
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Graphical Abstract
Abstract
Grain boundaries (GBs) represent one of the most important types of defects in solids and their instability leads to catastrophic failures in materials. Grain boundaries are challenging for theoretical studies because of their distorted atomic structure. Fortunately, quantum-mechanical methods can reliably compute their properties. We calculate and analyze (tensorial) anisotropic elastic properties of periodic approximants of interface states associated with GBs in one of the most important intermetallic compounds for industrial applications, Ni3Al, appearing in Ni-based superalloys. Focusing on the Σ5(210) GBs as a case study, we assess the mechanical stability of the corresponding interface states by checking rigorous elasticity-based Born stability criteria. The critical elastic constant is found three-/five-fold softer contributing thus to the reduction of the mechanical stability of Ni3Al polycrystals (experiments show their GB-related failure). The tensorial elasto-chemical complexity of interface states associated with the studied GBs exemplifies itself in high sensitivity of elastic constants to the GB composition. As another example we study the impact caused by Si atoms segregating into the atomic layers close to the GB and substituting Al atoms. If wisely exploited, our study paves the way towards solute-controlled design of GB-related interface states with controlled stability and/or tensorial properties.
1. Introduction
Grain boundaries (GBs) are important extended defects determining a number of material properties. For example, macroscopic strength of polycrystalline materials strongly depends on GB cohesion [Citation1–Citation5]. Quantum-mechanical computational methods are advantageous for theoretical studies of GBs because of their distorted atomic structure [Citation6–Citation20]. Properties of GBs are very sensitive to compositional changes [Citation21–Citation33]. It was found that the impurities even in very low concentrations (ppm) segregated at GBs can drastically change material properties (see e.g. [Citation34,Citation35]). For instance, they can completely suppress local magnetic moments at GBs in magnetic materials as shown by quantum-mechanical calculations for elemental Ni [Citation36,Citation37].
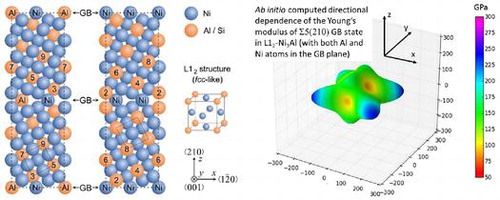
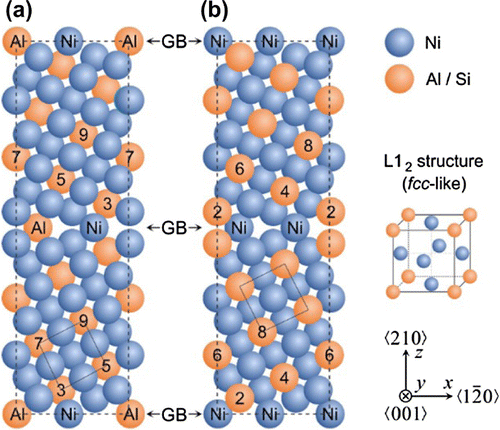
Figure 1. Supercells used in electronic structure calculations of interface states associated with the Σ5(210) GBs in Ni3Al with different interface stoichiometries, Σ5(210)Al,Ni with both Al and Ni atoms at the GB plane (a) and Σ5(210)Ni,Ni with only Ni atoms at the GB plane (b). The blue spheres represent Ni atoms and the orange ones Al (or Si) atoms. The numbers label the atomic layers with respect to the grain boundary (marked by number 1).
Considering the current interest in fabrication, characterization and use of nano-granular materials [Citation38], that can provide unprecedented properties not available in bulk samples, grain boundaries and associated interface states can become even more important than the grain interior. When reaching truly nano-scale grain sizes, even elastic properties (that are typically unaffected in case of larger grains) become dominated by GB states.
The principal goal of our work is to shed more light on tensorial, in particular elastic, properties of materialregions affected by grain boundaries. It is a critically important complement to a number of previous GB-related studies (e.g. [Citation39,Citation40]) that were focused mostly on scalar characteristics (energies, strength, changes in inter-atomic bonds) or electronic and atomic structure (see e.g. [Citation41–Citation54]). The fact that tensorial properties often require much more demanding approaches (see e.g. [Citation55]) is compensated by the wealth of insight that they provide. The knowledge of elastic properties allows for assessing the mechanical stability of the studied system (via generic Born stability criteria [Citation56]). They also play a crucial role in high-temperature thermodynamic stability as they determine long-wave phonon frequencies.
As a case study we selected one of the most important intermetallic compounds for industrial applications, Ni3Al, appearing e.g. as the phase in Ni-based superalloys, and the Σ5(210) GB (see Figure ), which is the smallest one that exhibits an additional volume compared to the bulk. Segregation-driven compositional changes will be exemplified by comparing properties with and without Si atoms substituting Al atoms (Si atoms were experimentally found on Al-sites in Ni3Al [Citation57]).
2. Methods
Our quantum-mechanical calculations within the framework of density functional theory [Citation58,Citation59] were performed using the Vienna ab initio simulation package [Citation60,Citation61]. The exchange and correlation energy was treated in the generalized gradient approximation as parametrized in [Citation62] and implemented in projector augmented wave pseudopotentials [Citation63]. We employed a plane-wave energy cutoff of 500 eV with a 5 17
3 Monkhorst-Pack k-point mesh for the 64-atom supercells, reducing the forces on atoms under 1 meV Å
. The crystal orbital Hamilton population (COHP) [Citation64] and density of states (DOS) analysis based on projection of plane waves to a local basis [Citation65] implemented in the program LOBSTER [Citation66,Citation67] have been used to understand the interatomic interaction in the studied systems.
The two different GB chemical compositions corresponding to either both Ni and Al or solely Ni atoms at the GB plane are shown in Figure (a) and (b). As periodic boundary conditions apply, the supercells are periodic approximants of the real GB-associated interface states. Elastic constants were computed by the stress–strain method outlined in [Citation68,Citation69] and all atomic positions were fully relaxed when simulating the application of external strains. Our tests show that the most critical shear elastic constant, , that is as low as 15 GPa in the case when only Ni atoms are located at the interface (see below), would be much higher (64 GPa) if these atomic relaxations are omitted.
As we study a magnetic material, we conveniently use the value of the local magnetic moment to differentiate the GB-affected material from the bulk, but e.g. inter-layer distances in the direction perpendicular to the grain boundary can be alternatively used (see them visualized as a part of the discussion below). Importantly, the size of the supercells is intentionally chosen so that the value of the local magnetic moment of Ni atoms in the bulk Ni3Al is reproduced in the 8th atomic layer that is the most distant from the GB interfaces (see Figure ).
Figure 2. Computed local magnetic moments of Ni atoms normalized with respect to their bulk value as functions of their positions within the computational supercells in the direction perpendicular to the simulated grain boundaries Σ5(210)Al,Ni and Σ5(210)Ni,Ni in Ni3Al. The GBs are located within the supercells at relative positions 0, 0.5 and 1.
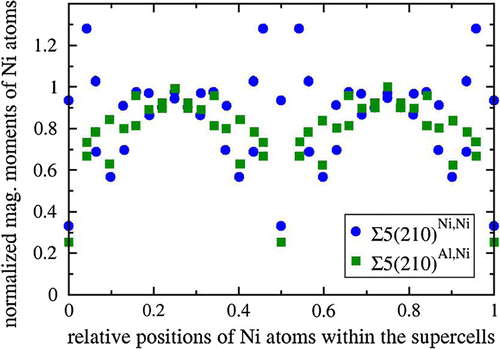
Figure 3. Directional dependences of the Young’s modulus of the studied Σ5(210)Al,Ni (a) and Σ5(210)Ni,Ni (b) configurations when the GB plane is the x–y plane. Displayed dependences were computed and visualized by the SC-EMA [Citation75–Citation77] library (scema.mpie.de) based on quantum-mechanically computed elastic constants (Table ).
![Figure 3. Directional dependences of the Young’s modulus of the studied Σ5(210)Al,Ni (a) and Σ5(210)Ni,Ni (b) configurations when the GB plane is the x–y plane. Displayed dependences were computed and visualized by the SC-EMA [Citation75–Citation77] library (scema.mpie.de) based on quantum-mechanically computed elastic constants (Table 2).](/cms/asset/c89a076c-86d5-4db2-93c2-cb66ef1affbd/tsta_a_1312519_f0003_oc.gif)
Table 1. The values of the two lattice parameters within the (210) plane of studied grain boundaries as obtained by the full relaxation of supercells in comparison with values obtained for the bulk. In particular, the lattice parameters along the x and y
directions (see Figure ) are listed together with their relative changes with respect to the bulk values.
In particular, in the case of the Si-segregated configurations, it is only the GB-affected material that is studied. It is an example of the Goldilocks principle when bigger supercells with more atomic layers would contain more bulk-like material, that we do not want to study, while smaller supercells would not contain all the material affected by the extended defect. Tensorial elastic properties of a bulk-like material in bigger supercells would also contribute into the overall elasticity and hinder our effort to determine the properties of the GB-affected material itself. A limiting case of only bulk material being present (and no GB-affected material included) is discussed later in case of an approximative linear-elasticity approach (see below). The supercells have the lattice parameters within the GB plane relaxed, but the values of the two lattice parameters within the (210) plane exhibit only negligible changes when compared with the corresponding bulk values (see Table ).
3. Results
Starting from the bulk Ni3Al with the L12 structure, our ab initio calculated lattice parameter, 3.571 Å, is in excellent agreement with the experimental value of 3.572 Å [Citation70]. Regarding Σ5(210) GBs in Ni3Al, our calculations show that the Σ5(210)Al,Ni with both types of atoms (Al and Ni) at the interface (Figure (a)) has a lower GB energy of 1.38 J m and an additional volume per unit GB area of 0.29 Å (Å3Å2) while the Σ5(210)Ni,Ni with only Ni atoms at the interface (Figure (b)) has a higher GB energy of 1.66 J m
and a larger additional volume per unit GB area of 0.32 Å. The latter values are in excellent agreement with the previous theoretical results of 1.7 J m
and 0.36 Å [Citation71].
It should be noted that the additional volume (expressed as a length parameter) is an averaged one when an additional volume obtained for the whole computational cell is divided by the total area of the two GBs inside of the supercell. It differs from vertical atomic-layer-resolved shifts that are analyzed, for example, in the recent study by Kumar et al. [Citation72]. The individual inter-layer distances significantly differ from the bulk equilibrium value (0.799 Å) with the extreme values of these deviations being (i) +0.33 Å for the distance between the GB plane and the first atomic layer off the GB plane and (ii) –0.26 Å between the first and second plane off the GB plane, respectively, in the Σ5(210)Al,Ni. The extreme values obtained for the Σ5(210)Ni,Ni variant of the interface are quite similar, +0.32 Å and –0.20 Å, respectively.
As far as magnetic properties are concerned, Figure shows local magnetic moments of Ni atoms across the computational supercells in the direction perpendicular to the GB interface. In case of Σ5(210)Al,Ni, all magnetic moments are reduced with the only exception being bulk-like values predicted for the atoms that are the most distant from the GB interface. The reduction of local magnetic moments is found also for a vast majority of atoms in case of Σ5(210)Ni,Ni GB variant, but, interestingly, the Ni atoms located in the second and third atomic layer away from the interface have the local magnetic moments increased (by up to 28%). As both GB variants exhibit additional volume with respect to the bulk, a simple magneto-volumetric argument cannot explain significant reduction of most of local magnetic moments within the studied GBs and the clarification will be a subject of future studies.
Focusing on the elastic properties, the three elastic constants computed for cubic Ni3Al at T = 0 K are in reasonable agreement with room-temperature experimental data (listed in parentheses [Citation73]): = 243 (224) GPa,
= 152 (149) GPa,
= 128 (123) GPa. Calculated elastic constants of both types of interface states affected by Σ5(210) GBs are given in Table together with bulk elastic constants in the same coordination system. In order to visualize how the interface states associated with GBs respond to uniaxial loading along different crystallographic directions, we exhibit directional dependences of the Young’s modulus in Figure .
Table 2. The ab initio computed elastic constants (all in GPa) of (i) the Ni3Al bulk in the coordination system of the studied GBs (shown in Figure ), (ii) two types of Ni3Al#x03A3;5(210) GBs with different atoms (see Figure ) at the interface, (iii) prediction based on the linear-elasticity approach from [Citation74], and (iv) systems with Si atoms substituting Al atoms in the second layer, Ni3(Al,) Σ5(210)Ni,Ni, and eighth layer, Ni3(Al,
) Σ5(210)Ni,Ni.
The periodic approximants of the GB-related interface states have an orthorhombic symmetry and comparison of Figure (a) and (b) for Σ5(210)Al,Ni and Σ5(210)Ni,Ni, respectively, shows how sensitive the elasticity is to compositional changes. The stiffest directions with the Young’s modulus of about 250 GPa (color-coded dark blue in Figure ) are within the same crystallographic plane as the GB interface while the softest directions with the Young’s modulus approaching only 50 GPa (red colors in Figure (b)) are inclined to the GB interface plane.
Analyzing the computed elastic constants listed in Table , it is important to note that the obtained shear elastic constants and
of interface states associated with the Σ5(210) GBs are considerably lower than those in the bulk. The bulk modulus is reduced, too, but only very weakly from 182 GPa (bulk Ni3Al) to 171 GPa (Σ5(210)Al,Ni). In order to rigorously assess these changes and their effect on the mechanical stability, we use Born stability criteria (see e.g. [Citation56]) that connect the mechanical stability with the positiveness of leading principal minors of the matrix of elastic constants. As a consequence, diagonal elements
,
and
must be positive and the drop predicted for
clearly identifies the weakest link.
The interface state associated with the higher-energy GB Σ5(210)Ni,Ni has an extremely low value of this crucial elastic constant, = 15 GPa, i.e. it is less mechanically stable when compared with the interface state related to the lower-energy GB variant Σ5(210)Al,Ni with
= 29 GPa. This fact is in line with the link between mechanical and thermodynamic stability identified so far only in bulk systems when thermodynamically less stable phases possess also lower mechanical stability (see e.g. [Citation78,Citation79]). Here we show this link between mechanical and thermodynamic stability also in case of interface states associated with GBs.
The predicted reduction of the shear elastic constant is also in line with previous findings reported for GBs in elemental face-centered cubic metals [Citation80,Citation81]. In particular, results of atomistic simulations were combined with a method [Citation81] that allows decomposition of an overall elasticity into that of different atomic layers and, similarly as in our study, the shear elastic constants were found reduced close to the GB plane. If the same trend of reduction of shear elastic constants is found also for other Ni3Al interface regions associated with GBs, it can shed new light on the classical problem related to Ni3Al, i.e. its experimentally found very low inter-granular cohesion (see e.g. [Citation82,Citation83]).
4. Discussion
Our quantum-mechanical study aims at tensorial properties of interface states associated with grain boundaries, i.e. material formed by only a few atomic layers of atoms close to a particular extended defect. In order to do so, the size of the supercells is carefully chosen to contain only a minimum amount of the bulk material (the grain interior). In particular, the value of the local magnetic moment of Ni atoms in the bulk Ni3Al is reproduced only in the eighth atomic layer that is the most distant from the GB planes. It is interesting to address another limit when there is such a high amount of bulk grains that the impact of the interface states related to grain boundaries becomes negligible. In order to do so we apply, in the following, a linear-elasticity approach derived for coherent multilayers (so-called superlattices) by Grimsditch and Nizzoli [Citation74].
The linear-elasticity method uses as the input the tensor of elastic constants for bulk Ni3Al (see the values in Table ) that is rotated [Citation84] by two different angles so as to mimic the rotations of the two interfacing grains forming the Σ5(210) grain boundary. The elastic constants obtained in this way correspond to a coherent two-phase multilayer system (stacked along the z-coordinate). They are listed in Table and visualized in Figure .
Figure 4. The directional dependence of the Young modulus (in GPa) of the studied interface states associated with grain boundary Σ5(210) as predicted by the linear elasticity theory by Grimsditch and Nizzoli [Citation74] using matrices of ab initio calculated elastic constants of bulk Ni3Al rotated [Citation84] in the same way as are rotated both grains forming the Σ5(210) grain boundary (the GBs interface is the x–y plane).
![Figure 4. The directional dependence of the Young modulus (in GPa) of the studied interface states associated with grain boundary Σ5(210) as predicted by the linear elasticity theory by Grimsditch and Nizzoli [Citation74] using matrices of ab initio calculated elastic constants of bulk Ni3Al rotated [Citation84] in the same way as are rotated both grains forming the Σ5(210) grain boundary (the GBs interface is the x–y plane).](/cms/asset/c86758e2-5d66-4a79-80bb-59a691c5a369/tsta_a_1312519_f0004_oc.gif)
The linear-elasticity prediction is different from quantum-mechanical results (Figure (a) and (b)) for a number of reasons. First, the linear-elasticity method does not take into account the actual interface properties including its chemical composition or atomic relaxations (only compatibility and equilibrium of normal stress apply). Therefore, Figure presents an approximation that cannot consider any of the two possible elastic characteristics shown in Figure (a) and (b) that do depend of the GB interface composition. Second, the linear elastic continuum model is length-scale independent while real atoms and real length dimensions are considered in quantum-mechanical calculations. Third, the input single-crystalline elastic constants were those computed for equilibrium lattice parameter of bulk Ni3Al whereas our quantum-mechanically computed elastic constants for interface states related to the Σ5(210) GBs have been determined for fully relaxedgrain-boundary supercells that exhibit additional volume (structural parameters corresponding to the result of the energy minimization).
Importantly, the linear-elasticity approach does not predict any significant reduction of the shear elastic constant . Knowing that the linear-elasticity method, in fact, ignores the actual GB interface with its specific properties including its possible weaknesses, we consider the above comparison as another proof that the softening of
is associated with the studied interface states connected with grain boundaries.
The use of the linear-elasticity method has, in fact, one more motivation. As periodic boundary conditions apply in our calculations, the predicted elasticity is that of a system containing periodically repeated GBs. It would be desirable to extract the elastic constants of a single interface state related to a single GB from our values. Linear-elasticity methods, which use elastic properties of two phases as the input and predict elastic properties of an infinite multilayer system with these two phases coherently co-existing, can be, in principle, inverted in order to meet this target. Unfortunately, the problem seems to be mathematically ill-defined. The number of unknown elastic constants is higher than the number of elastic parameters obtained from our calculations or, in other words, the symmetry of the interface state associated with a single GB is lower than that of its periodic approximant. Therefore, a method (such as in [Citation81,Citation85]), that would determine properties of a single interface state related to a single grain-boundary from results obtained for its periodic approximants remains a challenge for future studies.
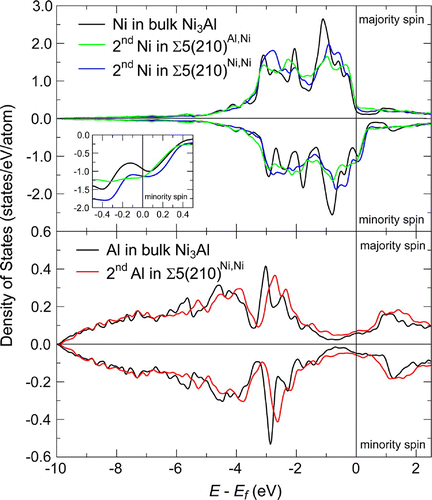
Our quantum-mechanical approach provides a number of other valuable insights. We have employed COHP and DOS analysis to identify the origin of the softening of the elastic constant discussed above. The atomic resolved DOS of atoms near the grain boundary as well as in bulk Ni3Al are shown in Figure . The nature of both Ni–Ni and Al–Ni bonds in the bulk material (bond length of 2.53 Å) arises from bonding overlaps of d-d and p-d bands approximately 3 eV below the Fermi level,
(see the vertical solid black lines in Figure ). The d-band corresponding to main peak in the DOS of Ni close to 1 eV below the Fermi energy does not participate in bonding. There is also an anti-bonding interaction of Ni d-bands lying just below the Fermi level in the majority spin channel and just above the Fermi energy in the minority spin channel. However, these contributions to bond strength are really small in the bulk material. The bonding behavior of atoms exactly at the grain boundary plane of both studied variants does not significantly differ from behavior of atoms in the bulk.
The origin of the lower mechanical stability can be found in atoms in the second layer and in their mutual interactions across the GB. The Σ5(210)Al,Ni contains two Ni–Ni bonds between atoms in the second layer which exhibit the shortest inter-atomic distances (2.23 and 2.25 Å) among all the bonds in the studied systems due to geometry of the GB. Shortening of distances results in a larger overlap of bonding as well as anti-bonding bands and subsequently in higher occupations of anti-bonding states at the Fermi energy in minority spin channel instead of non-bonding states (see green solid line), which destabilizes the whole system. A similar effect can be seen also for Ni–Ni bond across the GB in Σ5(210)Ni,Ni with inter-atomic distance equal to 2.23 Å (see solid blue line). The second bond across the boundary plane in this GB variant is between two Al atoms and is significantly longer (2.50 Å), because both Al atoms are strongly bonded to the Ni atoms in the third layer. The Al–Al interaction is weaker than interactions of Ni–Ni atoms across the boundary plane. The weaker Al–Al bond across the GB further destabilizes the whole system compared with the Σ5(210)Al,Ni case. The DOS of Al atom in the second layer (see solid red line) exhibits a significant shift of p-bands closer to the Fermi energy. Stronger bonds between Al atom in the second layer and Ni atoms in the third layer can also be recognized from inter-layer distances shown in Figure . The value of the distance between the second and third layer (marked as ‘2/3’ in Figure ) is in the case of atomic planes containing both Al and Ni atoms in Σ5(210)Ni,Ni GB significantly shorter than all other inter-planar distances between the second and third layers.
Figure 5. Ab initio computed atom- and spin-resolved electronic densities of states for Ni atoms (upper panel) in the bulk Ni3Al and in case of Ni atoms from the second atomic layer in both studied Ni3Al GBs, Σ5(210)Al,Ni and Σ5(210)Ni,Ni. The lower panel shows the atom- and spin-resolved densities of states of Al atoms in the bulk Ni3Al and in the second atomic layer of Σ5(210)Ni,Ni GB.
Further, to assess elasto-chemical aspects of interface states related to the studied GBs (and their potential for future solute-controlled design of GBs with specific tensorial properties) we further analyze the sensitivity of the elasticity to the chemical composition of the GB plane. We explore the segregation of Si atoms substituting Al atoms and associated elasticity changes. Testing nine different Al positions for Si substituents, the lowest energy was achieved when the Si atom is located at the Al position immediately next to the GB plane, Ni3(Al,), the position marked ‘2’ in Figure (b), in the case of the Σ5(210)Ni,Ni grain boundary that has only Ni atoms at the GB plane (see Figure ). The energy favors the Si atoms at position 2 over position 8 by 0.65 eV per Si atom, indicating strong segregation tendencies. The segregation favors the Σ5(210)Ni,Ni variant (with the Si substituting an Al atom at position 2, i.e. Ni3(Al,
)) over the Σ5(210)Al,Ni GB variant (with the Si atom substituting Al atom in any of the considered positions in the Σ5(210)Al,Ni supercell). It is worth discussing also the energy difference between two close-to-the-bulk states with Si atoms far away from the GB substituting Al atoms at positions 8 and 9, which is marked in Figure and is equal to 0.469 eV per one Si substituent. This energy difference translates into the energy difference of 0.938 eV per whole 64-atom computational cell (with two Si substituents) and this value is very close to the energy difference of 0.962 eV between the supercells modeling Si-free Σ5(210)Al,Ni and Σ5(210)Ni,Ni GBs.
Figure 6. Ab initio computed distances between the (210) atomic planes in the supercells modeling Σ5(210)Al,Ni and Σ5(210)Ni,Ni interface states compared with the value calculated for the bulk.
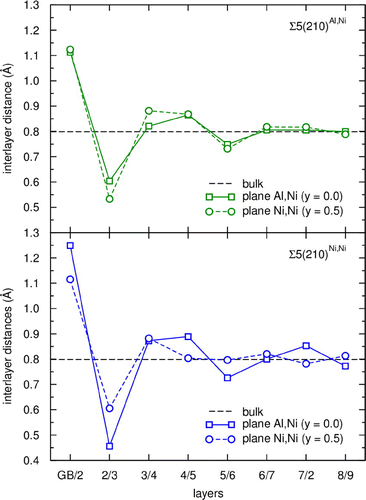
Figure 7. Ab initio computed energies of Si-substituted states for both Σ5(210)Al,Ni and Σ5(210)Ni,Ni GBs expressed as the energy difference with respect to the substituted state with the lowest energy (Ni3(Al,Si) and divided by the number of Si atoms in the supercells (there were two of them). The lines are added only to guide the eye.
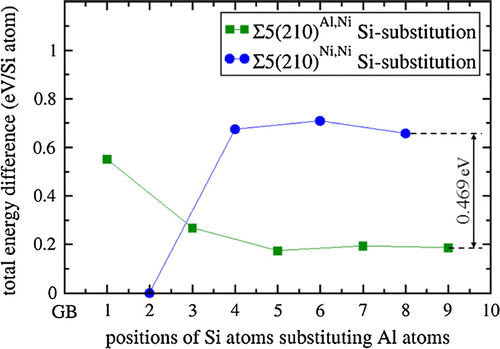
As the Si segregation alters the thermodynamic preference for the Σ5(210) GB composition variants in Ni3Al (Σ5(210)Al,Ni variant is preferred without Si atoms while Σ5(210)Ni,Ni variant is preferred when Si atoms are nearby), a solute-induced GB phase transformation may occur here.
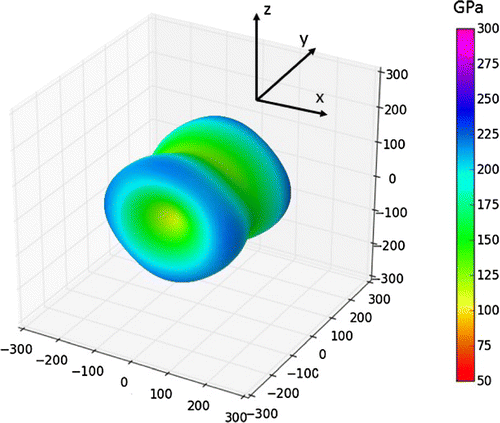
The elastic constants of the substituted Ni3(Al,) interface state close to the Σ5(210)Ni,Ni GB are given in Table and the corresponding directional dependence of the Young’s modulus isvisualized in Figure . In contrast to studied Si-free cases, the elastic response is nearly axially symmetric around the y axis, the
direction, reducing thus the directional anisotropy of elastic properties. Rather accidentally, the directional dependence at Figure shares certain similar features with the linear-elasticity prediction visualized for Ni3 Σ5(210) in Figure . Importantly, the simulated presence of Si atoms strongly reduces the softening of the shear elastic constant
that was discussed above in the case of the interface states associated with the Ni3AlΣ5(210) GBs. In particular, the replacement of an Al atom in the second layer by a Si atom completely changes the character of bonding across the GB. The Si–Al bond is stronger than the Al–Al bond and it is able to stabilize the grain boundary. If Al atoms are replaced by Si atoms in the bulk region instead of the GB region, the weak Al–Al bond still exists and the stability of Σ5(210)Ni,Ni GB is lower as in the case without Si. Therefore, the Si substitution (leading to the elimination of a weakest link) represents a neat example of options offered by future materials-design of GB interface states.
Our calculations further show that the impact of segregated Si atoms on the elasticity of GB-affected interface states is limited to a few atomic layers close to the GB interface. Putting Si atom at the Al position in the eighth layer, Ni3(Al,) Σ5(210)Ni,Ni, that has bulk-like properties, provides nearly identical elastic characteristics as the Si-free Σ5(210)Ni,Ni (compare the values in Table ).
Figure 8. The directional dependence of the Young’s modulus of the studied interface states in Ni3(Al, ) affected by Σ5(210)Ni,Ni.
5. Conclusions
We have applied first-principles calculations to study tensorial elastic properties of periodic approximants of the interface states in Ni3Al associated with the Σ5(210) grain boundary. The tensorial elasto-chemical aspects were illustrated by behavior of elastic constants computed for GB-related interface states with different atoms at the GB plane and we find to be strongly sensitive to the GB-plane chemical composition. A three-/five-fold reduction of the elastic constant
is identified as the crucial weakest link for the mechanical stability. Moreover, when comparing two GB-plane composition variants, we got compositional trends of
that indicate correlations between the mechanical and thermodynamic stability found previously in bulk systems. Our study thus clearly shows the importance of elastic-constant analysis for next studies of interface states close to GBs when determining their mechanical (in)stability. These should be ideally extended by phonon spectrum calculations, in-plane translation analysis (see e.g. [Citation86]) and simulations of larger deformations (see e.g. [Citation87–Citation92]) in future. The sensitivity of tensorial elasto-chemical properties was also illustrated by studying the impact caused by Si atoms substituting Al atoms in the atomic layers close to the GBs. Nevertheless, the influence of Si atoms at the eighth layer from the Σ5(210) GB in the Ni3Al is already negligible. A way towards a solute-controlled design of GB-associated interface states with controlled tensorial elastic properties and stability is thus paved. Finally, considering qualitative links between different tensorial properties postulated by the Neumann’s principle, our study aims at initiating future research addressing tensorial aspects of GB-related interface states.
Acknowledgements
We gratefully acknowledge Python software development contributions of Dr. T. Rebok, Bc. Š. Brynych and Bc. D. Lago from the Faculty of Informatics of the Masaryk University in Brno, Czech Republic, that allowed us to visualize the directional dependences of Young’s moduli in Figure . We also highly appreciate discussions with Prof. Ing. Pavel Lejček, DrSc., from the Institute of Physics of the Academy of Sciences of the Czech Republic in Prague, Czech Republic.
Additional information
Funding
Notes
No potential conflict of interest was reported by the authors.
References
- Duscher G, Chisholm MF, Alber U, et al. Bismuth-induced embrittlement of copper grain boundaries. Nat Mater. 2004 Sep;3(9):621–626.
- Lu GH, Deng SH, Wang TM, et al. Theoretical tensile strength of an Al grain boundary. Phys Rev B. 2004 Apr;69(13).
- Pokluda J, Čern\’{y} M, Šandera P, et al. Calculations of theoretical strength: state of the art and history. J Comp-Aided Mat Design. 2004 Jan;11(1):1–28.
- Kohyama M. Ab initio study of the tensile strength and fracture of coincidence tilt boundaries in cubic SiC: polar interfaces of the 122 Σ9 boundary. Phys Rev B. 2002 May 1;65(18).
- Ogata S, Umeno Y, Kohyama M. First-principles approaches to intrinsic strength and deformation of materials: perfect crystals, nano-structures, surfaces and interfaces. Model Sim Mat Sci Eng. 2009 Jan;17(1).
- Mishin Y, Mehl MJ, Papaconstantopoulos DA, et al. Structural stability and lattice defects in copper: ab initio, tight-binding, and embedded-atom calculations. Phys Rev B. 2001 Jun 1;63(22).
- Yazyev OV, Louie SG. Topological defects in graphene: dislocations and grain boundaries. Phys Rev B. 2010 May 15;81(19).
- Muller DA, Singh DJ, Silcox J. Connections between the electron-energy-loss spectra, the local electronic structure, and the physical properties of a material: a study of nickel aluminum alloys. Phys Rev B. 1998 Apr 1;57(14):8181–8202.
- Wang Y, Li J. Phase field modeling of defects and deformation. Acta Mater. 2010 Feb;58(4):1212–1235.
- Mrovec M, Gr\"{o}ger R, Bailey AG, et al. Bond-order potential for simulations of extended defects in tungsten. Phys Rev B. 2007 Mar;75(10).
- Yan YF, Noufi R, Al-Jassim MM. Grain-boundary physics in polycrystalline CuInSe2 revisited: experiment and theory. Phys Rev Lett. 2006 May 26;96(20).
- Lojkowski W, Fecht HJ. The structure of intercrystalline interfaces. Prog Mat Sci. 2000;45(5–6):339–568.
- Dawson I, Bristowe PD, Lee MH, et al. First-principles study of a tilt grain boundary in rutile. Phys Rev B. 1996 Nov 15;54(19):13727–13733.
- Imaeda M, Mizoguchi T, Sato Y, et al. Atomic structure, electronic structure, and defect energetics in [001](310) Σ5 grain boundaries of SrTiO3 and BaTiO3. Phys Rev B. 2008 Dec;78(24).
- Tanaka S, Kohyama M. Ab initio calculations of the 3C-SiC(111)/Ti polar interfaces. Phys Rev B. 2001 Dec 15;64(23).
- McKenna KP, Shluger AL. First-principles calculations of defects near a grain boundary in MgO. Phys Rev B. 2009 Jun;79(22).
- Kohyama M. Computational studies of grain boundaries in covalent materials. Model Sim Mater Sci Eng. 2002 May;10(3):R31–R59.
- Oba F, Nishitani SR, Adachi H, et al. Ab initio study of symmetric tilt boundaries in ZnO. Phys Rev B. 2001 Jan 15;63(4).
- Ochs T, Beck O, Elsässer C, et al. Symmetrical tilt grain boundaries in body-centred cubic transition metals: an ab initio local-density-functional study. Phil Mag A. 2000 Feb;80(2):351–372.
- Lejček P, Šob M, Paidar V. Interfacial segregation and grain boundary embrittlement: an overview and critical assessment of experimental data and calculated results. Progr Mat Sci. 2017 Jun;87:83–139.
- Ackland GJ, Mendelev MI, Srolovitz DJ, et al. Development of an interatomic potential for phosphorus impurities in α-iron. J Phys Cond Mat. 2004 Jul 14;16(27):S2629–S2642.
- Scheiber D, Pippan R, Puschnig P, Ruban A, Romaner L. Ab-initio search for cohesion-enhancing solute elements at grain boundaries in molybdenum and tungsten. Int J Refract. Met Hard Mater 2016;60:75-81.
- Yan M, \v{S}ob M, Luzzi DE, et al. Interatomic forces and atomic-structure of grain-boundaries in copper-bismuth alloys. Phys Rev B. 1993 Mar 1;47(10):5571–5582.
- Zhang L, Da Silva JLF, Li J, et al. Effect of copassivation of Cl and Cu on CdTe grain boundaries. Phys Rev Lett. 2008 Oct 10;101(15).
- Braithwaite JS, Rez P. Grain boundary impurities in iron. Acta Mater. 2005 May;53(9):2715–2726.
- Christensen M, Wahnstrom G. Co-phase penetration of WC(1010)/WC(110) grain boundaries from first principles. Phys Rev B. 2003 Mar 15;67(11).
- Domain C, Becquart CS. Diffusion of phosphorus in α-Fe: An ab initio study. Phys Rev B. 2005 Jun;71(21).
- Du YA, Ismer L, Rogal J, et al. First-principles study on the interaction of H interstitials with grain boundaries in α- and γ-Fe. Phys Rev B. 2011 Oct 31;84(14).
- Zhang L, Shu X, Jin S, et al. First-principles study of He effects in a bcc Fe grain boundary: site preference, segregation and theoretical tensile strength. J Phys Cond Mater. 2010 Sep 22;22(37).
- Asta M, Hoyt JJ. Thermodynamic properties of coherent interfaces in f.c.c.-based Ag-Al alloys: A first-principles study. Acta Mater. 2000 Mar 14;48(5):1089–1096.
- Thomson DI, Heine V, Payne MC, et al. Insight into gallium behavior in aluminum grain boundaries from calculation on Σ11 (113) boundary. Acta Mater. 2000 Sep 4;48(14):3623–3632.
- Wachowicz E, Ossowski T, Kiejna A. Cohesive and magnetic properties of grain boundaries in bcc Fe with Cr additions. Phys Rev B. 2010 Mar 1;81(9).
- Wachowicz E, Kiejna A. Effect of impurities on grain boundary cohesion in bcc iron. Comp Mater Sci. 2008 Oct;43(4):736–743.
- Sutton AP, Balluffi RW. Interfaces in crystalline materials. Oxford: Oxford University Press; 1995.
- Lejček P. Grain boundary segregation in metals. Heidelberg: Springer; 2010.
- Všianská M, Šob M. The effect of segregated sp-impurities on grain-boundary and surface structure, magnetism and embrittlement in nickel. Prog Mat Sci. 2011;56:817.
- Všianská M, Šob M. Magnetically dead layers at sp-impurity-decorated grain boundaries and surfaces in nickel. Phys Rev B. 2011;84:014418.
- Froseth A, Van Swygenhoven H, Derlet PM. The influence of twins on the mechanical properties of nc-Al. Acta Mater. 2004 May 3;52(8):2259–2268.
- Lu GH, et al. Origin of intergranular embrittlement of Al alloys induced by Na and Ca segregation: Grain boundary weakening. Phys Rev B. 2006;73:224115.
- Ogata S, Umeno Y, Kohyama M. Model Sim Mater Sci Eng. 2009;17:013001.
- Pang XY, Janisch R, Hartmaier A. Interplanar potential for tension-shear coupling at grain boundaries derived from ab initio calculations. Model Sim Mater Sci Eng. 2016 Jan;24(1).
- Razumovskiy VI, Ruban AV, Razumovskii IM, et al. The effect of alloying elements on grain boundary and bulk cohesion in aluminum alloys: an ab initio study. Scr Mater. 2011 Nov;65(10):926–929.
- Tahir AM, Janisch R, Hartmaier A. Hydrogen embrittlement of a carbon segregated Σ 5(310)[001] symmetrical tilt grain boundary in α-Fe. Mater Sci Eng A. 2014 Aug;26(612):462–467.
- Tahir AM, Janisch R, Hartmaier A. Ab initio calculation of traction separation laws for a grain boundary in molybdenum with segregated C impurites. Model Sim Mater Sci Eng. 2013 Oct;21(7).
- Razumovskiy VI, Vekilov YK, Razumovskii IM, et al. Effect of alloying elements and impurities on interface properties in aluminum alloys. Phys Sol State. 2011 Nov;53(11):2189–2193.
- Hristova E, Janisch R, Drautz R, et al. Solubility of carbon in α-iron under volumetric strain and close to the Σ5(310)[001] grain boundary: Comparison of DFT and empirical potential methods. Comp Mat Sci. 2011 Jan;50(3):1088–1096.
- Janisch R, Ahmed N, Hartmaier A. Ab initio tensile tests of al bulk crystals and grain boundaries: universality of mechanical behavior. Phys Rev B. 2010 May 1;81(18).
- Razumovskiy VI, Lozovoi AY, Razumovskii IM. First-principles-aided design of a new Ni-base superalloy: Influence of transition metal alloying elements on grain boundary and bulk cohesion. Acta Materialia. 2015 Jan;1(82):369–377.
- Scheiber D, Razumovskiy VI, Puschnig P, et al. Ab initio description of segregation and cohesion of grain boundaries in W-25 at.% Re alloys. Acta Mater. 2015;88:180–189.
- Janisch R, Elsässer C. Interstitial impurities at grain boundaries in metals: insight from atomistic calculations. Int J Mater Res. 2009 Nov;100(11):1488–1493.
- Janisch R, Elsässer C. Growth and mechanical properties of a MoC precipitate at a Mo grain boundary: An ab initio density functional theory study. Phys Rev B. 2008 Mar;77(9).
- Gemming S, Janisch R, Schreiber M, et al. Density-functional investigation of the (113)[-110] twin grain boundary in Co-doped anatase TiO2 and its influence on magnetism in dilute magnetic semiconductors. Phys Rev B. 2007 Jul;76(4).
- Janisch R, Els\"{a}sser C. Segregated light elements at grain boundaries in niobium and molybdenum. Phys Rev B. 2003 Jun 11;67(22).
- Razumovskiy VI, Lozovoi AY, Razumovskii IM. First-principles-aided design of a new Ni-base superalloy: Influence of transition metal alloying elements on grain boundary and bulk cohesion. Acta Mater. 2016 Mar;106:401–402.
- van de Walle A. A complete representation of structure-property relationships in crystals. Nat Mater. 2008;7:455.
- Mouhat F, Coudert FX. Necessary and sufficient elastic stability conditions in various crystal systems. Phys Rev B. 2014;90:224104.
- Jung SB, Minamino Y, Araki H, et al., Diffusion of Copper, Iron and Silicon in Ni3Al, Defect and diffusion forum. 1993;95–98:859.
- Hohenberg P, Kohn W. Inhomogeneous electron gas. Phys Rev. 1964;136:B864.
- Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys Rev. 1965;140:A1133.
- Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Phys Rev B. 1993 Jan;47(1):558–561.
- Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B. 1996 Oct;54(16):11169–11186.
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77:3865.
- Blöchl PE. Projector augmented-wave method. Phys Rev B. 1994 Dec;50(24):17953–17979.
- Dronskowski R, Blöchl PE. Crystal orbital hamilton populations (COHP). Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J Phys Chem. 1993;97:8617.
- Deringer VL, Tchougreeff AL, Dronskowski R. Crystal orbital hamilton population (COHP) analysis as projected from plane-wave basis sets. J Phys Chem A. 2011;115:5461.
- Maintz S, Deringer VL, Tchougreeff AL, et al. Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J Comput Chem. 2013;34:2557.
- Maintz S, Deringer VL, Tchougreeff AL, et al. LOBSTER: a tool to extract chemical bonding from plane-wave based DFT. J Comput Chem. 2016;37:1030.
- Yu R, Zhu J, Ye HQ. Calculations of single-crystal elastic constants made simple. Comp Phys Commun. 2010;181:671.
- Zhou L, Holec D, Mayrhofer PH. Ab initio study of the alloying effect of transition metals on structure, stability and ductility of CrN. J Appl Phys. 2013;113:043511.
- Smithells metals reference book. 6th ed. London: Butterworths; 1983.
- Lu G, Kioussis N, Wu R, et al. First-principles studies of the Σ5 tilt grain boundary in Ni3Al. Phys Rev B. 1999;59:891.
- Kumar A, Wang J, Tomé C. First-principles study of energy and atomic solubility of twinning-associated boundaries in hexagonal metals. Acta Mater. 2015;85:144–154.
- Frankel J, Vassiliou J, Jamieson JC, et al. The elastic-constants of Ni3Al to 1.4 GPa. Physica B+C. 1986;139–140:198.
- Grimsditch M, Nizzoli F. Effective elastic-constants of superlattices of any symmetry. Phys Rev B. 1986;33:5891.
- Titrian H, Aydin U, Friák M, et al. Self-consistent scale-bridging approach to compute the elasticity of multi-phase polycrystalline materials. Vol. 1524, MRS Proceedings; 2013.
- Friák M, Counts WA, Ma D, et al. Theory-guided materials design of multi-phase Ti-Nb alloys with bone-matching elastic properties. Materials. 2012;5:1853.
- Zhu LF, Friák M, Lymperakis L, et al. Ab initio study of single-crystalline and polycrystalline elastic properties of Mg-substituted calcite crystals. J Mech Behav Biomed Mat. 2013;20:296.
- Maisel SB, Hoefler M, Müller S. A canonical stability-elasticity relationship verified for one million face-centred-cubic structures. Nature. 2012 Nov 29;491(7426):740+.
- Zhu LF, Friák M, Dick A, et al. First-principles study of the thermodynamic and elastic properties of eutectic Fe-Ti alloys. Acta Mater. 2012 Feb;60(4):1594–1602.
- Wolf D, Lutsko J. Structurally induced supermodulus effect in superlattices. Phys Rev Lett. 1988;60:1170–1173.
- Kluge MD, Wolf D, Lutsko JF, et al. Formalism for the calculation of local elastic constants at grain boundaries by means of atomistic simulation. J Appl Phys. 1990;67:2370–2379.
- Aoki K, Izumi O. Improvement in room temperature ductility of the L12 type intermetallic compound Ni3Al by boron addition. J Jpn Inst Met. 1979;43:1190–1196.
- Jiao Z, Luan J, Liun C. Strategies for improving ductility of ordered intermetallics. Prog Nat Sci: Mater Int; 2016.
- Ting TCT. Anisotropic elasticity. New York (NY): Oxford University Press; 1996.
- Dingreville R, Qu J. A semi-analytical method to compute surface elastic properties. Acta Materialia. 2007 Jan;55(1):141–147.
- Salehinia I, Shao S, Wang J, et al. Interface structure and the inception of plasticity in Nb/NbC nanolayered composites. Acta Mater. 2015;86:331–340.
- Friák M, Šob M, Vitek V. Ab initio calculation of phase boundaries in iron along the bcc-fcc transformation path and magnetism of iron overlayers. Phys Rev B. 2001;63:052405.
- Legut D, Friák M, Šob M. Phase stability, elasticity, and theoretical strength of polonium from first principles. Phys Rev B. 2010;81:214118.
- Černý M, Pokluda J, Šob M, et al. Ab initio calculations of elastic and magnetic properties of Fe Co, Ni, and Cr crystals under isotropic deformation. Phys Rev B. 2003;67:035116.
- Šesták P, Černý M, Pokluda J. Influence of normal stress on shear strength along (110) and (112) deformation paths in B2 NiTi alloy from first principles. Mat Sci Eng A. 2008;481–482:247.
- Šesták P, Černý M, Pokluda J. Elastic properties of B19‘ structure of NiTi alloy under uniaxial and hydrostatic loading from first principles. Strength Mater. 2008;70:12.
- Černý M, Šesták P, Řehák P, et al. Ab initio tensile tests of grain boundaries in the fcc crystals of Ni and Co with segregated sp-impurities. Mat Sci Eng A. 2016;669:218.