
Disclaimer
As a service to authors and researchers we are providing this version of an accepted manuscript (AM). Copyediting, typesetting, and review of the resulting proofs will be undertaken on this manuscript before final publication of the Version of Record (VoR). During production and pre-press, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal relate to these versions also.1. Introduction
CHAPLE disease, also known as complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy (PLE), is a rare, severe multisystemic disorder. It is caused by biallelic loss-of-function mutations in the CD55 gene, also known as the decay-accelerating factor, which plays a crucial role in regulating the complement system [1]. CD55 helps regulate the C3- and C5-convertase enzymes of both the classical and alternative pathways. A genetic deficiency in CD55 leads to complement hyperactivation beyond the regulatory step. The defining feature of CHAPLE disease is primary intestinal lymphangiectasia (IL), marked by abnormal dilation of intestinal lymphatic vessels [2]. These dilated vessels allow lymph fluid to leak into the intestinal lumen, leading to serum protein depletion, known as PLE. In PLE, critical proteins such as albumin and immunoglobulins (Ig) are significantly reduced, along with various other serum proteins [3,4].
In CHAPLE disease, IL arises from inflammatory attacks initiated by the overactivation of the complement pathway, which also triggers subsequent immune responses. This inflammatory activity is largely confined to the gastrointestinal tract, likely due to the specific expression patterns of the CD55 protein. Studies have shown that CD59, a key regulator of the terminal complement complex, is found in blood vascular endothelial cells but is missing from lymphatic endothelium. Thus, in CHAPLE patients, the absence of CD55 renders the lymphatic endothelium susceptible to unchecked complement activation [5].
Besides PLE, CHAPLE patients face a significantly heightened risk of life-threatening systemic thrombosis. CHAPLE disease is regarded as one of the most severe thrombophilic disorders in childhood, with most individuals remaining at high risk for thrombosis as they age into adulthood. The underlying mechanisms of this serious complication are multifaceted; PLE itself may contribute by causing a loss of anticoagulatory proteins in the gut, thereby reducing their serum levels. However, a potentially more critical factor is the cross-activation of the coagulation pathway by complement mediators. This mechanism is also observed in systemic complement disorders such as atypical hemolytic uremic syndrome (aHUS) and paroxysmal nocturnal hemoglobinuria (PNH) [6,7]. Understanding the complex pathophysiology underlying these associations is crucial for the effective management of each condition.
CHAPLE disease presents with a wide range of severe clinical manifestations, including diarrhea, vomiting, abdominal pain, edema, loss of appetite, weight loss, ascites, and effusions in the pericardial and pleural spaces. Hypoproteinemia and intestinal malabsorption result in metabolic starvation, hindering growth. The associated Ig deficiency increases susceptibility to recurrent, sometimes severe systemic infections. Chronic gastrointestinal inflammation and ulceration can cause bowel obstruction, often requiring intestinal resection surgeries [3].
2. Anti-complement Therapies
Soon after the discovery of CHAPLE disease, complement inhibitor therapies were tested for clinical efficacy (). These treatments aim to control disease pathogenesis using a pathway-directed approach ().
Figure 1: Complement pathways and target of C5 inhibitors.
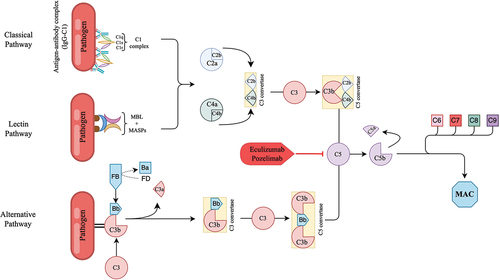
Table 1: Comparative analysis of eculizumab and pozelimab features.
2.1. Eculizumab
Eculizumab is a humanized monoclonal antibody that targets and inhibits complement component 5 (C5), preventing the formation of the terminal complement complex (C5b-9 or membrane attack complex) and thus blocking the harmful effects of uncontrolled complement activation [8].
CHAPLE patients treated with eculizumab showed rapid improvement in symptoms, hypoproteinemia, quality of life, and growth [3,9]. These improvements were linked to the normalization of serum proteomic and fecal microbiotal composition, endoscopic healing of IL, and radiologic normalization of structural features [3]. However, while eculizumab demonstrated remarkable efficacy in managing CHAPLE disease symptoms, the findings also highlighted the transient nature of its effects. Upon discontinuing eculizumab treatment, some patients experience symptom flare-ups, accompanied by serum albumin and Ig loss. This underscores the ongoing activation of complement and its innate immune and inflammatory effector mechanisms in CHAPLE disease pathogenesis. Unpublished observations suggest that another subset of patients may tolerate shorter-term off-therapy once a full and sustained remission is achieved. However, the factors determining immediate flare-ups remain unknown. Currently, it is considered that CHAPLE patients will need sustained C5 inhibition therapy to control its manifestations. Given its life-saving potential, eculizumab represents a significant therapeutic breakthrough in the management of CHAPLE disease.
It is well established that complement inhibitor therapy may result in fatal meningococcal infections. Therefore, meningococcal vaccination must be completed at least two weeks before the first dose of eculizumab treatment. Additionally, antibiotic prophylaxis against meningococcal infections should be seriously considered throughout the duration of treatment. Despite these precautions, meningococcal infections remain a significant concern. Patients should be well-trained to recognize the signs of such infections and instructed to report immediately to physicians for evaluation.
One of the significant challenges with eculizumab therapy is its mode of administration, requiring intravenous infusion every two weeks. This regimen can be burdensome for patients, particularly those living in regions with limited access to specialized medical facilities or in countries where the treatment is unavailable. The necessity for continuous use of this costly treatment further exacerbates its financial burden. Additionally, eculizumab has not received the US Food and Drug Administration (FDA) approval for this indication and is being used off-label in the treatment of CHAPLE disease, making inaccessibility to medication a serious concern for many patients.
2.2. Pozelimab
Pozelimab, a fully human IgG4 antibody administered via subcutaneous injection, targets complement component 5 (C5). IgG4P belongs to the IgG4 class of antibodies but includes a proline substitution to promote light chain stabilization, enhancing its stability and effectiveness. An earlier investigation demonstrated that pozelimab hinders the cleavage of complement component 5 and the formation of the membrane attack complex (MAC) [10]. The drug has been tested in clinical trials in various complement C5-mediated conditions ().
Table 2: Summary of completed trials of pozelimab monotherapy.
During the phase II/III clinical trial (NCT04209634) involving 10 patients diagnosed with CHAPLE disease, the administration of pozelimab resulted in rapid and significant improvements in total protein and Ig concentrations in patients [11]. The majority of participants achieved normal albumin levels (≥ 3.5 g/dL) by week 4, and all reached this level by week 12. These levels were maintained within the normal range for at least 72 weeks of treatment [11].
The high levels of fecal α-1-antitrypsin detected in patients serve as valuable indicators of enteric protein loss and active disease pathology in CHAPLE disease. Following pozelimab treatment, these elevated levels rapidly normalized, indicating a reduction in enteric protein loss and attenuation of disease activity. This normalization suggests that pozelimab effectively targets the underlying mechanisms contributing to protein loss in CHAPLE disease, thereby restoring intestinal barrier function and improving protein retention [11].
The clinical outcomes observed in all ten patients with CHAPLE disease at week 24 were notably positive, with each patient experiencing a complete resolution of the core signs and symptoms of the condition [11]. In addition to the objective clinical improvements observed, patients reported significant improvements in their overall quality of life, physical functioning, and emotional well-being. Furthermore, caregivers also noted positive changes in their well-being, including reduced stress and burden associated with caring for individuals with CHAPLE disease.
This first and only clinical trial presented compelling evidence regarding the therapeutic efficacy of pozelimab, a single-agent targeted therapy, in the context of CHAPLE disease. By specifically inhibiting complement overactivation, pozelimab demonstrates the capacity to ameliorate the diverse clinical manifestations associated with this severe and ultra-rare condition. The initial loading dose of pozelimab is administered via intravenous infusion, while maintenance doses are administered weekly by subcutaneous injection. The subcutaneous route of administration offers convenience and holds promise for improving treatment accessibility, especially in regions where resources are scarce, or healthcare infrastructure is limited. Also, treatment with pozelimab, a complement inhibitor, requires the administration of meningococcal vaccines and prophylactic antibiotics, as with eculizumab treatment.
As a result of these findings, FDA announced on February 21, 2023, that it had accepted for Priority Review the Biologics License Application (BLA) for pozelimab. The approved the use of the drug on August 18, 2023, as a treatment for adults and children younger than one year of age with CHAPLE disease [12]. Currently, pozelimab therapy is available through a compassionate use program, providing the medication at no cost to patients. This access has been a significant benefit for many patients worldwide.
Further research and clinical investigation are warranted to fully elucidate the long-term efficacy and safety profile of pozelimab, as well as its potential application in other complement-mediated disorders.
2.3. Other anti-complement therapies
The elucidation of the mechanism underlying CHAPLE disease has led to the trial of eculizumab, which has been used for a relatively long time in the treatment of other complement-mediated diseases such as PNH and aHUS. Eculizumab was the first targeted therapy tested in CHAPLE patients, resulting in rapid and significant clinical improvement due to its mechanism of action. Today, a growing number of anti-C5 therapeutic options have been developed through various production technologies (mAbs, RNAi, and PEGylated RNA aptamers), all sharing similar mechanisms of action [13]. It can be predicted that besides eculizumab and pozelimab, other anti-C5 therapies targeting terminal complement pathways, such as ravulizumab [14,15], crovalimab [16,17], tesidolumab [18], zilucoplan [19], and cemdisiran [20], which are either already in clinical use or undergoing clinical trials, could be effective in CHAPLE disease.
Besides C5-blockers, different classes of drugs that inhibit the central step of C3 activation and the alternative pathway amplification loop have been explored in PNH. Pegcetacoplan (Empaveli), a compstatin-based C3 inhibitor, received FDA approval for treating adults with PNH in May 2021 [21]. In 2023, an oral, selective small-molecule inhibitor of factor B inhibitor iptacopan (Febhalta) [22] and, in 2024, the factor D inhibitor danicopan (Voydeya) [23] received approval for the management of PNH. These inhibitors of the alternative complement pathway modulate the cleavage of C3, the generation of downstream effectors, and the amplification of the terminal complement cascade. By controlling complement activation at these early stages, both inhibitors could help alleviate symptoms associated with complement hyperactivation and mitigate tissue damage in other complement-mediated diseases, such as CHAPLE disease. Future studies should explore whether inhibiting the central step of C3 can be an effective strategy in CHAPLE disease similar to PNH.
3. Gene therapy for complement-mediated diseases
Although gene therapy has thus far been applied to only a limited number of specific diseases [24,25,26,27], it holds significant promise for the treatment of complement-mediated diseases. Preclinical and clinical research has demonstrated that gene therapy could potentially offer substantial therapeutic benefits by targeting the underlying genetic abnormalities associated with these disorders. Despite these promising advancements, no gene therapy-based treatments for complement-mediated diseases have yet progressed to the stage of clinical implementation [28].
Currently, gene therapy is in the experimental stages for conditions such as age-related macular degeneration (AMD) and PHN, with ongoing clinical trials aimed at evaluating its efficacy and safety [28]. These trials are anticipated to provide valuable insights and could lead to positive outcomes in the near future.
One of the major concerns associated with gene therapy is its irreversibility. In the context of the complement system, any disruptions caused by gene therapy could have profound and potentially detrimental effects on immune function. Comprehensive preclinical and clinical evaluations are necessary to balance the potential therapeutic benefits of gene therapy against the risks of adverse outcomes, ensuring that such treatments can be implemented safely and effectively in clinical practice.
4. Supportive and Other Therapies
Dietary modifications or pharmacological interventions may alleviate PLE symptoms in some cases. Pharmacological treatments such as octreotide, heparin analogs, antiplasmin therapy, glucocorticoids, sirolimus, and everolimus have been used. However, the evidence of their effectiveness is inconclusive.
4.1. Albumin replacement
When hypoalbuminemia becomes clinically significant and contributes to symptomatic complications such as edema, ascites, or impaired systemic circulation, albumin infusion may be warranted to alleviate symptoms and improve patient outcomes.
4.2. Ig replacement therapy (IgRT)
Patients with PLE exhibit a non-selective protein loss from the intestine and are more susceptible to infections due to low Ig levels and associated comorbidities. IgRT becomes necessary to address the deficiencies until protective Ig levels are restored through targeted treatment.
4.3. Immunosuppressive therapies
Corticosteroids are commonly used in treating PLEs, often initiated even when a definitive diagnosis is not immediately clear. These medications, with their potent anti-inflammatory and immunosuppressive properties, are crucial in mitigating the inflammatory component of PLE and reducing protein loss through the gastrointestinal tract. However, their long-term use poses significant risks to growth and development. Therefore, corticosteroids should be administered only as a temporary measure until the patient accesses a definitive treatment with C5-blocking agents.
4.4. Vaccination
Life-threatening and even fatal meningococcal infections have been reported in both vaccinated and unvaccinated patients who are undergoing treatment with complement inhibitors. It is crucial that meningococcal vaccines (for serogroup A, C, W, and Y [Men-ACWY] and serogroup B [MenB]) be applied at least two weeks before administering the first dose of complement inhibitor therapy.
4.5. Prophylactic antibiotics
If there is an indication for anti-complement therapy, antibacterial prophylaxis should be initiated in patients who have not yet received meningococcal vaccination or who have received meningococcal vaccination but have not elapsed two weeks since vaccination. In fully vaccinated individuals, there is insufficient evidence on which and for how long prophylactic antibiotics should be used in combination with anti-complement therapies.
4.6. Dietary changes
A high-protein and low-fat diet with a predominance of medium-chain and short-chain triglycerides may prove effective, especially when primary lymphatic flow disorders are implicated. This dietary modification aims to alleviate intestinal lymphatic pressure, as medium-chain triglycerides are directly transported to the portal circulation without relying on lymphatic transport via chylomicrons.
5. Expert Opinion
The management of CHAPLE disease requires a comprehensive and multidisciplinary approach integrating anti-complement therapies, supportive measures, and diligent monitoring for potential complications. The anti-complement component 5 therapy using pozelimab heralds a significant breakthrough in combating the basic pathophysiology of CHAPLE disease. Future studies should explore the potential of modulators targeting other complement components as viable alternatives to the C5-blockade strategy. Specifically, it would be valuable to test inhibitors that target the central step of C3 activation or the amplification loop of the alternative pathway.
In addition to pharmacotherapy, curative options like gene therapy are being explored for the treatment of various complement-mediated diseases. Recent advancements in gene editing technologies, such as CRISPR/Cas9, offer the potential for targeted correction of genetic mutations that contribute to disease pathology. Future studies should evaluate the feasibility of gene therapy approaches, considering the effectiveness of existing pharmacotherapeutics.
Supportive measures, including nutritional support, fluid and electrolyte management, and addressing specific organ complications, are essential components of care. Additionally, close monitoring for disease activity, treatment response, and potential adverse effects of therapy is crucial for optimizing outcomes and ensuring patient safety.
Despite the significant advancements in CHAPLE therapy to date, future research is crucial to fully understand the long-term efficacy and safety profiles of current treatment modalities. By advancing our knowledge and refining treatment approaches, we can further enhance patient outcomes and improve the quality of life for individuals affected by CHAPLE disease.
Declaration of interests
A Ozen is a consultant and steering committee member for Regeneron Pharmaceuticals; received sample analysis support for a previous study from Regeneron Pharmaceuticals; and has a pending patent on complement inhibitor treatment in CHAPLE disease.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
REFERENCES
- Ozen A, Comrie WA, Ardy RC, et al. CD55 Deficiency, Early-Onset Protein-Losing Enteropathy, and Thrombosis. N Engl J Med. 2017 Jul 6;377(1):52–61. doi:10.1056/NEJMoa1615887 •• This is the first study to describe CHAPLE disease by detailing its clinical and genetic features.
- Ozen A CHAPLE syndrome uncovers the primary role of complement in a familial form of Waldmann’s disease. Immunol Rev. 2019 Jan;287(1):20–32. doi:10.1111/imr.12715
- Ozen A, Kasap N, Vujkovic-Cvijin I, et al. Broadly effective metabolic and immune recovery with C5 inhibition in CHAPLE disease. Nat Immunol. 2021 Feb;22(2):128–139. doi:10.1038/s41590-020-00830-z •• This study demonstrates the efficacy of C5 inhibition in CHAPLE disease, highlighting significant improvements in both metabolic and immune recovery. The research provides critical insights into therapeutic strategies for this rare condition, contributing to the understanding of complement inhibition as a viable treatment approach for CHAPLE disease.
- Ozen A, Lenardo MJ, Longo DL Protein-Losing Enteropathy. N Engl J Med. 2023 Aug 24;389(8):733–748. doi:10.1056/NEJMra2301594
- Park SM, Angel CE, McIntosh JD, et al. Mapping the distinctive populations of lymphatic endothelial cells in different zones of human lymph nodes. PLoS One. 2014;9(4):e94781. doi: 10.1371/journal.pone.0094781
- Rother RP, Rollins SA, Mojcik CF, et al. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007 Nov;25(11):1256–1264. doi:10.1038/nbt1344
- Wong EK, Goodship TH, Kavanagh D. Complement therapy in atypical haemolytic uraemic syndrome (aHUS). Mol Immunol. 2013 Dec 15;56(3):199–212. doi:10.1016/j.molimm.2013.05.224
- Thomas TC, Rollins SA, Rother RP, et al. Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol Immunol. 1996 Dec;33(17–18):1389–1401. doi:10.1016/s0161-5890(96)00078-8
- Kurolap A, Eshach-Adiv O, Hershkovitz T, et al. Loss of CD55 in Eculizumab-Responsive Protein-Losing Enteropathy. N Engl J Med. 2017 Jul 6;377(1):87–89. doi:10.1056/NEJMc1707173
- Latuszek A, Liu Y, Olsen O, et al. Inhibition of complement pathway activation with pozelimab, a fully human antibody to complement component C5. PLoS One. 2020;15(5):e0231892. doi: 10.1371/journal.pone.0231892
- Ozen A, Chongsrisawat V, Sefer AP, et al. Evaluating the efficacy and safety of pozelimab in patients with CD55 deficiency with hyperactivation of complement, angiopathic thrombosis, and protein-losing enteropathy disease: an open-label phase 2 and 3 study. The Lancet. 2024 Feb 17;403(10427):645–656. doi:10.1016/S0140-6736(23)02358-9 •• This study investigates the efficacy and safety of pozelimab in treating patients with CD55 deficiency, specifically targeting those with complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy.
- FDA.gov [Internet].The USA. FDA, FDA, [cited 2024 July 17] Available from: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-first-treatment-cd55-deficient-protein-losing-enteropathy-chaple-disease.
- Mastellos DC, Hajishengallis G, Lambris JD. A guide to complement biology, pathology and therapeutic opportunity. Nat Rev Immunol. 2024 Feb;24(2):118–141. doi:10.1038/s41577-023-00926-1
- Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood. 2019 Feb 7;133(6):540–549. doi:10.1182/blood-2018-09-876805
- Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019 Feb 7;133(6):530–539. doi:10.1182/blood-2018-09-876136
- Liu H, Xia L, Weng J, et al. Efficacy and safety of the C5 inhibitor crovalimab in complement inhibitor-naive patients with PNH (COMMODORE 3): A multicenter, Phase 3, single-arm study. Am J Hematol. 2023 Sep;98(9):1407–1414. doi:10.1002/ajh.26998
- Dhillon S. Crovalimab: First Approval. Drugs. 2024 Jun;84(6):707–716. doi:10.1007/s40265-024-02032-5
- Nishimura JI, Ando K, Masuko M, et al. Tesidolumab (LFG316) for treatment of C5-variant patients with paroxysmal nocturnal hemoglobinuria. Haematologica. 2022 Jun 1;107(6):1483–1488. doi:10.3324/haematol.2020.265868
- Gorman DM, Lee J, Payne CD, et al. Chemical synthesis and characterisation of the complement C5 inhibitory peptide zilucoplan. Amino Acids. 2021 Jan;53(1):143–147. doi:10.1007/s00726-020-02921-5
- Gaya A, Munir T, Urbano-Ispizua A, et al. Results of a phase 1/2 study of cemdisiran in healthy subjects and patients with paroxysmal nocturnal hemoglobinuria. EJHaem. 2023 Aug;4(3):612–624. doi:10.1002/jha2.748
- Hoy SM. Pegcetacoplan: First Approval. Drugs. 2021 Aug;81(12):1423–1430. doi:10.1007/s40265-021-01560-8
- Syed YY. Iptacopan: First Approval. Drugs. 2024 May;84(5):599–606. doi:10.1007/s40265-024-02009-4
- Kang CD: Danicopan: First Approval. Drugs. 2024 May;84(5):613–618. doi:10.1007/s40265-024-02023-6
- Arlabosse T, Booth C, Candotti F Gene Therapy for Inborn Errors of Immunity. J Allergy Clin Immunol Pract. 2023 Jun;11(6):1592–1601. doi:10.1016/j.jaip.2023.04.001
- Sudhakar V, Richardson RM Gene Therapy for Neurodegenerative Diseases. Neurotherapeutics. 2019 Jan;16(1):166–175. doi:10.1007/s13311-018-00694-0
- Ducloyer JB, Le Meur G, Cronin T, et al. [Gene therapy for retinitis pigmentosa]. Med Sci (Paris). 2020 Jun-Jul;36(6–7):607–615. doi:10.1051/medsci/2020095
- Mahlangu J, Kaczmarek R, von Drygalski A, et al. Two-Year Outcomes of Valoctocogene Roxaparvovec Therapy for Hemophilia A. N Engl J Med. 2023 Feb 23;388(8):694–705. doi:10.1056/NEJMoa2211075
- Dreismann AK, Hallam TM, Tam LC, et al. Gene targeting as a therapeutic avenue in diseases mediated by the complement alternative pathway. Immunol Rev. 2023 Jan;313(1):402–419. doi:10.1111/imr.13149