKV) precedes and predicts the change in the estimated glomerular filtration rate (eGFR) [Citation2,Citation3]. A large phase 3 study of tolvaptan, a vasopressin V2 receptor (V2R) antagonist, in patients with ADPKD (TEMPO 3:4) revealed that tolvaptan reduced the rates of TKV growth and eGFR decline over 3 years and seemed to reconfirm that pharmacologic suppression of TKV growth results in amelioration of kidney function decline [Citation4]. Thereafter, the TEMPO 4:4 study was performed to examine an additional 2 years of data [Citation5]. The TEMPO 4:4, however, concluded unrelated effects of tolvaptan on TKV and eGFR, suggesting that tolvaptan may have renoprotective effects that are independent of changes in TKV.
When sodium delivery to the macula densa (MD) increases, the afferent arteriole (Af-art) constricts (denoted as tubuloglomerular feedback (TGF) phenomenon) [Citation6,Citation7] and renin secretion is inhibited, leading to the maintenance of sodium homeostasis at low glomerular blood flow and filtration pressure, thereby contributing to renoprotection. However, when the sodium load to the MD further increases, such as in advanced chronic kidney disease (CKD) and/or by pharmacotherapy, the Af-art dilates, increasing glomerular blood flow and sodium filtration (denoted as glomerular hyperfiltration) [Citation8], which results in increased sodium load to the MD and maintenance of sodium homeostasis at the expense of glomerular damage. These mechanisms (including the TGF phenomenon, glomerular hyperfiltration, and homeostatic mechanisms) are referred to as juxtaglomerular apparatus (JGA)-mediated homeostatic mechanisms.
Tolvaptan appears to retard the GFR decline, at least in part, through JGA-mediated homeostatic mechanisms in patients with ADPKD. This hypothesis is in accord with the association of hyperactivation of the vasopressin system with CKD progression [Citation9]. This review discusses the JGA-mediated homeostatic mechanisms that are related to the renoprotective effect of tolvaptan and how vasopressin is linked to CKD progression.
2. Tolvaptan and GFR
2.1. eGFR slope and tolvaptan in ADPKD
ADPKD is an inherited renal disorder characterized by the development of renal cysts and renal function decline, resulting in progression to end-stage renal disease [Citation1]. Adenosine-3′,5′-cyclic monophosphate (cAMP) plays a major stimulatory role in cystogenesis [Citation10], and V2R is the major cAMP agonist in the collecting duct (CD) [Citation11]. In mouse models of human ADPKD, administration of a V2R antagonist lowered renal cAMP levels and inhibited disease development [Citation12].
On the basis of these findings, a large phase 3 clinical study of tolvaptan in patients with ADPKD (TEMPO 3:4) was initiated. The results of TEMPO 3:4 showed that tolvaptan reduced the rates of TKV growth and eGFR decline over 3 years and confirmed that suppression of the TKV growth rate resulted in the amelioration of kidney function decline [Citation4].
The TEMPO 4:4 study was performed to examine an additional 2 years of data in patients who had completed TEMPO 3:4. A sustained disease-modifying effect of tolvaptan was observed on eGFR but not on TKV. The lack of a sustained treatment effect on TKV was attributed to limitations of the trial design [Citation5].
Interestingly, in both TEMPO 3:4 and 4:4, eGFR decreased by approximately 2.5 mL/min/1.73 m2 when tolvaptan treatment was initiated. Surprisingly, in TEMPO 3:4, the initial eGFR reduction was reversed by tolvaptan withdrawal even after 3 years of treatment [Citation5]. Tolvaptan resumption following several months of an off-treatment period caused reduced eGFR.
In a short-term study of a small sample of patients with ADPKD, the GFR decreased after 3 weeks of tolvaptan treatment and returned to baseline after tolvaptan cessation [Citation13].
The prompt occurrence of eGFR reduction and reversal by the initiation and withdrawal of tolvaptan, respectively, may be related to the short-term effect of tolvaptan but not the secondary effect of TKV changes because eGFR changed over a short period.
Moreover, the preservation of kidney function unrelated to TKV growth inhibition reported in the Japanese subpopulation analysis of the TEMPO 3:4 study suggested that the beneficial effect of tolvaptan on kidney function may not necessarily be secondary to TKV growth inhibition [Citation14].
2.2. Tolvaptan inhibition of Na+ reabsorption in the thick ascending limb
The thick ascending limb (TAL) reabsorbs approximately 20–25% of filtered sodium mainly through apical Na+-K+-2Cl− cotransporter 2 (NKCC2) and Na+/H+ exchanger 3 (NHE3) via the active extrusion of Na+ via Na-K-ATPase at the basal membrane. The activities of NKCC2, NHE3, and Na-K-ATPase in the TAL are mainly mediated by the cAMP/protein kinase A (cAMP/PKA) pathway [Citation15] ().
Figure 1. Putative V2R-mediated sodium transport via NKCC2 activation in the thick ascending limb.
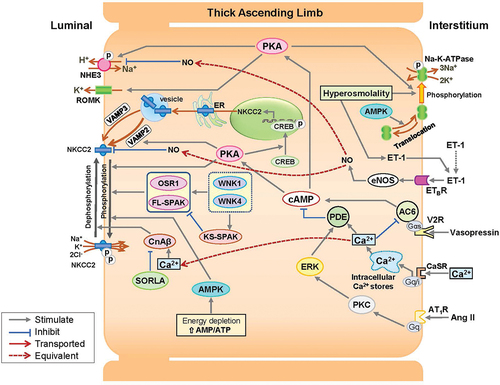
NKCC2 Na+ transport is mediated by two mechanisms: apical trafficking of NKCC2 and phosphorylation/dephosphorylation of the N-terminus of the NKCC2 protein, both of which are activated by the cAMP/PKA pathway [Citation16].
Apical trafficking is enhanced by vesicle-associated membrane protein 2 (VAMP2), which is involved in the cAMP/PKA pathway, and VAMP3, which mediates constitutive translocation [Citation17,Citation18]. The N-terminal of NKCC2 is phosphorylated by cAMP/PKA, the WNK1-SPAK/OSR1 pathway, and AMP-activated protein kinase (AMPK). Furthermore, NHE3 is phosphorylated via the cAMP/PKA pathway [Citation19–21].
V2R is located at the basolateral membrane of the TAL [Citation22–25] (), and vasopressin stimulates cAMP via V2R-mediated AC6 [Citation26]. The cAMP/PKA pathways in the TAL can be activated by substances other than vasopressin [Citation27]; however, it has been demonstrated that V2R-mediated cAMP/PKA pathways are the main determinants of sodium transport activity in the TAL [Citation24]. Thus, V2R inhibition is responsible for most of the decrease in sodium reabsorption in the TAL and the increase in sodium delivery to the MD, and it may account for an initial and reversible decrease in GFR in patients with ADPKD treated with tolvaptan [Citation5,Citation13].
Table 1. V2R distribution in renal tubules.
2.3. Adverse events of tolvaptan
In the TEMPO 4:3 study, tolvaptan-related adverse events were aquaresis-induced symptoms (polyuria, polydipsia, and nocturia) and elevation of liver enzymes, whereas those who received placebo had higher incidences of adverse events related to ADPKD (kidney pain, hematuria, urinary tract infection, and back pain) [Citation4]. In a longer-term open-label study (TEMPO 4:4) [Citation5] and other long-term observational studies at a single institute [Citation28,Citation29], similar adverse events were observed with tolvaptan.
3. JGA regulation of GFR
3.1. JGA anatomy
MD mediates both GFR and renin activity via signals to the vascular smooth muscle cells (VSMCs) of the glomerular arterioles and juxtaglomerular (JG) cells, respectively. Functional integration of the JGA is supported by the anatomically close arrangement of the MD, VSMCs of afferent (Af-art) and efferent (Ef-art) arterioles, extraglomerular mesangial cells, and JG cells [Citation30,Citation31].
The MD is located at the distal end of the TAL and forms a plaque of a monolayer epithelium comprising 35–40 cells arranged in the half of the distal end of the TAL that faces the parental Af-art [Citation31]. The Af-art has close anatomical relationships with the MD and connecting tubule (CNT) of the same nephron [Citation32]. Importantly, V2R is not expressed in the MD cells despite its clear presence in the cortical and medullary TAL [Citation25] ().
3.2. Ion transporters in MD and CNT
The NKCC2 is located at the apical end of the TAL and MD cells and comprises the main apical entry pathway for NaCl [Citation33]. The Na+/H+ exchanger 2 (NHE2) and exchanger 4 (NHE4) are expressed in the apical and basolateral membranes of the MD, respectively [Citation34]. The apical renal outer medullary K+ channel (ROMK) secretes K+, which is essential for the normal functioning of NKCC2 [Citation35]. Apical MD colonic H+-K+-ATPase functions as an Na+(H+)-K+-ATPase and regulates cytosolic Na+ concentration ([Na+]) via Na+ recycling [Citation36]. Basolateral Na+-K+ ATPase provides the driving force for these ion transporters [Citation15] ().
Figure 2. Putative signaling pathways in the regulation of afferent arteriole tonicity in JGA and CNT.
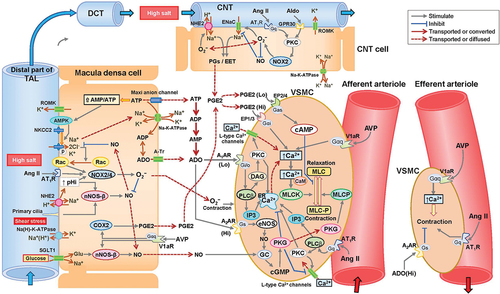
The epithelial Na+ channel (ENaC), NHE2, and ROMK are expressed in the apical membrane of the CNT. ENaC is sensitive to aldosterone and mediates the absorption of Na+, which is coupled with K+ secretion via the ROMK [Citation37].
3.3. Glomerular capillary pressure and arterioles tonicity
Glomerular filtration is a function of glomerular capillary pressure (PGC), plasma flow rate, plasma oncotic pressure, Bowman’s space pressure, the surface area available for filtration, and the filtration coefficient [Citation38]. The Af-art and Ef-art regulate the inflow and outflow of blood, respectively, and consequently are the major controllers of both the PGC and plasma flow rate. In a large number of TGF studies, single nephron GFR (SNGFR), stop-flow pressure, which is an estimation of PGC, or glomerular arteriole diameter was measured. However, the same time responses of Af-art and Ef-art have been observed in very few studies because such observations are technically difficult. Observation of living glomerulus using multiphoton excitation laser scanning fluorescence microscopy showed a ‘sphincter-like’ reaction of the terminal Af-art in response to increased perfusion rate to the MD; however, this response was absent in the Ef-art, suggesting the lack of vasoconstriction of the Ef-art during TGF [Citation39]. The increase in Af-art resistance is the result of an active process, whereas the increase in Ef-art resistance was considered a result of a passive process in TGF [Citation40,Citation41]. Therefore, Af-art diameter and tonicity are the main focus of this review.
3.4. NKCC2 mediates MD sensing
The NKCC2 inhibitor furosemide abolishes the vasocontraction response of the Af-art despite increased sodium delivery to the MD, indicating that NKCC2 in MD cells plays a primary role in the vasocontraction response to high-luminal [NaCl] [Citation42]
The NKCC2 isoforms NKCC2A and NKCC2B, but not NKCC2F, are found in the MD. NKCC2B has a higher affinity for NaCl than NKCC2A [Citation43,Citation44]. NKCC2A and NKCC2B are equally expressed in the MD, and salt restriction mainly increases NKCC2B [Citation45]. NKCC2B may be involved in Af-art contraction across a wide range of MD luminal NaCl concentrations ([NaCl]), and NKCC2A may antagonize the contraction response in the higher [NaCl] range [Citation46,Citation47].
3.5. Adenosine mediates Af-art tone
Adenosine formation is augmented when ATP hydrolysis surpasses ATP synthesis during negative energy balance, such as increased tubular sodium transport. Adenosine serves as a limiting factor in this context, exemplified by its ability to reduce blood supply [Citation48]. Extracellular adenosine is supplied by cellular adenosine release or extracellular breakdown of ATP, AMP, or cAMP [Citation49] ( and ).
Figure 3. Images of the putative intensity of MD(/CNT)-signalings to regulate the Af-art tone.
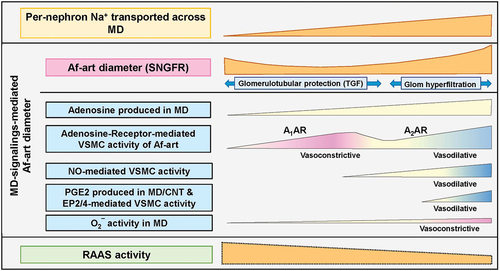
In response to increased MD luminal [NaCl], adenosine constricts the Af-art, Ef-art, and, possibly, the mesangial cells via A1AR receptor activation [Citation50]. A1AR Gi/o protein-coupled receptors activate phospholipase C (PLC)-β, leading to the production of 1,4,5-inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 induces Ca2+ release from the sarcoplasmic reticulum (SR) via IP receptor Ca2+ channels. DAG activates protein kinase C (PKC), which augments L-type Ca2+ channels and releases Ca2+ from the SR via ryanodine Ca2+ channels. The increased intracellular [Ca2+] binds to CaM and activates myosin light chain (MLC) kinase (MLCK), leading to MLC phosphorylation, actin polymerization, and vascular contraction [Citation51]. Increased cytosolic [Ca2+] promotes the CaM-activated PDE1 family, which lowers both cyclic guanylate monophosphate (cGMP) and cAMP, resulting in the antagonization of smooth muscle relaxation [Citation52] ().
Adenosine A2 receptor (A2AR) activation stimulates endothelial nitric oxide synthase (eNOS)-mediated nitric oxide (NO) production and results in Af-art relaxation, which becomes predominant at higher adenosine concentrations [Citation53–55] (). This biphasic response of the Af-art is eliminated by angiotensin II [Citation56].
3.6. NO dilates the Af-art
High MD luminal [Na+] stimulates NHE2, which elicits Na+ entry and H+ extrusion, resulting in the alkalinization of MD pHi [Citation57]. The increased pHi (alkalinization) in MD cells activates neuronal NOS (nNOS) and increases NO production, which diffuses easily to adjacent JGA tissues and affects their functions [Citation57,Citation58]. nNOS is also activated by primary cilia on the apical side of MD cells [Citation59] and tubular glucose via sodium glucose cotransporter type 1 [Citation60].
NO induces cGMP synthesis by activating guanylyl cyclase (GC). cGMP activates cGMP-dependent protein kinase (PKG or cGK), which decreases cytosolic [Ca2+], and activates MLC phosphatase, enhancing the dephosphorylation of phosphorylated MLC and promoting smooth muscle relaxation [Citation52].
nNOS and NHE2 do not affect Af-art diameter at low to average MD luminal [Na+] but dilate Af-art at high luminal [Na+] [Citation58,Citation61]. In MD cells, NO production is activated at a higher luminal Na+ level than that at which adenosine is activated [Citation62]. Thereby, NO is generated when MD luminal [Na+] is substantially elevated. In fact, the high luminal [Na+]-induced increase in GFR was reversed by NO inhibition in rats after subtotal nephrectomy and a high-salt diet [Citation63].
3.7. Angiotensin II modulates glomerular vascular tonicity
The functions of angiotensin II are mediated by angiotensin II type 1 (AT1R) and type 2 (AT2R) receptors, and AT1R mediates most of the known functions of angiotensin II [Citation64]. AT1R is found in mesangial cells, Af-art, Ef-art, JG cells, and renal interstitial cells but is not found in tubular cells, including MD and CNT cells [Citation65]. AT1R is predominantly coupled with Gq/11, which activates signal cascades that increase cytosolic [Ca2+], resulting in the enhancement of arteriole contraction [Citation64].
Transient adenosine treatment has a sustained effect on angiotensin II-induced constriction of isolated perfused Af-art. The adenosine effect is not calcium-dependent, and the involvement of mitogen-activated protein kinase (MAPK) activation has been suggested [Citation66].
3.8. Superoxide mediates Af-art tonicity
Nicotinamide adenine dinucleotide phosphate oxidases (NOXs) produce O2−, and NOX2 and NOX4 have been identified in the MD [Citation67]. An increase in [NaCl] in the MD perfusate from 10 mM to 80 mM induces O2− generation via NOX activation through apical translocation of Rac [Citation68]. O2− production is inhibited at both low and high intracellular pH [Citation69]. Aldosterone and angiotensin II stimulate O2− production via stimulation of NOX2 and NOX4 in the MD [Citation70,Citation71]. O2− constricts the Af-art, both directly and indirectly, by scavenging NO in the MD [Citation72].
3.9. CNT and Af-art
The CNT contacts the parental Af-art and regulates Af-art tonicity [Citation73]. When ENaC in the CNT is activated by high luminal [NaCl], activated ENaC stimulates the production of prostaglandins (PGS) and epoxyeicosatrienoic acids (EETS), which diffuse to and dilate the Af-art [Citation74]. PGE2 exerts a dual effect on renal vascular tone, inducing vasodilatation via prostaglandin E2 and E4 receptors (EP2 and EP4) at low PGE2 concentrations and vasocontraction via EP1 and EP3 at high concentrations [Citation75]. Accordingly, CNT may dilate the Af-art by releasing low-concentration PGE2 in response to high luminal [NaCl] ().
Angiotensin II and aldosterone enhance Af-art dilation by binding to the luminal AT1R and GPR30, respectively. The binding to these receptors leads to activation of ENaC and/or NHE2 through PKC/NOX2/O2− pathway activation in the CNT [Citation76,Citation77]. The CNT may play a final role in sensing luminal sodium in JGA-mediated homeostatic mechanisms.
4. Homeostatic renin–angiotensin system
In the renin–angiotensin system (RAS), angiotensin II is the main effector; renin acts as a rate-limiting enzyme in the formation of angiotensin II [Citation78]. The local (tubular, vascular, and neural) control of renin secretion has been systematically reviewed [Citation79,Citation80] ().
Figure 4. Putative signaling pathways in the regulation of renin secretion in JGA.
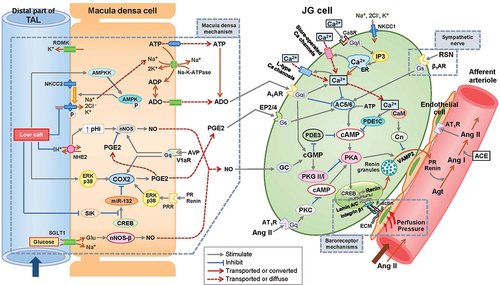
4.1. The cAMP/PKA pathway enhances renin release in JG cells
The cAMP/PKA pathway is the main stimulatory pathway of renin release from JG cells. cAMP synthesis and degradation are mediated by adenylate cyclase (AC) and phosphodiesterase (PDE), respectively. Thus, cAMP is regulated by either activation or inactivation of AC or PDE, and AC and PDE activity is controlled by MD-mediated signaling, including NO, PGE2, and adenosine, as well as by β1-adrenergic receptor (β1AR) signaling activated by circulating or locally released catecholamines [Citation81]. AC and PDE activities are also modulated by intracellular [Ca2+] in the JG cells through calcium-inhibitable AC subtypes 5 and/or 6 [Citation82] and calcium-activated PDE subtype 1 [Citation83]. Decreased [Ca2+] amplifies cAMP-stimulated renin secretion, and increases or decreases in intracellular [Ca2+] activate or inactivate calmodulin (CaM)-dependent calcineurin activity, respectively [Citation27,Citation84].
cAMP leads to the activation of PKA, followed by the phosphorylation of cAMP-responsive element-binding protein (CREB), a transcription factor that binds to the cAMP-responsive element of the renin gene (REN) and elicits REN transcription [Citation85].
The REN transcription product prorenin is either sorted to a vesicular network or directly released constitutively into circulation [Citation78]. Renin that remains stored in the vesicles is released via cAMP-stimulated exocytosis, which requires VAMP2 and synaptosome-associated protein [Citation86,Citation87]. Calcineurin and CaM suppress renin exocytosis independent of PKA [Citation84]. The complete process of renin release remains elusive [Citation88] ().
4.2. Intracellular calcium regulation in JG cells
Intracellular [Ca2+] in JG cells plays a fundamental role in modulating cAMP activity. JG cells express calcium-sensing receptor (CaSR), which senses and translates changes in extracellular [Ca2+] into changes in intracellular [Ca2+] [Citation89]. CaSR activation activates PLC, consequently activating IP3 and releasing calcium from the endoplasmic reticulum (ER) into the cytosol. The subsequent depletion of ER calcium stimulates several pathways that increase intracellular [Ca2+] in JG cells, including the activation of store-operated calcium channels, mobilization of calcium from the ER, and activation of L-type calcium channels [Citation27,Citation90].
4.3. MD-signaling pathways mediating cAMP/PKA activity in JG cells
Adenosine inhibits renin release by stimulating A1AR linked to cGMP [Citation91]. A1AR-dependent factors may play an inhibitory role in renin secretion in cases of renal perfusion pressure (RPP) elevation or decreased salt intake, and A1AR-dependent factors may not have such an inhibitory function under the conditions of suppressed RPP or high salt intake [Citation92,Citation93].
MD-dependent NO exerts dual inhibitory and stimulatory effects on renin release from JG cells. NO inhibits PDE3 by activating GC/cGMP, leading to the activation of the cAMP/PKA pathway and stimulating renin synthesis and release. In contrast, activation of cGMP, cGMP-dependent protein kinase II, and PKG I induced by NO is responsible for the NO inhibition of renin release in JG cells [Citation94–96]. NO production increases only when MD luminal [Na+] is markedly elevated [Citation62], suggesting that NO may have an inhibitory effect on renin release when sodium increases excessively.
PGE2 synthesis in MD is mediated by cyclooxygenase-2 (COX-2), which is regulated by intracellular chloride concentrations and depends on chloride uptake from the tubule lumen by NKCC2. Enhanced COX-2 transcription, triggered by phosphorylated p38 and Erk1/2 kinases, is responsible for decreased luminal [Cl−]-dependent PGE2 secretion [Citation97,Citation98]. PGE2-dependent stimulation of renin secretion is associated with EP2 and EP4 [Citation75].
4.4. Baroreceptors and β-adrenergic receptors in JG cells
Renin secretion is negatively regulated by RPP. Baroreceptors in the JG renin cells sense extracellular perfusion pressure via integrin β1 at the JG cell membrane. Integrin β1 transduces mechanical stimuli to the nuclear membrane and chromatin via lamin A/C, and subsequently regulates renin gene expression [Citation99]. Adenosine signaling via A1AR is critical for the inhibition of renin secretion induced by RPP elevation [Citation92].
Activation of β-adrenergic receptors increases renin secretion via activation of AC independently of renal vascular tone or MD signals [Citation100]. Tonic activation of β-adrenergic receptors is required for maintenance of the releasable renin pool size, and β-adrenergic receptors seem to maintain the magnitude of the change in plasma renin concentration caused by various renin stimulators [Citation81].
4.5. Angiotensin II: regulator of Na+ transport in tubules distal to MD
Angiotensin II and K+ are the main stimulators of adrenal aldosterone secretion. Aldosterone plays a dominant role in the activation of Na+-Cl− transport in the post-MD renal tubules, namely, the distal convoluted tubule (DCT), CNT, and CD, via a plethora of mechanisms that include activation of protein abundance of the thiazide-sensitive sodium-chloride cotransporter (NCC) located in the apical DCT and the amiloride-sensitive ENaC in the apical CNT and CD [Citation101,Citation102]. In addition, angiotensin II directly stimulates Na+ transport independently of aldosterone by mediating NCC and ENaC protein abundance [Citation103,Citation104].
The intra-adrenal RAS seems to work as a local amplifier of the systemic renin–angiotensin–aldosterone system (RAAS) [Citation105]. Angiotensin II-dependent upregulation of renin in the CD depends on PKC-α, which allows the augmentation of cAMP production and activation of the PKA/CREB pathway via AC6 [Citation106]. The reciprocal activation between angiotensin II and renin in the CD may augment the increase in Na+ absorption. The precise mechanisms underlying the regulation of CD-derived renin synthesis remain elusive [Citation107].
The results of angiotensin II function summarized in this section are heterogeneous with respect to the reported intrarenal AT1R distribution [Citation65].
5. Tradeoff between homeostatic adaptation and vulnerable glomerulus
JGA regulation of the Af-art tone may consist of two stages: glomerular protection during normal renal function and glomerular hyperfiltration during reduced renal function.
An increase in sodium delivery to the MD stimulates NKCC2, leading to a negative energy balance and adenosine formation in the MD [Citation48,Citation49]. At a wide range of lower adenosine concentrations, adenosine stimulates A1AR in the Af-art, leading to its contraction. At higher concentrations, adenosine stimulates A2AR, resulting in Af-art relaxation [Citation53–55].
In the glomerular protection stage, Af-art contraction protects the glomerular capillary by decreasing perfusion pressure and reduces tissue hypoxia risk by decreasing tubular sodium reabsorption [Citation108]. The systemic sodium surplus produced by low-set GFR is balanced by inhibition of the RAAS, partly through adenosine stimulation of A1AR in the JG cells [Citation93] ().
Figure 5. (a) Putative homeostatic mechanism of JGA-mediated glomerular regulation with RAAS the Af-art contracts in response to the increased sodium delivery to the MD, thereby mitigating the increase in glomerular blood flow. Therefore, homeostatic glomerular regulatory mechanisms result in glomerulotubular protection. Simultaneously, JGA signaling maintains systemic sodium homeostasis through RAAS suppression (see ‘5 Tradeoff between homeostatic adaptation and vulnerable glomerulus’). (b) Putative mechanism of vicious circle caused by glomerular hyperfiltration for sodium homeostasis in advanced CKD, the baseline per-nephron sodium load is high, and an additional sodium load, such as high salt intake or due to pharmacotherapy, further increases sodium delivery to the MD/CNT. High luminal [Na+] stimulates MD adenosine production, leading to Af-art dilation. A subsequent increase in glomerular sodium filtration itself augments glomerular sodium filtration in a short response (short-term vicious circle), resulting in glomerulotubular injury, leading to a decreased number of functioning nephrons in the long term (long-term vicious circle). Therefore, the per-nephron sodium load on MD/CNT further increases. Short- and long-term vicious circles lead to a trade-off between the acute need for sodium homeostasis and the progression of chronic glomerular sclerosis. (see ‘5 Tradeoff between homeostatic adaptation and vulnerable glomerulus’).
![Figure 5. (a) Putative homeostatic mechanism of JGA-mediated glomerular regulation with RAAS the Af-art contracts in response to the increased sodium delivery to the MD, thereby mitigating the increase in glomerular blood flow. Therefore, homeostatic glomerular regulatory mechanisms result in glomerulotubular protection. Simultaneously, JGA signaling maintains systemic sodium homeostasis through RAAS suppression (see ‘5 Tradeoff between homeostatic adaptation and vulnerable glomerulus’). (b) Putative mechanism of vicious circle caused by glomerular hyperfiltration for sodium homeostasis in advanced CKD, the baseline per-nephron sodium load is high, and an additional sodium load, such as high salt intake or due to pharmacotherapy, further increases sodium delivery to the MD/CNT. High luminal [Na+] stimulates MD adenosine production, leading to Af-art dilation. A subsequent increase in glomerular sodium filtration itself augments glomerular sodium filtration in a short response (short-term vicious circle), resulting in glomerulotubular injury, leading to a decreased number of functioning nephrons in the long term (long-term vicious circle). Therefore, the per-nephron sodium load on MD/CNT further increases. Short- and long-term vicious circles lead to a trade-off between the acute need for sodium homeostasis and the progression of chronic glomerular sclerosis. (see ‘5 Tradeoff between homeostatic adaptation and vulnerable glomerulus’).](/cms/asset/c944c89b-f936-4f59-85c4-ea0034fe0a1c/ieop_a_2357188_f0005_oc.jpg)
As renal function deteriorates, per-nephron sodium delivery to the MD increases further, leading to the activation of A2AR in the Af-art and subsequent Af-art relaxation [Citation54]. In addition, increased sodium load to the CNT stimulates the production of PGE2, leading to Af-art dilation [Citation74]. Moreover, angiotensin II and aldosterone enhance Af-art dilation via binding to their receptors at the CNT lumen [Citation76,Citation77,Citation109]. Thus, at the hyperfiltration stage, glomerular sodium filtration increases, which results in a further increase in sodium load to the MD; this short-term vicious circle leads to a tradeoff between the acute need for sodium homeostasis and augmented glomerular sodium filtration. In the long-term vicious circle, as the number of functioning nephrons decreases owing to glomerulotubular injury, the per-nephron sodium load increases ().
Single-nephron GFR increases to two or more times the normal levels, and suppression of renin release has been demonstrated in animals with subtotal nephrectomy and high-salt diets [Citation63,Citation110,Citation111]. In rats with subtotal nephrectomy, high-salt diets induce renal and cerebral RAS activation and promote oxidative stress, fibrosis, and progression of CKD [Citation112], suggesting that derangement of extrarenal homeostatic mechanisms is linked to renal injury at the glomerular hyperfiltration stage.
Treatment points for different CKD stages are summarized in 7.1 Targeting Pharmacotherapy of CKD ().
Table 2. Points of pharmacotherapy by CKD stages.
6. Conclusions
When sodium delivery to the MD is increased in a kidney with an average functioning nephron mass, the MD produces adenosine, contracts the Af-art, and simultaneously inhibits the RAS via the suppression of renin release from JG cells. Therefore, the kidney maintains sodium homeostasis at low glomerular filtration pressure.
In kidneys with decreased functioning nephron mass, such as in advanced CKD, per-nephron sodium delivery to the MD and MD-produced baseline adenosine concentrations are high. When the sodium load increases further, a high concentration of adenosine stimulates A2AR, dilates the Af-art, and increases sodium filtration to preserve sodium homeostasis at the expense of high filtration pressure (short-term vicious circle). The subsequent nephron mass reduction and enhancement of per-nephron sodium filtration results in a long-term vicious circle toward ESRD ().
Vasopressin increases sodium absorption in the TAL (), causing a decrease in sodium delivery to the MD and an increase in glomerular filtration pressure. The association of long-lasting elevated vasopressin levels with an increased incidence of CKD progression may be explained, at least partly, by vasopressin-stimulated sodium absorption in the TAL.
Pharmacological treatment to increase sodium delivery to the MD by inhibiting sodium reabsorption in the tubules upstream of the MD, while keeping the JGA-mediated homeostatic mechanisms intact, may reduce glomerular load with a low risk of enhancing systemic sodium excess. This may serve as a sustainable and effective way to maintain low glomerular filtration pressure and slow the progression of CKD, including ADPKD.
7. Expert opinion
7.1. Targeting pharmacotherapy of CKD
Blood pressure control, RAS inhibition, disease-specific treatments, and management of individual patient risk factors for CKD progression are the general therapeutic bases for controlling CKD progression. In addition, targeted therapy to reduce glomerular filtration pressure based on JGA-mediated homeostatic mechanisms consists of effective pharmacotherapies.
Sodium – glucose cotransporter 2 (SGLT2) inhibitors lower the risk of sustained decline in eGFR regardless of the presence or absence of diabetes [Citation113]. Although several mechanisms have been postulated for the renoprotective effects of SGLT2 inhibitors, an important mechanism is the inhibition of proximal tubular Na reabsorption, which increases Na supply to the MD and reduces TGF-mediated intraglomerular pressure [Citation114,Citation115]. The mechanism to slow the progression of eGFR decline by SGLT2 inhibitors may be linked to the JGA-mediated homeostatic mechanisms.
The core physiological factors for pharmacotherapy to elicit salutary effects on CKD progression based on JGA-mediated homeostatic mechanisms depend on the pharmacologically induced increase in per-nephron sodium load to the MD; however, this should not be high enough to evoke Af-art relaxation. Therapeutically managed sodium delivery to the MD is influenced by the patients’ CKD stage, the competence of pharmacotherapeutic compounds to inhibit sodium reabsorption in tubule(s) upstream of the MD, and the patients’ systemic sodium balance influenced by sodium intake and other factors, such as comorbidities or co-intake of medicine. The competence of pharmacotherapeutic agents, such as tolvaptan or SGLT2 inhibitors, is difficult to quantify and may vary according to the CKD stage and systemic sodium balance. In contrast, the kidney-protective effects of tolvaptan and SGLT2 inhibitors have been reported in later CKD stages [Citation12,Citation116–118]. Treatment points for different CKD stages may be summarized as follows ():
Ensuring glomerular protection at the stages of normal to moderately decreased kidney function (CKD G1 to G3 in the presence of evidence of kidney damage).
Both glomerular protection and prevention of glomerular hyperfiltration in the severely decreased kidney function stage (CKD G4).
Avoidance of glomerular hyperfiltration during kidney failure (CKD G5).
7.1.1. Pharmacological glomerular protection and avoidance of hyperfiltration
Pharmacological glomerular protection necessitates consideration of all points from 1 to 4 as discussed below, with point 5 elucidating the importance of avoiding glomerular hyperfiltration.
Direct or indirect pharmacological inhibition of sodium reabsorption at the tubular segment(s) upstream of the MD,
Preservation of the function to reabsorb sodium via NKCC2 located at the luminal membrane of the MD cells (main sensing),
Preservation of the function to generate and release adenosine from the MD cells (main signaling),
Preservation of the function of adenosine receptor (A1AR) and subsequent signaling pathways leading to myosin light chain contraction in the VSMC of the glomerular arterioles, and
The avoidance of a large sodium load on the tubular lumen of the MD, leading to the production of a high concentration of adenosine, which may reverse VSMC contraction to relaxation. Af-art relaxation results in diminished glomerular protection. This process is influenced by systemic sodium and fluid homeostasis in individual patients, which depend on kidney function, sodium intake, comorbidities, and co-prescribed medicines that influence JGA-mediated homeostatic mechanisms.
There have been no reports of clinical studies of combined use of tolvaptan and SGLT2 inhibitors for the treatment of ADPKD. The effect of such combined use may be beneficial or harmful depending on the stage of CKD. Increased sodium load to the MD may be beneficial in an early stage but become harmful even in moderately advanced stages. Investigation of the combined use of these drugs is needed to resolve an unmet need in the treatment of patients with ADPKD.
7.1.2. Points of pharmacological glomerular protection in CKD G4 to G5
In CKD stages G4 to G5, the baseline per-nephron glomerular sodium filtration is high because of the decreased number of functioning nephrons. This condition depends on the balance between systemic sodium homeostasis and glomerular injury. Under such conditions, pharmacological enhancement of the sodium load in the MD may amplify Af-art dilatation and augment glomerular hyperfiltration and damage. However, SGLT2 inhibitors and tolvaptan may be beneficial for treating CKD G4 [Citation116–118].
7.2. Diuretics related to JGA-mediated homeostatic mechanisms
7.2.1. Loop diuretics
Loop diuretics are another risk factor for CKD progression, the mechanism of which is JGA-mediated homeostasis. Loop diuretics, including furosemide, bumetanide, and ethacrynic acid, inhibit NKCC2 in the TAL and MD [Citation42]. As sodium reabsorption is inhibited in the MD, the signaling of adenosine in the MD is blunted, allowing for the dilatation of the Af-art to a level corresponding to zero sodium load on the MD (). This may cause an increase in glomerular filtration and lead to glomerular injury in the long term.
Article highlights
When sodium delivery to the macula densa (MD) increases, the MD increases adenosine production, resulting in contraction of the afferent arteriole (Af-art). Simultaneously adenosine inhibits the renin-angiotensin system, leading to maintenance of sodium homeostasis at low glomerular blood flow.
High salt intake causes excessive per-nephron sodium load to the MD in patients with advanced chronic kidney disease (CKD), and the MD increases adenosine production to dilate the Af-art, leading to increased glomerular sodium filtration. This process leads to a vicious circle of sodium load to the MD and causes glomerular injury.
The juxtaglomerular apparatus (JGA)-mediated homeostatic mechanism maintains sodium homeostasis at low energy consumption and protects the kidney in most stages of CKD, but in patients with reduced nephron mass this mechanism maintains sodium homeostasis at the expense of vulnerable glomeruli.
Tolvaptan inhibits sodium reabsorption by suppressing NKCC2 co-transporters in the thick ascending limb and increases sodium delivery to the MD. Thus, in addition to the secondary effect of cyst growth inhibition, tolvaptan may also protect glomerular capillaries via the JGA mechanism.
Sodium-glucose cotransporter 2 inhibitors slow CKD progression probably via the JGA mechanism.
Declaration of interest
T Harada is an employee of Otsuka Pharmaceutical Co., Ltd., Japan. H Fukuhara has received research funding from Otsuka Pharmaceutical Co., Ltd.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Author contributions
E Higashihara designed and wrote the manuscript; T Harada assisted in the preparation of the manuscript; and T Harada and H Fukuhara provided constructive advice for improving the manuscript.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
We would like to thank Yoshihisa Yamada, Hiroyuki Fujiki, and Yukinobu Takeshita of Otsuka Pharmaceutical Co., Ltd., Japan for their constructive advice regarding this manuscript. We thank Diane Williams, Ph.D., of Edanz (www.edanz.com) for providing editing support, which was funded by Otsuka Pharmaceutical Co., Ltd.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Chebib FT, Torres VE. Autosomal dominant polycystic kidney disease: core curriculum 2016. Am J Kidney Dis. 2016;67(5):792–810. doi: 10.1053/j.ajkd.2015.07.037
- Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354(20):2122–2130. doi: 10.1056/NEJMoa054341
- Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015;26(1):160–172. doi: 10.1681/ASN.2013101138
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407–2418. doi: 10.1056/NEJMoa1205511
- Torres VE, Chapman AB, Devuyst O, et al. Multicenter, open-label, extension trial to evaluate the long-term efficacy and safety of early versus delayed treatment with tolvaptan in autosomal dominant polycystic kidney disease: the TEMPO 4: 4 Trial. Nephrol Dial Transplant. 2018;33(3):477–489. doi: 10.1093/ndt/gfx043
- Thurau K, Schnermann J. Die Natriumkonzentration an den Macula densa-Zellen als regulierender Faktor für das Glomerulumfiltrat (Mikropunktionsversuche) [The sodium concentration in the macula densa cells as a regulating factor for glomerular filtration (micropuncture experiments)]. Klin Wochenschr. 1965;43:410–413. German. doi: 10.1007/BF01483845
- Schnermann J, Briggs JP. Synthesis and secretion of renin in mice with induced genetic mutations. Kidney Int. 2012;81(6):529–538. doi: 10.1038/ki.2011.451
- Cortinovis M, Perico N, Ruggenenti P, et al. Glomerular hyperfiltration. Nat Rev Nephrol. 2022;18(7):435–451. doi: 10.1038/s41581-022-00559-y
- Sholokh A, Klussmann E. Local cyclic adenosine monophosphate signalling cascades-Roles and targets in chronic kidney disease. Acta Physiol (Oxf). 2021;232(1):e13641. doi: 10.1111/apha.13641
- Yamaguchi T, Pelling JC, Ramaswamy NT, et al. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int. 2000;57(4):1460–1471. doi: 10.1046/j.1523-1755.2000.00991.x
- Yasuda G, Jeffries WB. Regulation of cAMP production in initial and terminal inner medullary collecting ducts. Kidney Int. 1998;54(1):80–86. doi: 10.1046/j.1523-1755.1998.00990.x
- Gattone VH 2nd, Wang X, Harris PC, et al. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003;9(10):1323–1326. doi: 10.1038/nm935
- Boertien WE, Meijer E, de Jong PE, et al. Short-term renal hemodynamic effects of tolvaptan in subjects with autosomal dominant polycystic kidney disease at various stages of chronic kidney disease. Kidney Int. 2013;84(6):1278–1286. doi: 10.1038/ki.2013.285
- Horie S, Muto S, Kawano H, et al. Preservation of kidney function irrelevant of total kidney volume growth rate with tolvaptan treatment in patients with autosomal dominant polycystic kidney disease. Clin Exp Nephrol. 2021;25(5):467–478. doi: 10.1007/s10157-020-02009-0
- Palmer LG, Schnermann J. Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol. 2015;10(4):676–687. doi: 10.2215/CJN.12391213
- Giménez I, Forbush B. Short-term stimulation of the renal Na-K-Cl cotransporter (NKCC2) by vasopressin involves phosphorylation and membrane translocation of the protein. J Biol Chem. 2003;278(29):26946–26951. doi: 10.1074/jbc.M303435200
- Caceres PS, Mendez M, Ortiz PA. Vesicle-associated membrane protein 2 (VAMP2) but Not VAMP3 mediates cAMP-stimulated trafficking of the renal Na±K±2Cl- co-transporter NKCC2 in thick ascending limbs. J Biol Chem. 2014;289(34):23951–23962. doi: 10.1074/jbc.M114.589333
- Caceres PS, Mendez M, Haque MZ, et al. Vesicle-associated membrane protein 3 (VAMP3) mediates constitutive trafficking of the Renal Co-transporter NKCC2 in thick ascending limbs: role in renal function and blood pressure. J Biol Chem. 2016;291(42):22063–22073. doi: 10.1074/jbc.M116.735167
- Fraser SA, Gimenez I, Cook N, et al. Regulation of the renal-specific Na±K±2Cl- co-transporter NKCC2 by AMP-activated protein kinase (AMPK). Biochem J. 2007;405(1):85–93. doi: 10.1042/BJ20061850
- Richardson C, Sakamoto K, de Los Heros P, et al. Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J Cell Sci. 2011;124(Pt 5):789–800. doi: 10.1242/jcs.077230
- Gunaratne R, Braucht DW, Rinschen MM, et al. Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc Natl Acad Sci USA. 2010;107(35):15653–15658. doi: 10.1073/pnas.1007424107
- Nonoguchi H, Owada A, Kobayashi N, et al. Immunohistochemical localization of V2 vasopressin receptor along the nephron and functional role of luminal V2 receptor in terminal inner medullary collecting ducts. J Clin Invest. 1995;96(4):1768–1778. doi: 10.1172/JCI118222
- Terada Y, Tomita K, Nonoguchi H, et al. Different localization and regulation of two types of vasopressin receptor messenger RNA in microdissected rat nephron segments using reverse transcription polymerase chain reaction. J Clin Invest. 1993;92(5):2339–2345. doi: 10.1172/JCI116838
- Mutig K, Paliege A, Kahl T, et al. Vasopressin V2 receptor expression along rat, mouse, and human renal epithelia with focus on TAL. Am J Physiol Renal Physiol. 2007;293(4):F1166–77. doi: 10.1152/ajprenal.00196.2007
- Mutig K, Borowski T, Boldt C, et al. Demonstration of the functional impact of vasopressin signaling in the thick ascending limb by a targeted transgenic rat approach. Am J Physiol Renal Physiol. 2016;311(2):F411–23. doi: 10.1152/ajprenal.00126.2016
- Rieg T, Tang T, Uchida S, et al. Adenylyl cyclase 6 enhances NKCC2 expression and mediates vasopressin-induced phosphorylation of NKCC2 and NCC. Am J Pathol. 2013;182(1):96–106. doi: 10.1016/j.ajpath.2012.09.014
- Jesus Ferreira MC D, Bailly C. Extracellular Ca2+ decreases chloride reabsorption in rat CTAL by inhibiting cAMP pathway. Am J Physiol. 1998;275(2):F198–F203. doi: 10.1152/ajprenal.1998.275.2.F198
- Edwards ME, Chebib FT, Irazabal MV, et al. Long-term administration of tolvaptan in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2018;13(8):1153–1161. doi: 10.2215/CJN.01520218
- Higashihara E, Nutahara K, Itoh M, et al. Long-term outcomes of longitudinal efficacy study with tolvaptan in autosomal dominant polycystic kidney disease. Kidney Int Rep. 2022;7(2):270–281. doi: 10.1016/j.ekir.2021.11.034
- Beierwaltes WH. The role of calcium in the regulation of renin secretion. Am J Physiol Renal Physiol. 2010;298(1):F1–F11. doi: 10.1152/ajprenal.00143.2009
- Lorenzi T, Graciotti L, Sagrati A, et al. Normal human macula densa morphology and cell turnover: a histological, ultrastructural, and immunohistochemical investigation. Anat Rec. 2020;303(11):2904–2916. doi: 10.1002/ar.24465
- Romero CA, Carretero OA. Tubule-vascular feedback in renal autoregulation. Am J Physiol Renal Physiol. 2019;316(6):F1218–26. doi: 10.1152/ajprenal.00381.2018
- Castrop H, Schiessl IM. Physiology and pathophysiology of the renal Na-K-2Cl cotransporter (NKCC2). Am J Physiol Renal Physiol. 2014;307(9):F991–F1002. doi: 10.1152/ajprenal.00432.2014
- Peti-Peterdi J, Chambrey R, Bebok Z, et al. Macula densa Na(+)/H(+) exchange activities mediated by apical NHE2 and basolateral NHE4 isoforms. Am J Physiol Renal Physiol. 2000;278(3):F452–63. doi: 10.1152/ajprenal.2000.278.3.F452
- Wang WH. Regulation of ROMK (Kir1.1) channels: new mechanisms and aspects. Am J Physiol Renal Physiol. 2006;290(1):F14–9. doi: 10.1152/ajprenal.00093.2005
- Peti-Peterdi J, Bebok Z, Lapointe JY, et al. Novel regulation of cell [Na(+)] in macula densa cells: apical Na(+) recycling by H-K-ATPase. Am J Physiol Renal Physiol. 2002;282(2):F324–9. doi: 10.1152/ajprenal.00251.2001
- Reilly RF, Ellison DH. Mammalian distal tubule: physiology, pathophysiology, and molecular anatomy. Physiol Rev. 2000;80(1):277–313. doi: 10.1152/physrev.2000.80.1.277
- Briggs JP. A simple steady-state model for feedback control of glomerular filtration rate. Kidney Int Suppl. 1982;12:S143–S150.
- Peti-Peterdi J, Morishima S, Bell PD, et al. Two-photon excitation fluorescence imaging of the living juxtaglomerular apparatus. Am J Physiol Renal Physiol. 2002;283(1):F197–F201. doi: 10.1152/ajprenal.00356.2001
- Thomson S, Vallon V, Blantz RC. Asymmetry of tubuloglomerular feedback effector mechanism with respect to ambient tubular flow. Am J Physiol. 1996;271(6 Pt 2):F1123–30. doi: 10.1152/ajprenal.1996.271.6.F1123
- Ren Y, Garvin JL, Carretero OA. Efferent arteriole tubuloglomerular feedback in the renal nephron. Kidney Int. 2001;59(1):222–229. doi: 10.1046/j.1523-1755.2001.00482.x
- Wright FS, Schnermann J. Interference with feedback control of glomerular filtration rate by furosemide, triflocin, and cyanide. J Clin Invest. 1974;53(6):1695–1708. doi: 10.1172/JCI107721
- Gimenez I, Isenring P, Forbush B. Spatially distributed alternative splice variants of the renal Na-K-Cl cotransporter exhibit dramatically different affinities for the transported ions. J Biol Chem. 2002;277(11):8767–8770. doi: 10.1074/jbc.C200021200
- Plata C, Meade P, Vazquez N, et al. Functional properties of the apical Na±K±2Cl- cotransporter isoforms. J Biol Chem. 2002;277(13):11004–11012. doi: 10.1074/jbc.M110442200
- Schiessl IM, Rosenauer A, Kattler V, et al. Dietary salt intake modulates differential splicing of the Na-K-2Cl cotransporter NKCC2. Am J Physiol Renal Physiol. 2013;305(8):F1139–48. doi: 10.1152/ajprenal.00259.2013
- Oppermann M, Mizel D, Huang G, et al. Macula densa control of renin secretion and preglomerular resistance in mice with selective deletion of the B isoform of the Na,K,2Cl co-transporter. J Am Soc Nephrol. 2006;17(8):2143–2152. doi: 10.1681/ASN.2006040384
- Oppermann M, Mizel D, Kim SM, et al. Renal function in mice with targeted disruption of the a isoform of the Na-K-2Cl co-transporter. J Am Soc Nephrol. 2007;18(2):440–448. doi: 10.1681/ASN.2006091070
- Beach RE, Watts BA 3rd, Good DW, et al. Effects of graded oxygen tension on adenosine release by renal medullary and thick ascending limb suspensions. Kidney Intern. 1991;39(5):836–842. doi: 10.1038/ki.1991.105
- Vallon V, Osswald H. Adenosine receptors and the kidney. In: Wilson C Mustafa S, editors. Adenosine receptors in health and disease. Handbook of experimental pharmacology. Vol. 193. Berlin (Heidelberg): Springer; 2009. p. 443–470.
- Brown R, Ollerstam A, Johansson B, et al. Abolished tubuloglomerular feedback and increased plasma renin in adenosine A1 receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2001;281(5):R1362–67. doi: 10.1152/ajpregu.00470.2001
- Touyz RM, Alves-Lopes R, Rios FJ, et al. Vascular smooth muscle contraction in hypertension. Cardiovasc Res. 2018;114(4):529–539. doi: 10.1093/cvr/cvy023
- Francis SH, Busch JL, Corbin JD, et al. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62(3):525–563. doi: 10.1124/pr.110.002907
- Li L, Lai EY, Huang Y, et al. Renal afferent arteriolar and tubuloglomerular feedback reactivity in mice with conditional deletions of adenosine 1 receptors. Am J Physiol Renal Physiol. 2012;303(8):F1166–75. doi: 10.1152/ajprenal.00222.2012
- Carlström M, Wilcox CS, Welch WJ. Adenosine A2A receptor activation attenuates tubuloglomerular feedback responses by stimulation of endothelial nitric oxide synthase. Am J Physiol Renal Physiol. 2011;300(2):F457–64. doi: 10.1152/ajprenal.00567.2010
- Nishiyama A, Inscho EW, Navar LG. Interactions of adenosine A1 and A2a receptors on renal microvascular reactivity. Am J Physiol Renal Physiol. 2001;280(3):F406–14. doi: 10.1152/ajprenal.2001.280.3.F406
- Lai EY, Patzak A, Steege A, et al. Contribution of adenosine receptors in the control of arteriolar tone and adenosine-angiotensin II interaction. Kidney Int. 2006;70(4):690–698. doi: 10.1038/sj.ki.5001650
- Liu R, Carretero OA, Ren Y, et al. Increased intracellular pH at the macula densa activates nNOS during tubuloglomerular feedback. Kidney Int. 2005;67(5):1837–1843. doi: 10.1111/j.1523-1755.2005.00282.x
- Wang H, Carretero OA, Garvin JL. Inhibition of apical Na+/H+ exchangers on the macula densa cells augments tubuloglomerular feedback. Hypertension. 2003;41(3 Pt 2):688–691. doi: 10.1161/01.HYP.0000048863.75711.B2
- Wang L, Shen C, Liu H, et al. Shear stress blunts tubuloglomerular feedback partially mediated by primary cilia and nitric oxide at the macula densa. Am J Physiol Regul Integr Comp Physiol. 2015;309(7):R757–66. doi: 10.1152/ajpregu.00173.2015
- Zhang J, Wei J, Jiang S, et al. Macula densa SGLT1-NOS1-tubuloglomerular feedback pathway, a new mechanism for glomerular hyperfiltration during hyperglycemia. J Am Soc Nephrol. 2019;30(4):578–593. doi: 10.1681/ASN.2018080844
- Lorenz JN, Dostanic-Larson I, Shull GE, et al. Ouabain inhibits tubuloglomerular feedback in mutant mice with ouabain-sensitive alpha1 Na,K-ATPase. J Am Soc Nephrol. 2006;17(9):2457–2463. doi: 10.1681/ASN.2006040379
- Kovacs G, Komlosi P, Fuson A, et al. Neuronal nitric oxide synthase: its role and regulation in macula densa cells. J Am Soc Nephrol. 2003;14(10):2475–2483. doi: 10.1097/01.ASN.0000088737.05283.2B
- Thomson SC. Nitric oxide mediates anomalous tubuloglomerular feedback in rats fed high-NaCl diet after subtotal nephrectomy. Am J Physiol Renal Physiol. 2019;316(2):F223–30. doi: 10.1152/ajprenal.00066.2018
- de Gasparo M, Catt KJ, Inagami T, et al. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52(3):415–472.
- Schrankl J, Fuchs M, Broeker K, et al. Localization of angiotensin II type 1 receptor gene expression in rodent and human kidneys. Am J Physiol Renal Physiol. 2021;320(4):F644–53. doi: 10.1152/ajprenal.00550.2020
- Patzak A, Lai EY, Fahling M, et al. Adenosine enhances long term the contractile response to angiotensin II in afferent arterioles. Am J Physiol Regul Integr Comp Physiol. 2007;293(6):R2232–42. doi: 10.1152/ajpregu.00357.2007
- Sedeek M, Nasrallah R, Touyz RM, et al. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol. 2013;24(10):1512–1518. doi: 10.1681/ASN.2012111112
- Liu R, Juncos LA. GTPase-rac enhances depolarization-induced superoxide production by the macula densa during tubuloglomerular feedback. Am J Physiol Regul Integr Comp Physiol. 2010;298(2):R453–8. doi: 10.1152/ajpregu.00622.2009
- Liu R, Carretero OA, Ren Y, et al. Intracellular pH regulates superoxide production by the macula densa. Am J Physiol Renal Physiol. 2008;295(3):F851–6. doi: 10.1152/ajprenal.90204.2008
- Zhang J, Chandrashekar K, Lu Y, et al. Enhanced expression and activity of Nox2 and Nox4 in the macula densa in ANG II-induced hypertensive mice. Am J Physiol Renal Physiol. 2014;306(3):F344–50. doi: 10.1152/ajprenal.00515.2013
- Zhu X, Rd M Jr, Lu D, et al. Aldosterone stimulates superoxide production in macula densa cells. Am J Physiol Renal Physiol. 2011;301(3):F529–35. doi: 10.1152/ajprenal.00596.2010
- Liu R, Ren Y, Garvin JL, et al. Superoxide enhances tubuloglomerular feedback by constricting the afferent arteriole. Kidney Int. 2004;66(1):268–274. doi: 10.1111/j.1523-1755.2004.00727.x
- Ren Y, Garvin JL, Liu R, et al. Crosstalk between the connecting tubule and the afferent arteriole regulates renal microcirculation. Kidney Int. 2007;71(11):1116–1121. doi: 10.1038/sj.ki.5002190
- Ren Y, D’Ambrosio MA, Garvin JL, et al. Prostaglandin E2 mediates connecting tubule glomerular feedback. Hypertension. 2013;62(6):1123–1128. doi: 10.1161/HYPERTENSIONAHA.113.02040
- Schweda F, Klar J, Narumiya S, et al. Stimulation of renin release by prostaglandin E2 is mediated by EP2 and EP4 receptors in mouse kidneys. Am J Physiol Renal Physiol. 2004;287(3):F427–33. doi: 10.1152/ajprenal.00072.2004
- Ren Y, D’Ambrosio MA, Wang H, et al. Mechanisms of angiotensin II-enhanced connecting tubule glomerular feedback. Am J Physiol Renal Physiol. 2012;303(2):F259–65. doi: 10.1152/ajprenal.00689.2011
- Ren Y, Janic B, Kutskill K, et al. Mechanisms of connecting tubule glomerular feedback enhancement by aldosterone. Am J Physiol Renal Physiol. 2016;311(6):F1182–8. doi: 10.1152/ajprenal.00076.2016
- Kurtz A. Control of renin synthesis and secretion. Am J Hypertens. 2012;25(8):839–847. doi: 10.1038/ajh.2011.246
- Castrop H, Hocherl K, Kurtz A, et al. Physiology of kidney renin. Physiol Rev. 2010;90(2):607–673. doi: 10.1152/physrev.00011.2009
- Sequeira-Lopez MLS, Gomez RA. Renin cells, the kidney, and hypertension. Circ Res. 2021;128(7):887–907. doi: 10.1161/CIRCRESAHA.121.318064
- Kim SM, Chen L, Faulhaber-Walter R, et al. Regulation of renin secretion and expression in mice deficient in beta1- and beta2-adrenergic receptors. Hypertension. 2007;50(1):103–109. doi: 10.1161/HYPERTENSIONAHA.107.087577
- Aldehni F, Tang T, Madsen K, et al. Stimulation of renin secretion by catecholamines is dependent on adenylyl cyclases 5 and 6. Hypertension. 2011;57(3):460–468. doi: 10.1161/HYPERTENSIONAHA.110.167130
- Ortiz-Capisano MC, Liao TD, Ortiz PA, et al. Calcium-dependent phosphodiesterase 1C inhibits renin release from isolated juxtaglomerular cells. Am J Physiol Regul Integr Comp Physiol. 2009;297(5):R1469–76. doi: 10.1152/ajpregu.00121.2009
- Madsen K, Friis UG, Gooch JL, et al. Inhibition of calcineurin phosphatase promotes exocytosis of renin from juxtaglomerular cells. Kidney Int. 2010;77(2):110–117. doi: 10.1038/ki.2009.418
- Gomez RA, Sequeira-Lopez MLS. Renin cells in homeostasis, regeneration and immune defence mechanisms. Nat Rev Nephrol. 2018;14(4):231–245. doi: 10.1038/nrneph.2017.186
- Mendez M, Gross KW, Glenn ST, et al. Vesicle-associated membrane protein-2 (VAMP2) mediates cAMP-stimulated renin release in mouse juxtaglomerular cells. J Biol Chem. 2011;286(32):28608–28618. doi: 10.1074/jbc.M111.225839
- Mendez M, Gaisano HY. Role of the SNARE protein SNAP23 on cAMP-stimulated renin release in mouse juxtaglomerular cells. Am J Physiol Renal Physiol. 2013;304(5):F498–04. doi: 10.1152/ajprenal.00556.2012
- Mendez M. Renin release: role of SNAREs. Am J Physiol Regul Integr Comp Physiol. 2014;307(5):R484–6. doi: 10.1152/ajpregu.00175.2014
- Ortiz-Capisano MC, Ortiz PA, Garvin JL, et al. Expression and function of the calcium-sensing receptor in juxtaglomerular cells. Hypertension. 2007;50(4):737–743. doi: 10.1161/HYPERTENSIONAHA.107.095158
- Ortiz-Capisano MC, Reddy M, Mendez M, et al. Juxtaglomerular cell CaSR stimulation decreases renin release via activation of the PLC/IP(3) pathway and the ryanodine receptor. Am J Physiol Renal Physiol. 2013;304(3):F248–56. doi: 10.1152/ajprenal.00451.2012
- Vallon V, Muhlbauer B, Osswald H. Adenosine and kidney function. Physiol Rev. 2006;86(3):901–940. doi: 10.1152/physrev.00031.2005
- Schweda F, Segerer F, Castrop H, et al. Blood pressure-dependent inhibition of renin secretion requires A1 adenosine receptors. Hypertension. 2005;46(4):780–786. doi: 10.1161/01.HYP.0000183963.07801.65
- Schweda F, Wagner C, Kramer BK, et al. Preserved macula densa-dependent renin secretion in A1 adenosine receptor knockout mice. Am J Physiol Renal Physiol. 2003;284(4):F770–7. doi: 10.1152/ajprenal.00280.2002
- Sayago CM, Beierwaltes WH. Nitric oxide synthase and cGMP-mediated stimulation of renin secretion. Am J Physiol Regul Integr Comp Physiol. 2001;281(4):R1146–51. doi: 10.1152/ajpregu.2001.281.4.R1146
- Kurtz A, Gotz KH, Hamann M, et al. Stimulation of renin secretion by nitric oxide is mediated by phosphodiesterase 3. Proc Natl Acad Sci USA. 1998;95(8):4743–4747. doi: 10.1073/pnas.95.8.4743
- Gambaryan S, Wagner C, Smolenski A, et al. Endogenous or overexpressed cGMP-dependent protein kinases inhibit cAMP-dependent renin release from rat isolated perfused kidney, microdissected glomeruli, and isolated juxtaglomerular cells. Proc Natl Acad Sci USA. 1998;95(15):9003–9008. doi: 10.1073/pnas.95.15.9003
- Yang T, Park JM, Arend L, et al. Low chloride stimulation of prostaglandin E2 release and cyclooxygenase-2 expression in a mouse macula densa cell line. J Biol Chem. 2000;275(48):37922–37929. doi: 10.1074/jbc.M006218200
- Cheng HF, Wang JL, Zhang MZ, et al. Role of p38 in the regulation of renal cortical cyclooxygenase-2 expression by extracellular chloride. J Clin Invest. 2000;106(5):681–688. doi: 10.1172/JCI10318
- Watanabe H, Belyea BC, Paxton RL, et al. Renin cell baroreceptor, a nuclear mechanotransducer central for homeostasis. Circ Res. 2021;129(2):262–276. doi: 10.1161/CIRCRESAHA.120.318711
- DiBona GF, Kopp UC. Neural control of renal function. Physiol Rev. 1997;77(1):75–197. doi: 10.1152/physrev.1997.77.1.75
- Loffing J, Kaissling B. Sodium and calcium transport pathways along the mammalian distal nephron: from rabbit to human. Am J Physiol Renal Physiol. 2003;284(4):F628–43. doi: 10.1152/ajprenal.00217.2002
- Mount DB. Transport of sodium, chloride, and potassium. In: Skorecki K, Chertow G Marsden P, editors, et al. Brenner and rector’s the kidney. 10th ed. Amsterdam: Elsevier; 2015.
- Beutler KT, Masilamani S, Turban S, et al. Long-term regulation of ENaC expression in kidney by angiotensin II. Hypertension. 2003;41(5):1143–1150. doi: 10.1161/01.HYP.0000066129.12106.E2
- Brooks HL, Allred AJ, Beutler KT, et al. Targeted proteomic profiling of renal Na(+) transporter and channel abundances in angiotensin II type 1a receptor knockout mice. Hypertension. 2002;39(2 Pt 2):470–473. doi: 10.1161/hy02t2.102959
- Spat A, Hunyady L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev. 2004;84(2):489–539. doi: 10.1152/physrev.00030.2003
- Gonzalez AA, Liu L, Lara LS, et al. PKC-alpha-dependent augmentation of cAMP and CREB phosphorylation mediates the angiotensin II stimulation of renin in the collecting duct. Am J Physiol Renal Physiol. 2015;309(10):F880–8. doi: 10.1152/ajprenal.00155.2015
- Prieto MC, Gonzalez AA, Visniauskas B, et al. The evolving complexity of the collecting duct renin-angiotensin system in hypertension. Nat Rev Nephrol. 2021;17(7):481–492. doi: 10.1038/s41581-021-00414-6
- Evans RG, Gardiner BS, Smith DW, et al. Intrarenal oxygenation: unique challenges and the biophysical basis of homeostasis. Am J Physiol Renal Physiol. 2008;295(5):F1259–70. doi: 10.1152/ajprenal.90230.2008
- Ren Y, D’Ambrosio MA, Garvin JL, et al. Angiotensin II enhances connecting tubule glomerular feedback. Hypertension. 2010;56(4):636–642. doi: 10.1161/HYPERTENSIONAHA.110.153692
- Singh P, Deng A, Blantz RC, et al. Unexpected effect of angiotensin AT1 receptor blockade on tubuloglomerular feedback in early subtotal nephrectomy. Am J Physiol Renal Physiol. 2009;296(5):F1158–65. doi: 10.1152/ajprenal.90722.2008
- Hostetter TH, Olson JL, Rennke HG, et al. Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol. 1981;241(1):F85–F93. doi: 10.1152/ajprenal.1981.241.1.F85
- Cao W, Li A, Wang L, et al. A salt-induced reno-cerebral reflex activates renin-angiotensin systems and promotes CKD progression. J Am Soc Nephrol. 2015;26(7):1619–1633. doi: 10.1681/ASN.2014050518
- Perkovic V, Jardine MJ, Neal B, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. 2019;380(24):2295–2306. doi: 10.1056/NEJMoa1811744
- Heerspink HJL, Kosiborod M, Inzucchi SE, et al. Renoprotective effects of sodium-glucose cotransporter-2 inhibitors. Kidney Int. 2018;94(1):26–39. doi: 10.1016/j.kint.2017.12.027
- Palmer BF, Clegg DJ. Kidney-protective effects of SGLT2 inhibitors. Clin J Am Soc Nephrol. 2023;18(2):279–289. doi: 10.2215/CJN.09380822
- Bakris G, Oshima M, Mahaffey KW, et al. Effects of canagliflozin in patients with baseline eGFR <30 ml/min per 1.73 m2: subgroup analysis of the randomized CREDENCE trial. Clin J Am Soc Nephrol. 2020;15(12):1705–1714. doi: 10.2215/CJN.10140620
- Chertow GM, Vart P, Jongs N, et al. Effects of dapagliflozin in stage 4 chronic kidney disease. J Am Soc Nephrol. 2021;32(9):2352–2361. doi: 10.1681/ASN.2021020167
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med. 2017;377(20):1930–1942. doi: 10.1056/NEJMoa1710030