
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Renewable hydrogen production is a sustainable method for the development of next-generation energy technologies. Utilising solar energy and photocatalysts to split water is an ideal method to produce hydrogen. In this review, the fundamental principles and recent progress of hydrogen production by artificial photosynthesis are reviewed, focusing on hydrogen production from photocatalytic water splitting using organic–inorganic composite-based photocatalysts.
Classification:
1. Introduction
Renewable hydrogen production is key for establishing clean energy systems because hydrogen can be used as a clean energy source in hydrogen fuel cells and a hydrogen-driven society [Citation1,2]. However, the only practical hydrogen production source at present is fossil fuels [Citation3]. Fossil fuels are finite and burning them releases carbon dioxide, which has impacts on the global environment such as global warming. To accomplish renewable energy production, a novel hydrogen production method is required. The ideal source is water. Water can be split into hydrogen (H2) and oxygen (O2) through chemical reactions including electrolysis, biomass, thermal, thermochemical and photochemical reactions, among which electrolysis is the most practical method nowadays. In the early 2000s, 95% of hydrogen production was achieved by reforming fossil fuels, and 5% was obtained from water by electrolysis [Citation3]. Biomass, thermal and thermochemical processes are still in the demonstration stage. On the other hand, hydrogen production using solar energy with photocatalysts is an ideal ‘holy grail’ generation method because: (i) it only involves a simple and clean reaction that directly converts solar energy to hydrogen; (ii) only a photocatalyst, sunlight and water are required; (iii) the reaction occurs under mild conditions (even at room temperature); and (iv) water is split into hydrogen without producing carbon dioxide as a by-product. Although this method has many environmental advantages, many challenges must be overcome before photocatalytic water splitting can be practically implemented. The most challenging issues with photocatalysts are how to take advantage of light beyond the ultraviolet (UV) region and how to improve charge extraction in photosynthesis [Citation4−11]. A photocatalyst that can utilize visible light would be more effective because UV light accounts for only 5–7% of sunlight, whereas visible light accounts for 47%, and the remaining energy falls in the near-infrared (IR) region of the electromagnetic spectrum [Citation12]. Here, we focus on semiconductor-based photocatalysts and their photocatalytic water-splitting performance. One method for using visible light is to employ organic semiconductors with reduced band gaps compared with conventional inorganic semiconductors to obtain the driving force for charge separation in photosynthesis. Therefore, various types of organic–inorganic composites for hydrogen production by water splitting are reviewed herein.
2. Mechanism of photocatalytic water splitting
The first example of water splitting was demonstrated by Honda and Fujishima [Citation13]. They conducted water splitting using a rutile titanium oxide (TiO2) electrode as the anode and a platinum (Pt) electrode as the cathode in an electrochemical cell system in 1972. When the photon energy was absorbed by semiconducting TiO2, electron–hole pairs were separated at the interface between TiO2 (hole) and Pt (electron). When the light is irradiated on TiO2, the absorbed light energy makes it possible to decompose water into hydrogen and oxygen. This phenomenon is called the Honda–Fujishima effect [Citation13]. Bard reported a photocatalysis system using Pt-loaded TiO2 powders for water splitting in 1980 [Citation14]. The success of this system indicates that cell-type systems such as a TiO2–Pt system [Citation15] are not required for photocatalytic water splitting, and photocatalytic reactions can proceed with powder-type photocatalysts, such as a TiO2–ruthenium oxide (RuO2) powder system [Citation16]. These findings have inspired many scientists to investigate hydrogen gas production by photocatalytic water splitting.
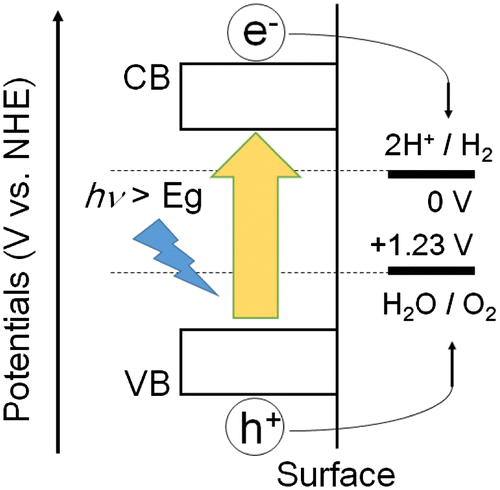
The mechanism of photocatalytic water splitting is shown in Figure . First, when the optical band gap (Eg) in the photocatalyst is irradiated, electrons in the valence band are excited to the conduction band (CB), and holes are generated in the valence band (VB). The photo-generated charges then move to the surface of the catalyst, the electrons at the conduction band reduce protons (H+) to generate hydrogen while water is oxidized by the holes at the valence band to produce oxygen. The hydrogen production reaction requires a potential more negative than 0 V vs the standard hydrogen electrode (SHE) for the H+/H2 half-reaction (pH = 0), and oxygen production requires a potential more positive than 1.23 V vs SHE for the O2/H2O half-reaction (pH = 0). The reactions involved in water splitting can be expressed by equations (Equation1(1)
(1) , Equation2
(2)
(2) ).
(1)
(1)
(2)
(2)
Figure 1. Schematic of photocatalytic water splitting. NHE: the normal hydrogen electrode. CB: conduction band. VB: valence band. Eg: band gap.
In the case of powder-type photocatalysts, electron–hole pairs are produced after light absorption. Almost all electrons and holes recombine before photochemical reactions occur. For example, Bahnemann et al. [Citation17] reported that 80% of charge carriers recombined within 200 ns in 2.4 nm TiO2 particles after 10 mJ/pulse band-gap excitation, and Serpone et al. [Citation18] found that 100% recombined within 10 ns in 2.1 nm TiO2 particles and 90% in 13 nm TiO2 particles after 2.5 mJ/pulse band-gap excitation. The unrecombined electrons and holes are transported to the surface, and water is decomposed to hydrogen and oxygen by electrons and holes, respectively. In the case of TiO2 particles, Tang et al. [Citation19] reported that free holes and electrons are generated picoseconds after excitation, and the hydrogen production reaction occurs on the order of microseconds. The water oxidation reaction is much slower than the hydrogen production reaction because the former reaction requires four electrons to be transferred. These results suggest that charge extraction plays an important role in photocatalytic water splitting.
3. Visible-light-driven semiconductor materials as photocatalysts
In the sunlight spectrum (AM 1.5), the UV region (280–380 nm) is narrower than other regions [Citation12]. The UV component of sunlight is only 5–7%, whereas the visible-light (VL) contribution is 47%. If VL could be used, the efficiency of water splitting would improve dramatically. However, one of the key issues for water splitting is the difficulty in using visible light.
Typical photocatalysts are presented in Figure [Citation20]. Metal oxide semiconductors that are stable and easy to synthesize, such as tungsten oxide (WO3), bismuth vanadium oxide (BiVO4) and tin dioxide (SnO2), can be potential candidates for visible-light-driven photocatalysts. The band gaps of WO3 [Citation21, 22] and BiVO4 [Citation23, 24] are 2.8 eV (440 nm) and 2.4 eV (517 nm), respectively; however, they could not be used for hydrogen production due to their deep conduction band (CB) levels at potentials more positive than 0 V vs NHE (pH = 0). Several visible-light-responsive photocatalysts have been reported [Citation4–6]. Non-oxide semiconductor of cadmium sulfide (CdS, 2.4 eV, 517 nm) showing a visible-light-responsive property has an ideal band level for water splitting; however, cadmium is toxic and decomposes during photoreaction [Citation25–27].
Figure 2. Energy potentials of semiconductors and redox energies for hydrogen production and water oxidation at pH = 7. Reprinted with permission from Ref. [Citation20]. Copyright 2016 American Chemical Society.
![Figure 2. Energy potentials of semiconductors and redox energies for hydrogen production and water oxidation at pH = 7. Reprinted with permission from Ref. [Citation20]. Copyright 2016 American Chemical Society.](/cms/asset/181b5650-9732-440b-9067-0a3e0e8ea683/tsta_a_1375376_f0002_b.gif)
The valence band (VB) of oxynitrides is higher than that of the corresponding metal oxides, whereas the conduction band (CB) of oxynitrides is almost not affected to bottom down the potential due to p - d repulsion [Citation5]. Oxynitrides such as TaON [Citation28−30], Ta3N5 [Citation31,32] and LaTiO2N [Citation33−35] show visible light responses and are remarkably stable when used as photocatalysts for water splitting. Solid solutions have also been used as visible-light-responsive photocatalysts [Citation36−39]. For example, solid solution nanocrystals of gallium nitride−zinc oxide (GaN:ZnO, band gap of 2.6 eV) with a co-catalyst was demonstrated for water splitting under visible-light irradiation at 420–440 nm [Citation40]. Although some semiconductors exhibited high visible-light-driven photocatalytic activity, challenges still remain such as improving the water-splitting hydrogen production efficiency and the stability of catalysts by semiconductor engineering [Citation41−43].
Dye-sensitized photocatalytic water-splitting systems (dye–inorganic semiconductor) may be an alternative for achieving hydrogen production under visible light irradiation. By using the visible-light absorption of dye molecules, photo-excited electrons can be efficiently transferred to inorganic semiconductors, and hydrogen can be produced using these electrons. Since the dye molecules are used as the light absorber, the inorganic semiconductors only need to be UV-responsive materials (>3.0 eV). As it is possible to separate charges from the dye to the inorganic semiconductor over a long distance, charge separation is easier than that in a photocatalyst. In addition, since the photon energies absorbed can be changed by modification of the dye molecule, various inorganic semiconductors can be used in dye-sensitized photocatalytic water-splitting systems.
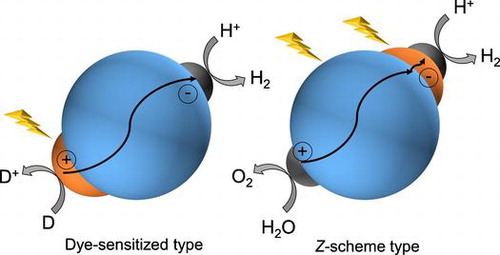
For organic–inorganic composite photocatalysts, charge transfer can be controlled by a combination of materials. Figure shows two types of organic–inorganic composite photocatalysts. The mechanism of dye-sensitized photocatalysts is generally similar to that of dye-sensitized solar cells. The dye reaches the excited state by photoexcitation. After that, electrons are injected from the excited state of oxidation potential of the dye to the CB of an inorganic semiconductor within a timescale of several hundred femtoseconds [Citation44−47]. The injected electrons travel to the surface of the inorganic semiconductor (or move to the co-catalyst) and react with protons to produce hydrogen (Figure (a)).
Figure 3. Two types of water-splitting system using organic–inorganic composites. (a) Dye-sensitized photocatalysts for hydrogen production and (b) Z-scheme-type photocatalysts for hydrogen and oxygen production.
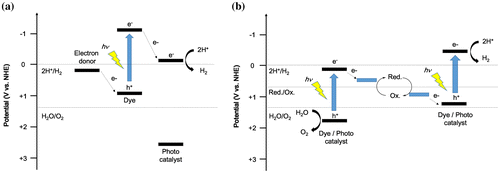
The other composite system is a Z-scheme photocatalytic reaction system, which mimics the process of plant photosynthesis (PSI and PSII). In the Z-scheme system, electrons that are separated after photoexcitation are transferred from a dye or photocatalyst to another dye or photocatalyst via a mediator. These previously excited electrons can regain high energy by photoexcitation. As a result, the transferred electrons contribute to hydrogen production and the holes contribute to oxygen production (Figure (b)).
4. Dye-sensitized photocatalysts
4.1. Metal complex-based dye-sensitized photocatalysts for hydrogen production
4.1.1. Effects of anchoring groups of ruthenium-complex dyes
A typical dye-sensitized photocatalyst system is fabricated using an organic dye and a metal oxide. TiO2 in various phases such as rutile, anatase, P25 and N-TiO2 have been used, with different morphologies such as particle, mesoporous and nanotube structures [Citation48]. The CB energy of TiO2 (anatase) is −0.5 V vs NHE. Therefore, organic dyes with the excited state of oxidation have potential energies higher that can inject electrons to TiO2 (anatase). One advantage of a dye-sensitized photocatalyst system is that it allows us to use more light than visible light even in catalytic systems where the catalysts only absorb ultraviolet light. The first attempt used a system comprising colloidal Pt nanoparticles and RuO2 [Citation49−51]. This system can be cycled by methyl viologen (MV) as redox mediator with tris(bipyridine)ruthenium(II) complex system. Ruthenium complexes have been employed as typical sensitizers for dye-sensitized-type hydrogen production in water.
The structure of the ruthenium complexes can affect hydrogen production [Citation52]. To gain sufficient light absorption and promote charge transfer from Ru-complex to semiconductor, modification of ligands are required to improve the hydrogen production rate. Tris(2,2′-bipyridine)ruthenium sensitizer (Ru(bpy)3), tris(2,2′-bipyridine-4,4′-dicarboxylic acid)ruthenium(II) sensitizer (Ru(dcbpy)3) and tris(bipirimidine)ruthenium(II) sensitizer (Ru(bpym)3) have been loaded on Pt/TiO2 to evaluate the hydrogen production rate. The yield of hydrogen gas by irradiating with a xenon (Xe) lamp (500 W) with 0.1 M ethylenediaminetetraacetic acid (EDTA) as electron donor reagents was 23.3 μL/h with 1.00 mM Ru(bpy)32+, and a similar value of 17.8 μL/h was obtained with 0.25 mM Ru(dcbpy)34−. The chemical structure of Ru(bpym)32+ contains more nitrogen, which showed a higher affinity to the TiO2 surface than Ru(bpy)32+ and thus resulted in enhanced charge transfer. The results also indicated that the bipyrimidine structure showed the highest hydrogen production rate of 80.1 μL/h [Citation53]. The electron injection rate from Ru(bpy)2(dcbpy)2+ to TiO2 was estimated to be in the range 1.0–5.5 × 108 s-l [Citation54]. The effects of the anchoring groups (carboxylate and phosphoric acid) of Ru-complexes on photocatalytic hydrogen production were evaluated by Choi et al. in detail [Citation55,56]. The yields of hydrogen gas after 3 hours with various numbers of anchoring groups were in the order: P2 (ca. 130 μmol) > P6 (ca. 110 μmol) > C6 (ca. 30 μmol) > C2 (ca. 12 μmol) > (C4 ca. 10 μmol), suggesting that the phosphoric acid group showed a higher hydrogen production capacity (Figure (a)) [Citation55, 56].
Figure 4. Ruthenium complexes for dye-sensitized photocatalytic hydrogen production. (a) Ruthenium complexes with multiple anchoring groups. Reprinted with permission from Ref. [Citation56]. Copyright 2006 American Chemical Society. (b) Hybrids of a ruthenium(II) polypyridyl complex and a metal oxide nanosheet for hydrogen production. Reprinted with permission from Ref. [Citation58]. Copyright 2015 American Chemical Society. (c) Di-nuclear ruthenium-complex systems for effective dye-sensitized photocatalytic hydrogen production. Reprinted with permission from Ref. [Citation63]. Copyright 2012 John Wiley & Sons.
![Figure 4. Ruthenium complexes for dye-sensitized photocatalytic hydrogen production. (a) Ruthenium complexes with multiple anchoring groups. Reprinted with permission from Ref. [Citation56]. Copyright 2006 American Chemical Society. (b) Hybrids of a ruthenium(II) polypyridyl complex and a metal oxide nanosheet for hydrogen production. Reprinted with permission from Ref. [Citation58]. Copyright 2015 American Chemical Society. (c) Di-nuclear ruthenium-complex systems for effective dye-sensitized photocatalytic hydrogen production. Reprinted with permission from Ref. [Citation63]. Copyright 2012 John Wiley & Sons.](/cms/asset/c5364e26-41f1-49f8-a4f0-b9815b9be05f/tsta_a_1375376_f0004_oc.gif)
The adsorption mechanism of bis(tetrabutylammonium) cis-bis(thiocyanato)bis(2,2′-bypiridine-4,4′-dicarboxylato)ruthenium(II) (N719) on TiO2 was investigated by Lee et al. [Citation57]. Fourier transform infrared spectroscopy and Raman spectroscopy analyses of the anchoring mode of N719 at the TiO2 interface showed that two carboxylic acid groups were anchored on the TiO2 surface at a TiOH group, and the carboxylic group exhibited electrostatic bonding (H-bonding) and covalent bonding with a bidentate bridging mode on TiOH groups [Citation57].
4.1.2. Effects of ligand structure of Ru-complex dyes on hydrogen production
Ligands also influence hydrogen production capacity. Maeda et al. investigated a variety of nanosheets that were sensitized by different Ru(II) complexes [Citation58]. The dye-sensitized layered perovskite niobate (KCa2Nb3O10) nanosheet showed effective hydrogen production with turnover numbers (TONs) of 1550, 960 and 12 for Ru−CH3, Ru-H and Ru−CF3, respectively, under visible-light irradiation. The reason for this result came from the differences between the oxidation potentials of the metal to ligand charge transfer (MLCT) excited states of the Ru(II) complexes (Eox*) and the CB of nanosheets. The MLCT excited state potentials were −1.18 V, −1.08 V and −0.67 V vs Ag/AgNO3 for Ru-CH3, Ru-H and Ru-CF3, respectively, and that of the reference HCa2Nb3O10 nanosheet was −1.13 V vs silver/silver nitrate (Ag/AgNO3). The energy potential (LUMO) of Ru-CF3 was too negative for the CB of the nanosheet, suggesting that ligands have a strong effect on the LUMO’s energy potential and hydrogen production rate (Figure (b)) [Citation58]. Other Ru-complexes have also been investigated. Zheng et al. [Citation59] investigated a 2-hydroxyl-5-(imidazo-[4,5-f]-1,10-phenanthrolin) benzoic acid linked ruthenium-complex. This system showed a hydrogen production rate of 2578 μmol/h under optimal conditions.
4.1.3. Di-nuclear Ru-complex system for hydrogen production
Multi-nuclear Ru-complex systems are also effective for hydrogen production. The design of multi-nuclear Ru-complexes was suggested by Amadelli et al [Citation60]. Multi-nuclear complexes can increase the electron injection rate by adopting an antenna-sensitizer molecular device structure. Based on this system, Peng et al. investigated a binuclear Ru-complex system for photocatalytic hydrogen production [Citation61–64]. The Pt-loaded TiO2 showed effective hydrogen production with TONs of 874.3, 561.3 and 271.5 for Ru2(bpy)4L1–PF6 (L = μ-4,4-azo-benzene carboxylic acid), Ru(bpy)2(him)2-NO3 (him = imidazole) and N719, respectively, under visible-light irradiation. This result suggested that the bi-nuclear Ru-complex acted as an ‘antenna’ to improve photo-excited charge transfer from the dye into TiO2, while preventing backward charge transfer from TiO2 to the dye because the electron donor part of the Ru-complex is far away from TiO2 [Citation61,62]. A di-Ru-complex, [Ru2(bpy)4(BL)](ClO4)2 (BL = bridging ligand), demonstrated efficient hydrogen production with a TON of 2364 and an apparent quantum efficiency (AQY) of 16.8%. On the other hand, when the ligand of [Ru2(bpy)4(BL)](ClO4)2 had a carboxylic acid group, i.e. [Ru2(dcbpy)4(BL)](ClO4)2, a TON of 317 and an AQY of 1.7% were obtained. Experiments with N719 under the same conditions showed similar performance with a TON of 306 and an AQY of 1.6%. During the photocatalytic reaction, the photo-generated electrons were injected from ruthenium complexes to TiO2. The injected electrons can be used for hydrogen production, and they can also be involved in the backward electron reaction from the TiO2 surface to the Ru-complex through carboxylic acids such as in [Ru2(dcbpy)4(BL)](ClO4)2 and N719. In the case of [Ru2(bpy)4(BL)](ClO4)2, the coordination structure between Ru-complex and TiO2 was varied as the valence mode of RuII was changed after electron injection into TiO2. Therefore, the backward electron reaction and/or charge recombination in [Ru2(bpy)4(BL)](ClO4)2 could be reduced. As a result, the electron injection and hydrogen production efficiencies were increased (Figure (c)) [Citation63]. The yield of hydrogen for [Ru2(bpy)4(BL)](ClO4)2 (1420 μmol) was higher than that for mono-nuclear Ru-complex [Ru(bpy)2(L)](ClO4)2 (803 μmol), suggesting the antenna effects could enhance the hydrogen production rate [Citation64].
4.2. Other metal complexes
Ruthenium dyes can use a long charge lifetime associated with a triplet state for hydrogen production, but on the other hand, weak light absorption is a disadvantage [Citation65]. The porphyrin skeleton which is also found in photosynthetic systems has a high light absorption efficiency and a wide light absorption band in the ultraviolet–visible light region, and thus, we will review dye-sensitized photocatalysts utilizing this property. Malinka et al. [Citation66] investigated zinc porphyrin-coated TiO2 for photocatalytic hydrogen production, and the catalyst showed a visible range absorbance up to 700 nm. The tris(N-methyl-4-pyridyl)-substituted zinc porphyrin (ZnP3+) with 0.4 wt% Pt-loaded TiO2 particles and EDTA showed efficient photocatalytic hydrogen production with a TON of 182. The rate of electron injection into TiO2 for zinc tetra-carboxyphenyl porphyrin is on the order of femtoseconds (kinj ~ 1012 s−1), whereas for zinc cytochrome c (Zn-Cyt-c) enzyme, the electron injection rate was three to four orders of magnitude slower. This efficient electron injection from the triplet state of Zn Cyt-c into TiO2 electrodes can be used for effective hydrogen production, which was estimated to have a quantum efficiency of 10 ± 5% per absorbed photon (Figure (a)) [Citation67,68].
Figure 5. Porphyrin-related organic–inorganic composite systems for photocatalytic hydrogen production. (a) Zinc-treated Cyt-c enzyme-sensitized photocatalyst. Reprinted with permission from Ref. [Citation68]. Copyright 2005 American Chemical Society. (b) Sn-porphyrin–TiO2 hybrid system. Reprinted from Ref. [Citation69] with permission from The Royal Society of Chemistry. (c) Porphyrin nanocylinder-packed TiO2 system. NADH: nicotinamide adenine dinucleotide. Reprinted with permission from Ref. [Citation71]. Copyright 2013 American Chemical Society. (d) Porphyrin–graphene composite photocatalyst. Reprinted with permission from Ref. [Citation73]. Copyright Elsevier.
![Figure 5. Porphyrin-related organic–inorganic composite systems for photocatalytic hydrogen production. (a) Zinc-treated Cyt-c enzyme-sensitized photocatalyst. Reprinted with permission from Ref. [Citation68]. Copyright 2005 American Chemical Society. (b) Sn-porphyrin–TiO2 hybrid system. Reprinted from Ref. [Citation69] with permission from The Royal Society of Chemistry. (c) Porphyrin nanocylinder-packed TiO2 system. NADH: nicotinamide adenine dinucleotide. Reprinted with permission from Ref. [Citation71]. Copyright 2013 American Chemical Society. (d) Porphyrin–graphene composite photocatalyst. Reprinted with permission from Ref. [Citation73]. Copyright Elsevier.](/cms/asset/7d8fdfad-c237-483b-9cad-b91a498aa684/tsta_a_1375376_f0005_oc.gif)
If the porphyrin itself has a long charge lifetime, efficient photoreaction can occur without loading it to a photocatalyst. Kim et al. [Citation69] investigated tin porphyrin (SnP)-sensitized photocatalytic hydrogen production in a TiO2 with EDTA electron donor system. The SnP could not be adsorbed on TiO2 as a physical/chemical driving force in the solution process. However, effective hydrogen production (TON of 440, AQY = 35% at 550 nm) was found during photoreaction. A photo-induced long-lived radical anion species (SnP•-) was generated by a coupled electron donor and had a lifetime long enough to react with the TiO2 surface from the bulk solution under photo-irradiation (Figure b) [Citation69].
Zhu et al. [Citation70] reported a catalyst with porphyrin directly anchored on platinum nanoparticles. The catalyst comprised 2.2 nm platinum particles, porphyrin dye and weakly bound pyrene via hydrogen bonding with porphyrin. The catalyst raised the porphyrin to the excited state using energy transferred by charge transfer from the absorption of pyrene. Electrons were transferred from the porphyrin to platinum, and hydrogen was produced. The TON and quantum yields of hydrogen were 6311 and 2.65%, respectively.
Hasobe et al. [Citation71] reported a hybrid photocatalyst with a self-assembled cylinder-like structure comprising porphyrin cage and TiO2 particles (Figure (c)). The nanocylinder structure enhanced charge separation and, thus, drastic enhancement in the hydrogen production rate was observed. The hydrogen production rate for Pt/TiO2−ZnP(Py)4 nanocylinders was ∼60 mol/g Pt, whereas that for an unencapsulated system comprising Pt/TiO2 and ZnP(Py)4/Pt nanocylinders was ∼0.3 mol/g Pt.
Zhu et al. [Citation72] reported a 5,10,15,20-tetrakis(4-(hydroxyl)phenyl) porphyrin (TPPH)-adsorbed reduced graphene oxide (RGO) hybrid catalyst. The porphyrin was non-covalently adsorbed on the RGO. The TPPH/RGO/Pt catalyst showed an effective photocatalytic activity with a hydrogen production rate of 5.29 mmol/g and an AQY of 1.7%; the hydrogen production rate with P25-TiO2 was 1.52 mmol/g. Aggregation of TPPH/RGO/Pt occurred during photosynthesis, which caused charge quenching. The addition of a surfactant, cetyltrimethylammonium bromide (CTAB), further improved the stability and catalytic hydrogen production rate owing to prevention of TPPH/RGO/Pt aggregation, and a hydrogen production rate of 11.2 mmol/g and an AQY of 3.6% were obtained.
Li et al. [Citation73] investigated chromium (Cr)-loaded reduced graphene oxide (RGO) with 5,15-diphenyl-10,20-di(4-pyridyl)porphyrin (DPyP). The pyridine group of DPyP can be coordinated to the Cr cluster, and thus, DPyP-Cr-RGO with a sandwich-like layered structure was fabricated. Without adding other co-catalysts, RGO, DPyP-RGO and DPyP-Cr-RGO showed visible-light-driven hydrogen production, with yields of 137, 686 and 928 μmol/g, respectively (Figure (d)).
Yuan et al. and Li et al. evaluated other porphyrin sensitizers and composites. Yuan et al. [Citation74] reported tetrakis(4-carboxyphenyl)porphyrin dye-sensitized molybdenum sulfide (MoS2)/ZnO. This system showed a TON of 516 at > 420 nm. Li et al. [Citation75] reported a 5,10,15,20-tetraphenylporphyrin (THPP)-coated copper (I) oxide (Cu2O) photocatalyst. The Cu2O/THPP photocatalyst yielded a 5% photocatalytic activity, evolving 1.3 mmol/g after 6 hours, which suggested that high dye-sensitized photocatalytic activity was achieved with a Cu2O system.
5. Metal-free dye-sensitized hydrogen production
In the case of metal-free organic dye-sensitized photocatalysts, charge injection is inefficient due to easily occurring charge recombination or backward electron reaction. For coumarin pigments, the addition of 4-tert-butylpyridine (TBP) is necessary to prevent charge recombination in order to demonstrate performance equivalent to Ru-dye [Citation76]. This result indicates that the charge recombination lifetime of metal-free dyes is shorter than that of Ru-dyes, which will reduce the hydrogen production rate. To increase the hydrogen production rate when using metal-free organic dyes, precise molecular design is required to ensure efficient charge transfer. To investigate this system, covalently adsorbed metal-free dyes on metal oxides such TiO2 were investigated. Houlding and Grätzel [Citation77] reported hydrogen generation under visible light using an 8-hydroxy-orthoquinoline metal-free dye with Pt-loaded anatase TiO2. The hydrogen production rate was as high as 750–1000 μL/h, and the TON was over 40. Other organic dyes also showed dye-sensitized photocatalytic properties such as xanthene dyes [Citation78−81]. Eosin Y demonstrated a high hydrogen production rate for the xanthene dyes. A variety of Eosin Y dye-sensitized TiO2 photocatalysts were investigated with various TiO2 systems, such as rhodium (Rh)-TiO2 [Citation82], copper(II) oxide (CuO)/TiO2 [Citation83], mesoporous TiO2 [Citation84], TiO2–SiO2 mixed oxide [Citation85] and aeroxide TiO2 [Citation86]. Among these combinations, aeroxide TiO2 showed the highest hydrogen production rate with a 12.2% AQY by optimization of irradiation energy and intensity.
Electron donors such as EDTA, triethanolamine (TEOA), ascorbic acid, oxalic acid and alcohol have been investigated. The concentration and pH were shown to strongly affect the hydrogen production rate [Citation66].
Coumarin and merocyanine derivatives could also be used for effective visible-light-driven photocatalysts. A merocyanine dye together with an I2 sacrificial reagent showed high photocatalytic activity [Citation87]. This system showed a higher hydrogen production rate than an EDTA system. External quantum efficiencies (EQYs) of 1.8% and 2.5% were obtained with NK-2045 (merocyanine dye) at 517 nm and C-343 (Coumarin) at 440 nm, respectively [Citation88].
Ikeda et al. [Citation89] reported benzyl alcohol (Bn(OH)2)-type photocatalysts for visible-light-driven photocatalytic hydrogen production. The phenol moieties of alcohol groups were chemically bonded to a TiO2 surface and showed hydrogen production under visible-light irradiation, which indicated effective charge transfer. Zhang and Choi [Citation90] investigated phenolic resin as a VL sensitizer. The resin can be coated successively on TiO2, and it showed a new absorption band up to 650 nm (Figure (a)). This ligand-to-metal charge transfer showed effective hydrogen production. Kamegawa et al. [Citation91] investigated calixarenes as a sensitizer. This sensitizer has a sulfonic group on the phenol moiety, the negative charge of which could accept the cationic thiazole orange. The photocatalyst–calixarene–cationic dye three-component system broadened the absorption spectra and showed effective charge transfer from the cationic dye to the photocatalyst through calixarene. The AQY was up to 10.4% at 460 nm (Figure (b)).
Figure 6. Phenol–anchored polymer/oligomer structure and TiO2 composite systems for photocatalytic hydrogen production. (a) A hybrid system of TiO2 and phenolic resin acting as a visible-light sensitizer. LMCT: ligand to metal charge transfer. PR: phenolic resin. Reprinted from Ref. [Citation90] with permission from The Royal Society of Chemistry. (b) Photocatalyst–calixarene–cationic dye three-component system. Reprinted with permission from Ref. [Citation91]. Copyright 2013 John Wiley & Sons.
![Figure 6. Phenol–anchored polymer/oligomer structure and TiO2 composite systems for photocatalytic hydrogen production. (a) A hybrid system of TiO2 and phenolic resin acting as a visible-light sensitizer. LMCT: ligand to metal charge transfer. PR: phenolic resin. Reprinted from Ref. [Citation90] with permission from The Royal Society of Chemistry. (b) Photocatalyst–calixarene–cationic dye three-component system. Reprinted with permission from Ref. [Citation91]. Copyright 2013 John Wiley & Sons.](/cms/asset/918d6c63-37b5-40fe-9f42-78b1e7740672/tsta_a_1375376_f0006_oc.gif)
To increase the hydrogen production rate of metal-free dye-sensitized photocatalysts, the design of organic dyes can play a critical role in improving charge extraction during photosynthesis. In metal-free dye-sensitized solar cells, sensitizers of a donor–bridge–acceptor-type were used [Citation92]. The donor–acceptor structure could separate charge upon photoexcitation. The bridge moiety could increase light absorption over a wide wavelength region due to reduction of the HOMO–LUMO band gap and increase in the charge separation lifetime. The donor part acts as a light harvesting moiety, which determines the light harvesting efficiency of the dye. Choi et al. [Citation93] investigated a triphenyl amine (D)–thiophene (B)–acrylic acid (A) system for visible-light-driven photocatalytic reactions. The multiple anchoring groups could increase the extinction coefficient of the CT transition state due to an increase in the donor–acceptor interaction between π-chromophores. This system showed an increase in the hydrogen production rate with an increasing number of anchoring groups. The multi-anchoring groups also showed better stability during hydrogen production. This result indicated that multi-anchoring groups could improve the hydrogen production rate.
Lee et al. [Citation94] investigated the hydrophilic effects using a triphenyl amine (D)–dithiophene (B)–acrylic acid (A) system (Figure (a)). When the impact of hydrophilicity was increased by changing the number of ethylene glycol moieties, the hydrogen production was improved by as much as three times. The AQY of this system at 426 nm was 0.27% [Citation95]. The water molecules and hydrophilic groups interacted at the excited state, which led to a slow charge recombination rate. This slow charge recombination improves the hydrogen production. On the other hand, hydrophobic groups also affect the hydrogen production rate. This phenomenon has been reported by Lee group and our group [Citation96,97]. We investigated the effect of the number of alkoxy chains that was varied in a carbazole (D)–terthiophene (B)–carboxyacrylic acid (A) moiety. Introducing hydrophobic effects also improved the hydrogen production rate by two times compared with bare conditions. The highest TON value with dye CAR16 was 3094 after 24 hours, whereas a TON of 1497 was obtained under bare conditions (CAR1). The differences arose from the interface between the dye and the water medium. The diameters of the largest semicircle in the impedance spectra decreased in the order CAR16 (293.4 Ω) > CAR22 (249.3 Ω) > CAR6 (222.7 Ω) > CAR1 (205.2 Ω), suggesting that the interface between the organic layer and the water medium trapped electrons when the hydrophobicity decreased. These results also indicated that the alkoxy group impacted the hydrogen production (Figure (b)) [Citation96]. Lee et al. [Citation97], Manfredi et al. [Citation98] and Tiwari et al. [Citation99] also reported similar phenomena. Manfredi et al. [Citation98] compared the effects of hydrophilic groups and hydrophobic groups on hydrogen production, and the results showed higher hydrogen production rate with hydrophobic groups (PTZ-ALK dye: 1060 μmol after 20 hours) than with hydrophilic groups (PTZ-TEG dye: 421 μmol after 20 hours).
Figure 7. (a) Hydrophilic triphenylamine-thiophene–TiO2 hybrid system. Reprinted with permission from Ref. [Citation94]. Copyright 2010 American Chemical Society. (b) Hydrophobic carbazole-thiophene–TiO2 hybrid system. Reprinted from Ref. [Citation96] with permission from The Royal Society of Chemistry.
![Figure 7. (a) Hydrophilic triphenylamine-thiophene–TiO2 hybrid system. Reprinted with permission from Ref. [Citation94]. Copyright 2010 American Chemical Society. (b) Hydrophobic carbazole-thiophene–TiO2 hybrid system. Reprinted from Ref. [Citation96] with permission from The Royal Society of Chemistry.](/cms/asset/1d61408c-8a75-4f40-a591-e14b2f857312/tsta_a_1375376_f0007_oc.gif)
The hydrogen production rate was lower than that in our case, probably because the bridge group reduced the charge recombination rate. To solve this issue, we have systematically investigated a phenothiadine (D)–oligothiophene (B)–aclilcarboxilic acid (A) system. The system without oligothiophene showed a TON of only 483 after 16 hours and an EQY of 0.03% at 420 nm [Citation97].
However, the hydrogen production rate was dramatically increased when a thiophene bridge was inserted between the donor and the acceptor. A TON as high as 4460 and an EQY of 1.65% at 420 nm were obtained, which were almost nine times higher than those without the thiophene bridge group. These results indicated that the bridge group increased the number of extracted electrons in photosynthesis. The transient absorption spectra of these photocatalysts showed different charge recombination rates. The MW1 dye showed a shorter decay charge recombination component of τ1 (0.18 ps), suggesting the charge was shortly quenched after electron injection into the TiO2. On the other hand, when the thiophene rings were inserted in the sensitizer, longer components in the decays of MW2 (47.0 ps) and MW3 (85.4 ps) were found. This fact suggested that the thiophene bridge slowed down recombination between the dye and the TiO2. Therefore, MW1 showed a lower TON and quantum efficiency. The longer decay components can provide long-lived electrons in TiO2, and thus the rate of hydrogen production can be increased (Figure (a)) [Citation100].
Figure 8. Bridge and anchor effects on dye-sensitized photocatalytic hydrogen production. (a) Phenothiazine-thiophene (MW1–MW3)–TiO2 hybrid systems, and their transient absorption decay profiles. Reprinted from Ref. [Citation100] with permission from The Royal Society of Chemistry. (b) Cyanoacrylic acid (YFT-1) and triazole (YFT-2) as anchoring groups and the corresponding photocurrent decay profiles. Reprinted with permission from Ref. [Citation103]. Copyright Elsevier.
![Figure 8. Bridge and anchor effects on dye-sensitized photocatalytic hydrogen production. (a) Phenothiazine-thiophene (MW1–MW3)–TiO2 hybrid systems, and their transient absorption decay profiles. Reprinted from Ref. [Citation100] with permission from The Royal Society of Chemistry. (b) Cyanoacrylic acid (YFT-1) and triazole (YFT-2) as anchoring groups and the corresponding photocurrent decay profiles. Reprinted with permission from Ref. [Citation103]. Copyright Elsevier.](/cms/asset/be3ce0f0-e851-42bc-917a-ea2205eee53b/tsta_a_1375376_f0008_oc.gif)
Pal et al. [Citation101] have developed DBA-based dyes using triphenylamine as a donor.When a hydrophobic group such as butoxy group was used for triphenylamine derivatives, hydrogen production capacity was improved. Furthermore, when cyclopentadithiophene derivatives were used as bridges, the performance was improved.
Narayanaswamy et al. [Citation102] developed dyes that used dithiafulvalenes as a donor and reported that a TON of up to 9664 was obtained. Yu et al. [Citation103] investigated the influence of anchoring groups by using cyanoacrylic acid (YFT-1) and triazole (YFT-2). The dye-sensitized solar cell performance was 7.58% for YFT-1 and 4.43% for YFT-2, and the hydrogen production rate was 62.3 μmol/h for YFT-1 and 83.5 μmol/h for YFT-2. YFT-2/TiO2 showed a higher photocurrent value than YFT-1/TiO2 in a TEOA solution, suggesting efficient charge transfer in the triazole system (Figure (b)). Thus, with new substituents, the reactivity of molecules can change significantly even with metal-free dyes. These results mean that more efficient dye-sensitized photocatalysts can be developed by designing new dyes.
6. Organic semiconductor-type photocatalysts
Carbon materials have attracted much attention for use in photocatalytic hydrogen production because the carbon source is abundant on earth. Carbon nitride (C3N4) with a carbon-nitride polymeric structure showed a small band gap of less than 3.0 eV and effective photocatalytic activity in water medium for hydrogen production [Citation20]. Pt or RuO2 catalysts can be supported by C3N4, which improved the catalytic activity for water splitting due to the co-catalytic activity for H2 or O2 formation [Citation104].
Due to the wide band gap (ca. 2.7 eV), C3N4 does not absorb light above 600 nm. To overcoming this issue, band-gap engineering of C3N4 was performed to tune the band gap of C3N4 in a wide range (400–700 nm) and change the thermal polymerization temperature from precursors such as melanin, cyanamide, dicyanamide, urea and thiourea [Citation20, 105]. A method for improving the hydrogen production rate by coexistence with a dye and C3N4 was reported by Takanabe et al [Citation106]. They used phthalocyanato-magnesium (PcMg) as an organic sensitizer, which was shown to absorb light from 400 to 1100 nm. The dye-sensitized-type photocatalytic hydrogen production was demonstrated using this system, which showed an AQY of 0.07% at 660 nm with TEOA as sacrificial reagents. Yu et al. [Citation107] and Zhang et al. [Citation108] used Zn-Pc as a sensitizer, with carboxylic acids as the anchoring group. This sensitizer showed a sharp peak at 700 nm in solution, and a similar absorption spectrum was obtained after it was anchored to C3N4 powder, suggesting that the sensitizer self-assembled on C3N4. The dye-loaded C3N4 showed effective hydrogen production with an AQY of as high as 1.85% at 700 nm. Min and Lu [Citation109] reported a variety of organic sensitizers with C3N4 of photocatalytic activity as 19.4% at 550 nm. Compared with Rose Bengal, fluorescein, Rhodamine B and Ru(bpy)3Cl2, the Eosin Y showed the highest photocatalytic activity. Yan and Huang [Citation110] reported polymer-sensitized C3N4. Polythiophene (P3HT) is a well-known photo-responsive semiconducting material. P3HT-coated C3N4 showed a 300 times increase in the hydrogen production rate when 3 wt% of P3HT was used, and an AQY of 2.9% at 420 nm was obtained with Na2S sacrificial reagents, suggesting effective charge transport during the photoreaction. Ascorbic acid showed a smoother electron transfer pathway than Na2S and, therefore, it showed a remarkably higher hydrogen production rate with an EQY as high as 77.4% at 420 nm [Citation111].
Chen et al. [Citation112] reported a photocatalyst based on a graphitized polyacrylonitrile (g-PAN)/C3N4 composite, where the graphitized polyacrylonitrile was directly grown on the layered C3N4. This composite showed good charge separation and transfer between g-PAN and C3N4, and thus efficient hydrogen evolution was achieved; the hydrogen production rate was 37 μmol/h.
The photocatalytic activity of C3N4 can also be enhanced by combining with inorganic photocatalysts. Li et al. [Citation113] reported a composite of TaON and C3N4, which showed that electron transfer from the CB of C3N4 (−1.12 eV vs SHE) to the CB of TaON (−0.34 eV vs SHE) occurred during VL photoexcitation. Photodegradation of rhodamine B by TaON-C3N4 composite was demonstrated under visible-light irradiation. Although this system was not used as a photocatalyst for hydrogen production, it showed the possibility of charge separation between C3N4 and semiconductor materials.
Yan and Yang [Citation114] reported a C3N4/TiO2 photocatalyst. Reasonably fast electron transfer from the CB of C3N4 (−1.12 eV vs SHE) to the CB of TiO2 (−0.29 eV vs SHE) occurred during VL photoexcitation. The particle size of TiO2–50 wt% C3N4 composite materials was estimated to be approximately in the range 30–40 nm. The composite structure can efficiently separate electron–hole pairs between TiO2 and C3N4; therefore, a hydrogen production rate as high as 22.5 μmol/h was obtained. The structure of semiconductors also affects charge extraction in photosynthesis. TiO2 nanotubes facilitated charge separation in the orthogonal array shown in Figure . The combination of C3N4 and TiO2 nanotubes increased the hydrogen production rate to 15.62 μL/h cm2, whereas the hydrogen production rate for the device containing only TiO2 nanotubes was 0.16 μL/h cm2 (Figure ) [Citation115].
Figure 9. Scanning electron microscopy images showing (a) top and (b) side view of TiO2 nanotube arrays, (c) TiO2 nanotube/C3N4 and (d) TiO2 nanotube/C3N4/Pt. Reprinted with permission from Ref. [Citation115]. Copyright 2016 John Wiley & Sons.
![Figure 9. Scanning electron microscopy images showing (a) top and (b) side view of TiO2 nanotube arrays, (c) TiO2 nanotube/C3N4 and (d) TiO2 nanotube/C3N4/Pt. Reprinted with permission from Ref. [Citation115]. Copyright 2016 John Wiley & Sons.](/cms/asset/672ba3bb-b127-4085-ba9e-ad24f0f922b5/tsta_a_1375376_f0009_oc.gif)
7. Co-catalysts in organic–inorganic composites
Co-catalysts are important for improving the water-splitting activity of organic–inorganic composites. The co-catalyst serves as a reaction site that catalyzes the reaction. In addition, it promotes efficient charge separation between co-catalyst and catalyst to achieve efficient hydrogen production [Citation116]. Platinum is commonly used for photocatalytic hydrogen production due to its low overpotential and excellent kinetics. However, platinum is an expensive and rare metal, which will become a bottleneck in the commercialization stage. To overcome this issue, research and development of low-cost catalysts is urgently required. Trasatti [Citation117, 118] reported that the hydrogen production reaction at the electrode depends on the binding energy between the metal surface and hydrogen and that there is a volcanic relationship. Both acidic and basic platinum, rhodium, gold, nickel and ruthenium have shown high co-catalytic ability. A similar tendency was also observed with a powdery co-catalyst, and Pt, Rh, gold (Au), nickel oxide (NiO) and RuO2 exhibited high activity [Citation4].
Platinum metal complex-based organic dyes have been investigated for hydrogen production. Pt(alizarin)2 dye-loaded TiO2 particles showed a higher hydrogen production rate than alcidine yellow-coated Pt-TiO2 [Citation119]. Zhang et al. [Citation120] reported a co-catalyst system comprising platinum(II) diimine dithiolate sensitizer-loaded Pt–TiO2 particles. The Pt-1 sample showed the highest hydrogen production rate, with a TON of as high as 84, whereas samples 2, 3 and 4 showed TONs of 72, 5 and 7, respectively; samples 3 and 4 were not bound to TiO2. Jarosz et al. [Citation121] also investigated a platinum(II) terpyridyl acetylide-type sensitizer with TiO2 for photocatalytic hydrogen production. In contrast to the di-imine dithiolate system, terpyridyl acetylides with carboxylic anchoring groups showed a lower hydrogen production of TON = 16 than that without carboxylic anchoring groups (TON = 115), due to oxidative quenching occurring during the reaction.
Among non-noble metals, nickel shows the highest activity. Ni-metal organic complexes have been investigated for use as co-catalysts for hydrogen production. Wilson et al. and Jacobsen et al. investigated a 1,3,5,7-tetraphenyl-1,5-diaza-3,7-diphosphacyclooctane ligand-based Ni-complex [Citation122−124]. This Ni-complex showed electrocatalytic activity with trifluoroacetic acid (TFA) or triethylamine (TEA) as a proton donor, suggesting the nickel (Ni)-complex could be used as a reaction centre for hydrogen production. Chen et al. [Citation125] reported a Ni-thiourea complex for photocatalytic reaction. After the Ni-thiourea complex was mixed with a C3N4 semiconductor, the excited electrons were transferred from C3N4 to the Ni-thiourea complex (Figure (a)). Das et al. [Citation126] reported a water-soluble ligand-capped CdSe photocatalyst with a Ni-complex dye for co-catalytic hydrogen production. A TON of > 280000 was achieved with ascorbic acid as sacrificial reagent at pH 4.5. Reynal et al. [Citation127] reported dye-sensitized-type photocatalytic hydrogen production with a Ni-complex as co-catalyst. The combination of ruthenium-complex/TiO2/Ni-complex system showed that the electrons were transferred from the ruthenium-complex sensitizer to the Ni-complex co-catalyst through TiO2 particles. The optimized system showed a TON of 524 after 30 hours.
Figure 10. Organic nickel and cobalt complexes as co-catalysts for photocatalytic hydrogen production. (a) Structure of a nickel-complex–TiO2 hybrid system. Reprinted from Ref. [Citation125] with permission from The Royal Society of Chemistry. (b, c) Ruthenium-complex sensitizer–TiO2–cobalt-complex system for photocatalytic hydrogen production. Reprinted with permission from Ref. [Citation130]. Copyright 2012 John Wiley & Sons.
![Figure 10. Organic nickel and cobalt complexes as co-catalysts for photocatalytic hydrogen production. (a) Structure of a nickel-complex–TiO2 hybrid system. Reprinted from Ref. [Citation125] with permission from The Royal Society of Chemistry. (b, c) Ruthenium-complex sensitizer–TiO2–cobalt-complex system for photocatalytic hydrogen production. Reprinted with permission from Ref. [Citation130]. Copyright 2012 John Wiley & Sons.](/cms/asset/73eec4c5-2117-4b0f-a462-e914b9085b4d/tsta_a_1375376_f0010_b.gif)
Organic cobalt (Co)-complexes also showed a catalytic hydrogen production rate [Citation128]. Reisner et al. [Citation129] developed a dimethylglyoxime-ligand-type Co-complex for dye-sensitized-type photocatalytic reaction. The combination of TiO2/Co-complex with UV irradiation showed effective hydrogen production at 6.07% (AQY) after 4 hours, which suggested that the Co-complex could be used as a co-catalyst for photocatalytic reactions. The combination of RuP/CoP-TiO2 showed visible-light-driven hydrogen production at 2.29% (AQY) at > 420 nm after 8 hours (Figure (b)) [Citation130]. The mechanism involved two-electron reductions of cobalt-complexes by TiO2 particles that were then used for hydrogen production [Citation131]. In the system comprising Ru-dye/TiO2/Co-complex, the electrons were injected into TiO2 nanoparticles from the Ru-dye within 180 ps. The electrons in the TiO2 then migrate to the Co-complex within 10 μs (Figure (c)) [Citation130]. Modifying the ligand of Co-complexes can also improve the hydrogen production rate. For a cobalt complex with a pyridyl ligand, visible-light-driven photocatalytic hydrogen production was achieved with an AQY of 0.35% at 465 nm [Citation132].
8. Z-scheme-type photocatalysts
Another type of photocatalyst is the Z-scheme-type photocatalyst, which mimics the process in plant photosynthesis (PSI and PSII). In 1979, a two-step water-splitting system involving excitation of a combination of two semiconductors was proposed by Bard [Citation133]. In this system, the charges are separated between two n-type semiconductors via a mediator, which can suppress recombination. Therefore, the efficiency of water splitting can be increased. The system contains photocatalysts for O2 evolution and H2 evolution and an electron mediator. The mediator passes the electron on to the H2 generation photocatalyst, and then a second charge separation process occurs due to light absorption. After the Z-scheme-type photocatalytic reaction system was developed, several p- and n-type semiconductors were developed for utilizing this reaction mechanism. The p-type semiconductors such as WO3, TiO2, strontium titanate (SrTiO3) and BiVO4 can be used for the oxidation reaction to produce O2, and the n-type semiconductors such as TiO2, SrTiO3, TaON, zirconium oxide (ZrO2) and C3N4 are used for the reduction reaction to generate H2 [Citation134,135].
Grätzel [Citation136] discussed the Z-scheme-type electron transfer in a complete water-splitting system, using a dye-sensitized photocatalyst. This system used a mesoporous WO3 film as a catalyst for oxygen production, and a dye-sensitized mesoporous TiO2 film as a catalyst for hydrogen generation. The powder-type photocatalytic Z-scheme reaction was reported by Abe et al. [Citation137, 138]. A coumarin-based dye-sensitized Z-scheme water-splitting system, with iridium oxide (IrO2)-Pt/WO3 and Pt/H4Nb6O17 as photocatalysts and I-/I3- as the mediator, showed complete water splitting under VL irradiation. The AQY was 0.05% at 480 nm (Figure a).
Figure 11. Z-scheme-type organic–inorganic composite system for photocatalytic hydrogen production. (a) Dye-sensitized nanosheet with WO3 hybrid system. Reprinted with permission from Ref. [Citation138]. Copyright 2013 American Chemical Society. (b) RGO-mediated C3N4–BiWO4 hybrid Z-scheme-type photocatalyst. Reprinted with permission from Ref. [Citation140]. Copyright Elsevier. (c) Au-mediated C3N4–P3HT hybrid Z-scheme-type photocatalyst. Reprinted from Ref. [Citation145] with permission from The Royal Society of Chemistry. (d) Hexaphyrin–GaN:ZnO composite photocatalyst. Reprinted from Ref. [Citation153] with permission from The Royal Society of Chemistry.
![Figure 11. Z-scheme-type organic–inorganic composite system for photocatalytic hydrogen production. (a) Dye-sensitized nanosheet with WO3 hybrid system. Reprinted with permission from Ref. [Citation138]. Copyright 2013 American Chemical Society. (b) RGO-mediated C3N4–BiWO4 hybrid Z-scheme-type photocatalyst. Reprinted with permission from Ref. [Citation140]. Copyright Elsevier. (c) Au-mediated C3N4–P3HT hybrid Z-scheme-type photocatalyst. Reprinted from Ref. [Citation145] with permission from The Royal Society of Chemistry. (d) Hexaphyrin–GaN:ZnO composite photocatalyst. Reprinted from Ref. [Citation153] with permission from The Royal Society of Chemistry.](/cms/asset/988aaf9d-5389-439a-9bca-b5e72ca38ac1/tsta_a_1375376_f0011_oc.gif)
Solid-state materials can also be used as mediators. A mixture of Ru/SrTiO3:Rh and BiVO4 particles was co-loaded on RGO, and effective charge transfer occurred from BiVO4 to SrTiO3:Rh, similar to a Z-scheme reaction [Citation139]. This result indicates that RGO can be used as a mediator for photocatalysts. The composite of C3N4/RGO/bismuth tungsten oxide (Bi2WO6) also showed effective electron transfer, although this system did not show hydrogen production (Figure (b)) [Citation140]. Similar systems have also been investigated by using combinations of C3N4/BiVO4 [Citation141] and C3N4/AgPO4 [Citation142]. Katsumata et al. [Citation143] investigated a WO3/C3N4 composite, and this composite with TEA as an electron donor was demonstrated for Z-scheme-type hydrogen production (AQY: 0.9% at 405 nm). Martin et al. [Citation144] used Pt-WO3 and Pt-C3N4 with NaI as the redox mediator. This system was capable of complete water splitting, with a H2/O2 yield of 74/37 μmol/h. An Ag-C3N4/Au/P3HT/Pt system also showed VL Z-scheme hydrogen production from water, although complete water splitting was not achieved. In this system, C3N4 and P3HT were separated by gold nanoparticles. Under photo-irradiation, the electrons were transferred from C3N4 to Au, and the holes in P3HT moved to Au. The electron–hole pairs reacted on the Au due to quenching, and the electrons in P3HT can thus react with protons to produce hydrogen on a platinum co-catalyst (Figure (c)) [Citation145].
Organic materials can also be used as mediators. Hagiwara et al. [Citation146−148] reported zirconium (Zr)-doped potassium tantalate (KTaO3) modified with an organic molecule, which completely decomposed water via Z-scheme electron transfer. Due to the wide band gap of KTa(Zr)O3 (3.0 eV), it cannot absorb visible light without being coated with an organic dye. When the Pt/organic dye/KTa(Zr)O3 powder was suspended in pure water and shone with UV-Vis light, both hydrogen and oxygen were produced [Citation147,148]. This result indicated that complete water splitting was achieved. In this system, the UV light was used to excite KTa(Zr)O3 and charge was separated between the CB and the VB. The separated electrons were transferred to the organic dye due to lower energy potentials relative to the CB of KTa(Zr)O3. The electrons on the organic dye were promoted again by a second excitation. These electrons finally migrated to the surface of the Pt co-catalyst, and hydrogen was then produced. The separated holes were used to produce oxygen.
The decay profile of the photo-induced voltage for KTa(Zr)O3 showed a half-life of 11.2 μs. After being treated with organic molecules such as chromate porphyrin (Cr-TCPP), the photo-induced voltage half-life increased to 100 μs. This result suggested that photo-excited charge separation of holes and electrons was enhanced by coating the semiconductors with organic molecules to enable a Z-scheme-type reaction [Citation146].
The organic molecules that can be used include vitamin B12, porphyrin, phthalocyanine, Ru(bpy)3, Cu(bpy)3, pelerine, coronene and xanthene [Citation149−151]. The LUMO energy of the organic molecules plays an important role in the reaction. After examining many dyes, it is found that when the LUMO energy has a potential balance just between the CB of inorganic oxides and platinum, electrons pumped up by the pigment can efficiently migrate to platinum and the hydrogen production efficiency is maximized [Citation149,150].
Modification of the organic molecule showed a positive effect on the water-splitting activity. Hagiwara et al. [Citation152] estimated the aggregation structure of porphyrins on photocatalysts. The parent Cr-porphyrin-treated KTa(Zr)O3 showed characteristic absorption spectra around 613–531 nm for Q bands; however, the alkylated Cr-porphyrin-treated catalysts showed red-shifted and broad peaks around 644–532 nm. These results suggested that the alkylated Cr-porphyrins were J-aggregated and accumulated on the semiconductor. A J-aggregation geometry will not only enhance the absorption range, which can enhance the frequency of excitation, but also enhance charge transport. An increased water-splitting efficiency for hydrogen production was achieved. As a result, the gas generation rate of H2/O2 was 53.7/29.4 μmol/h, which is higher than that with the parent Cr-TPP (40.0/22.7 μmol/h). To increase the light to energy conversion efficiency, harvesting and utilization of visible light are important. By modifying the structure of the porphyrin, it is possible to adjust the HOMO–LUMO gap by extending the structure of the ring. The absorption spectrum shows a wide absorption band from ultraviolet to the near-infrared region. In addition, the choice of semiconductor also affects the photocatalytic activity. The band gap of a solid solution of GaN:ZnO is 2.58–2.76 eV, which showed visible-light-driven photocatalytic activity for complete water splitting. The absorbance spectrum of GaN:ZnO after coating with porphyrins showed a new band around 550–1100 nm, suggesting composites of porphyrin/GaN:ZnO formed successively. After optimizing the photocatalysts and GaN:ZnO, RhOx, NiO /porphyrin/IrO2-GaN:ZnO showed effective fluorescence quenching with a 0.94 ns half-life, which was significantly shorter than that with IrO2-GaN:ZnO (2.9 ns half-life). This result suggested that the energy of GaN:ZnO was readily transferred to the porphyrins. When the RhOx, NiO/porphyrin/IrO2-GaN:ZnO was irradiated with a combination of 300 nm and 600 nm wavelength light, efficient hydrogen (8.3 μmol/h) and oxygen (3.9 μmol/h) production were achieved, which was higher than that obtained without a porphyrin photocatalyst (hydrogen: 2.2 μmol/h, oxygen: 1.1 μmol/h), suggesting the Z-scheme-type photocatalytic reaction (Figure (d)) [Citation153].
9. Summary
In this work, various organic–inorganic hybrid photocatalysts for hydrogen production by water splitting have been reviewed. The organic–inorganic hybrid photocatalyst system has many advantages: (i) the absorption range can be improved, even if the band gap of the semiconductor is > 3.0 eV, by using organic dye molecules that absorb light in the visible to near IR region; (ii) the HOMO–LUMO energy of the molecules can be adjusted by modifying their molecular structure; (iii) charge separation can be enhanced and surface modes can be modified; and (iv) it can be used for co-catalysts instead of noble metals. For dye-sensitized photocatalytic hydrogen production from water, the design of molecular structures is important for achieving visible-light-driven photocatalytic activity. Metal complexes such as ruthenium and donor–bridge–acceptor-type complexes have shown enhanced responses in the visible range. The anchoring group of the dye can affect its orientation on the photocatalyst, and this can increase the charge recombination lifetime. The longer charge recombination lifetime increases the number of electrons available for photosynthesis. Organic molecules can also be used to modify the surface of photocatalysts, which will prevent electron leakage due to charge recombination. Metal complexes have been effectively used as co-catalysts for hydrogen production. For example, platinum, cobalt, nickel and iron metal complexes have been used as reaction centres for hydrogen production, together with a combination of typical semiconductors such as TiO2, visible-light-responsive CdX (X = S, Se, or Te) and the organic photocatalyst C3N4. The combination of Z-scheme-type organic–inorganic photocatalysts has also been discussed. The Z-scheme-type system requires a p–n-type photocatalyst with a redox mediator. Both semiconductors and organic dyes can be selected in the visible light response system. The system could efficiently split water to produce hydrogen and oxygen, indicating effective charge separation in the system. Inorganic semiconductors coated by organic molecules can also be used for Z-scheme-type photocatalytic water splitting.
This review has listed organic materials that have high potential for use as visible-light-driven photocatalysts and co-catalysts. The organic materials can also be used in dye-sensitized-type systems for hydrogen production and Z-scheme-type systems for complete water splitting, indicating that the organic–inorganic system has potential for various applications, similar to inorganic composite systems.
Thus, the advantages of hydrogen production using a hybrid system have been demonstrated. However, designing organic molecules or systems to increase a hydrogen production rate beyond that obtained by natural systems is still a challenging issue. In addition, many hybrid organic sensitizer systems required sacrificial electron donor reagents as it is difficult to oxidize water by organic dye itself. Difficult choices about the specific type of organic hybrid system to be used as a photocatalyst also still remain. This flexible system can be used in other applications such as organic transformation systems. In the future, organic–inorganic hybrid materials will be used to build photocatalyst systems exhibiting higher efficiencies.
Funding
This work was supported by a Grant-in-Aid for Science Research [grant numbers JP17H04888; JP17K19123] from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). The author acknowledges support from I2CNER, funded by the World Premier International Research Centre Initiative (WPI), Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), Japan.
Disclosure statement
No potential conflict of interest was reported by the author.
References
- Bard AJ, Fox MA. Artificial photosynthesis: solar splitting of water to hydrogen and oxygen. Acc Chem Res. 1995;28:141–145.10.1021/ar00051a007
- Arakawa H, Aresta M, Armor JN, et al. Catalysis research of relevance to carbon management: progress, challenges, and opportunities. Chem Rev. 2001;101:953–996.10.1021/cr000018s
- Ogden JM. Prospects for building a hydrogen energy infrastructure. Ann Rev Energy Environ. 1999;24:227–279.10.1146/annurev.energy.24.1.227
- Kudo A, Miseki Y. Heterogeneous photocatalyst materials for water splitting. Chem Soc Rev. 2009;38:253–278.10.1039/B800489G
- Maeda K, Domen K. Solid Solution of GaN and ZnO as a stable photocatalyst for overall water splitting under visible light. Chem Mater. 2010;22:612–623.10.1021/cm901917a
- Tong H, Ouyang S, Bi Y, et al. Nano-photocatalytic Materials: Possibilities and Challenges. Adv Mater. 2012;24:229–251.10.1002/adma.201102752
- Sang Y, Liu H, Uamr A. Photocatalysis from UV/Vis to near-infrared light: towards full solar-light spectrum activity. Chem Cat Chem. 2015;7:559–573.10.1002/cctc.201402812
- Jafari T, Moharreri E, Amin AS, et al. Photocatalytic water splitting—the untamed dream: a review of recent advances. Molecules. 2016;21:900.10.3390/molecules21070900
- Maeda K, Domen K. Photocatalytic water splitting: recent progress. J Phys Chem Lett. 2010;1:2655–2661.10.1021/jz1007966
- Takata T, Pan C, Domen K. Recent progress in oxynitride photocatalysts for visible-light-driven water splitting. Sci Technol Adv Mater. 2015;16:033506.10.1088/1468-6996/16/3/033506
- Yuan L, Han C, Yang MQ, et al. Photocatalytic water splitting for solar hydrogen generation: fundamentals and recent advancements. Int Rev Phys Chem. 2016;35:1–36.10.1080/0144235X.2015.1127027
- Bird RE, Hulstrom RL, Lewis LJ. Terrestrial solar spectral data sets. Solar Energy. 1983;30:563–573.10.1016/0038-092X(83)90068-3
- Honda K, Fujishima A. Electrochemical photolysis of water at a semiconductor electrode. Nature. 1972;238:37–38.
- Bard AJ. Photoelecrochemisty. Science. 1980;207:139–144.
- Sato S, White JM. Photodecomposition of water over Pt/TiO2 catalysts. Chem Phys Lett. 1980;72:83–86.10.1016/0009-2614(80)80246-6
- Kawai T, Sakata T. Photocatalytic decomposition of gaseous water over TiO2 and TiO2—RuO2 surfaces. Chem Phys Lett. 1980;72:87–89.10.1016/0009-2614(80)80247-8
- Bahnemann DW, Hilgendorff M, Memming R. Charge carrier dynamics at TiO2 particles: reactivity of free and trapped holes. J Phys Chem B. 1997;101:4265–4275.10.1021/jp9639915
- Serpone N, Lawless D, Khairutdinov R. Subnanosecond relaxation dynamics in TiO2 colloidal sols (particle sizes Rp = 1.0-13.4 nm). relevance to heterogeneous photocatalysis. J Phys Chem. 1995;99:16655–16661.10.1021/j100045a027
- Tang J, Durrant JR, Klug DR. Mechanism of photocatalytic water splitting in TiO2. Reaction of water with photoholes, importance of charge carrier dynamics, and evidence for four-hole chemistry. J Am Chem Soc. 2008;130:13885–13891.10.1021/ja8034637
- Ong WJ, Tan LL, Ng YH, et al. Graphitic carbon nitride (g-C3N4) - based photocatalysts for Artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability? Chem Rev. 2016;116:7159–7329.10.1021/acs.chemrev.6b00075
- Dong P, Hou G, Xi X, et al. WO3 -based photocatalysts: morphology control, activity enhancement and multifunctional applications. Environ Sci Nano. 2017;4:539–557.10.1039/C6EN00478D
- Zheng H, Ou JZ, Strano MS, et al. Nanostructured tungsten oxide – properties, synthesis, and applications. Adv Funct Mater. 2011;21:2175–2196.10.1002/adfm.v21.12
- Li R, Zhang F, Wang D, et al. Spatial separation of photogenerated electrons and holes among {010} and {110} crystal facets of BiVO4. Nat Commun. 2013;4:1432.10.1038/ncomms2401
- Suarez CM, Hernández S, Russo N. BiVO4 as photocatalyst for solar fuels production through water splitting: a short review. Appl Cat A: General. 2015;504:158–170.10.1016/j.apcata.2014.11.044
- Bao N, Shen L, Takata T, et al. Self-templated synthesis of nanoporous CdS nanostructures for highly efficient photocatalytic hydrogen production under visible light. Chem Mater. 2008;20:110–117.10.1021/cm7029344
- Amirav L, Alivisatos AP. Photocatalytic hydrogen production with tunable nanorod heterostructures. J Phys Chem Lett. 2010;1:1051–1054.10.1021/jz100075c
- Kalisman P, Nakibli Y, Amirav L. Perfect photon-to-hydrogen conversion effciency. Nano Lett. 2016;16:1776–1781.10.1021/acs.nanolett.5b04813
- Maeda K, Terashima H, Kase K, et al. Surface modification of TaON with monoclinic ZrO2 to produce a composite photocatalyst with enhanced hydrogen evolution activity under visible light. Bull Chem Soc Jpn. 2008;81:927–937.10.1246/bcsj.81.927
- Hitoki G, Takata T, Kondo JN, et al. An oxynitride, TaON, as an efficient water oxidation photocatalyst under visible light irradiation (λ ≤ 500 nm). Chem Commun. 2002; 1698–1699.10.1039/B202393H
- Hara M, Nunoshige J, Takata T, et al. Unusual enhancement of H2 evolution by Ru on TaON photocatalyst under visible light irradication. Chem Commun. 2003; 3000–3001.
- Hitoki G, Ishikawa A, Tanaka T, et al. Ta3N5 as a novel visible light-driven photocatalyst (λ < 600 nm). Chem Lett. 2002;31:736–737.10.1246/cl.2002.736
- Lee Y, Nukumizu K, Watanabe T, et al. Effect of 10 MPa ammonia treatment on the activity of visible light responsive Ta3N5 photocatalyst. Chem Lett. 2006;35:352–353.10.1246/cl.2006.352
- Kasahara A, Nukumizu K, Hitoki G, et al. Photoreactions on LaTiO2N under visible light irradiation. J Phys Chem A. 2002;106:6750–6753.10.1021/jp025961+
- Kasahara A, Nukumizu K, Takata T, et al. LaTiO2N as a visible-light (≤ 600 nm)-driven photocatalyst (2). J Phy Chem B. 2003;107:791–797.10.1021/jp026767q
- Matsukawa M, Ishikawa R, Hisatomi T, et al. Enhancing photocatalytic activity of LaTiO2N by removal of surface reconstruction layer. Nano Lett. 2014;14:1038–1041.10.1021/nl404688 h
- Maeda K, Takata T, Hara M, et al. GaN:ZnO solid solution as a photocatalyst for visible-light-driven overall water splitting. J Am Chem Soc. 2005;127:8286–8287.10.1021/ja0518777
- Maeda K, Teramura K, Takata T, et al. Overall water splitting on (Ga1-xZnx)(N1-xOx) solid solution photocatalyst: relationship between physical properties and photocatalytic activity. J Phys Chem B. 2005;109:20504–20510.10.1021/jp053499y
- Maeda K, Teramura K, Lu D, et al. Photocatalyst releasing hydrogen from water. Nature. 2006;440:295.10.1038/440295a
- Sun X, Maeda K, Faucheur ML, et al. Preparation of (Ga1−xZnx) (N1−xOx) solid-solution from ZnGa2O4 and ZnO as a photo-catalyst for overall water splitting under visible light. Appl Catal A. 2007;327:114–121.10.1016/j.apcata.2007.05.005
- Maeda K, Teramura K, Domen K. Effect of post-calcination on photocatalytic activity of (Ga1−xZnx)(N1−xOx) solid solution for overall water splitting under visible light. J Catal. 2008;254:198–204.10.1016/j.jcat.2007.12.009
- Abe R. Recent progress on photocatalytic and photoelectrochemical water splitting under visible irradiation. J Photochem Photobiol C: Photochem Rev. 2010;11:179–209.10.1016/j.jphotochemrev.2011.02.003
- Maeda K. Photocatalytic water splitting using semiconductor particles: history and recent developments. J Photochem Photobiol C: Photochem Rev. 2011;12:237–268.10.1016/j.jphotochemrev.2011.07.001
- Chen X, Shen S, Guo L, et al. Semiconductor-based photocatalytic hydrogen generation. Chem Rev. 2010;110:6503–6570.10.1021/cr1001645
- Furube H, Katoh R, Hara H, et al. Ultrafast stepwise electron injection from photoexcited Ru-complex into nanocrystalline ZnO film via intermediates at the surface. J Phys Chem B. 2003;107:4162–4166.10.1021/jp034039c
- Huber R, Spörlein S, Moser JE, et al. The role of surface states in the ultrafast photoinduced electron transfer from sensitizing dye molecules to semiconductor colloids. J Phys Chem B. 2000;104:8995–9003.10.1021/jp9944381
- Kallioinen J, Benkö G, Sundström V. Electron transfer from the singlet and triplet excited states of Ru(dcbpy)2(NCS)2 into nanocrystalline TiO2 thin films. J Phys Chem B. 2002;106:4396–4404.10.1021/jp0143443
- Benkö G, Kallioinen J, Korppi-Tommola JEI. Photoinduced ultrafast dye-to-semiconductor electron injection from nonthermalized and thermalized donor states. J Am Chem Soc. 2002;124:489–493.10.1021/ja016561n
- Pelaez M, Nolan NT, Pillai SC, et al. A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl Catal B: Environ. 2012;125:331–349.10.1016/j.apcatb.2012.05.036
- Kiwi J, Grätzel M. Hydrogen evolution from water induced by visible light mediated by redox catalysis. Nature. 1979;281:657–658.10.1038/281657a0
- Kiwi J, Borgarello E, Pelizzetti E, et al. Cyclic water cleavage by visible light: drastic improvement of yield of H2 and O2 with bifunctional redox cataltsts. Angew Chem Int Ed Engl. 1980;19:646–648.10.1002/(ISSN)1521-3773
- Duonghong D, Borgarello E, Grätzel M. Dynamics of light-induced water cleavage in colloidal systems. J Am Chem Soc. 1981;103:4685–4690.10.1021/ja00406a004
- Borgarello E, Kiwi J, Pelizzetti E, et al. Photochemical cleavage of water by photocatalysis. Nature. 1981;289:158–160.10.1038/289158a0
- Hirano K, Suzuki E, Ishikawa A, et al. Sensitization of TiO2 particles by dyes to achieve H2 evolution by visible light. J Photochem Photobiol A: Chem. 2000;136:157–161.10.1016/S1010-6030(00)00342-7
- Vinodgopal K, Hua X, Dahlgren RL, et al. Photochemistry of Ru(bpy)2(dcbpy)2+ on A12O3 and TiO2 surfaces. an insight into the mechanism of photosensitization. J Phys Chem. 1995;99:10883–10889.10.1021/j100027a032
- Bae E, Choi W, Park J, et al. Effects of surface anchoring groups (carboxylate vs phosphonate) in ruthenium-complex-sensitized TiO2 on visible light reactivity in aqueous suspensions. J Phys Chem B. 2004;108:14093–14101.10.1021/jp047777p
- Bae R, Choi W. Effect of the anchoring group (carboxylate vs phosphonate) in ru-complex-sensitized TiO2 on hydrogen production under visible light. J Phys Chem B. 2006;110:14792–14799.10.1021/jp062540+
- Lee KE, Gomez MA, Elouatik S, et al. Further Understanding of the adsorption mechanism of N719 sensitizer on anatase TiO2 films for DSSC applications using vibrational spectroscopy and confocal Raman imaging. Langmuir. 2010;26:9575–9583.10.1021/la100137u
- Maeda K, Sahara G, Eguchi M, et al. Hybrids of a ruthenium(II) polypyridyl complex and a metal oxide nanosheet for dye-sensitized hydrogen evolution with visible light: effects of the energy structure on photocatalytic activity. ACS Catal. 2015;5:1700–1707.10.1021/acscatal.5b00040
- Zheng HQ, Yong H, Ou-Yang T. A new photosensitive coordination compound [RuL(bpy)2](PF6)2 and its application in photocatalytic H2 production under the irradiation of visible light. Int J Hydro Energy. 2013;38:12938–12945.10.1016/j.ijhydene.2013.04.138
- Amadelli R, Argazzi R, Bignozzi CA, et al. Design of antenna-sensitizer polynuclear complexes. Sensitization of titanium dioxide with [Ru(bpy)2(CN)2]2Ru(bpy(COO)2)22-. J Am Chem Soc. 1990;112:7099–7103.10.1021/ja00176a003
- Peng T, Ke D, Cai P, et al. Influence of different ruthenium(II) bipyridyl complex on the photocatalytic H2 evolution over TiO2 nanoparticles with mesostructures. J Power Sources. 2008;180:498–505.10.1016/j.jpowsour.2008.02.002
- Peng T, Ke D, Yi H, et al. Photosensitization of different ruthenium(II) complex dyes on TiO2 for photocatalytic H2 evolution under visible-light. Chem Phys Lett. 2008;460:216–219.10.1016/j.cplett.2008.06.001
- Zhang X, Veikko U, Mao J, et al. Visible-light-induced photocatalytic hydrogen production over binuclear RuII–bipyridyl dye-sensitized TiO2 without noble metal loading. Chem Eur J. 2012;18:12103–12111.10.1002/chem.201200725
- Veikko U, Zhang X, Peng T, et al. The synthesis and characterization of dinuclear ruthenium sensitizers and their applications in photocatalytic hydrogen production. Spectrochimica Acta A: Mol Biomol Spect. 2013;105:539–544.10.1016/j.saa.2012.12.061
- Ladomenou K, Natali M, Iengo E, et al. Photochemical hydrogen generation with porphyrin-based systems. Coord Chem Rev. 2015;304-305:38–54.10.1016/j.ccr.2014.10.001
- Malinka EA, Kamalov GL, Vodzinskii SV, et al. Hydrogen production from water by visible light using zinc porphyrin- sensitized platinized titanium dioxide. J Photochem Photobiol A: Chem. 1995;90:153–158.10.1016/1010-6030(95)04093-U
- Topoglidis E, Campbell CJ, Palomares E, et al. Photoelectrochemical study of Zn cytochrome-c immobilised on a nanoporous metal oxide electrode. Chem Commun. 2002; 1518–1519.10.1039/b203448d
- Astuti Y, Palomares E, Haque SA. Triplet state photosensitization of nanocrystalline metal oxide electrodes by zinc-substituted cytochrome c: application to hydrogen evolution. J Am Chem Soc. 2005;127:15120–15126.10.1021/ja0533444
- Kim W, Tachikawa T, Majima T, et al. Tin-porphyrin sensitized TiO2 for the production of H2 under visible light. Energy Environ Sci. 2010;3:1789–1795.10.1039/c0ee00205d
- Zhu M, Lu Lu Y, Du Y, et al. Photocatalytic hydrogen evolution without an electron mediator using a porphyrinepyrene conjugate functionalized Pt nanocomposite as a photocatalyst. Int J Hydro Energy. 2011;36:4298–4304.10.1016/j.ijhydene.2011.01.007
- Hasobe T, Sakai H, Mase K, et al. Remarkable enhancement of photocatalytic hydrogen evolution efficiency utilizing an internal cavity of supramolecular porphyrin hexagonal nanocylinders under visible-light irradiation. J Phys Chem C. 2013;117:4441–4449.10.1021/jp400381 h
- Zhu M, Li Z, Xiao B, et al. Surfactant assistance in improvement of photocatalytic hydrogen production with the porphyrin noncovalently functionalized graphene nanocomposite. ACS Appl Mater Interfaces. 2013;5:1732–1740.10.1021/am302912v
- Ge R, Li X, Kang SZ, et al. Highly efficient graphene oxide/porphyrin photocatalysts for hydrogen evolution and the interfacial electron transfer. Appl Cat B: Environ. 2016;187:67–74.10.1016/j.apcatb.2016.01.024
- Yuan YJ, Tu JR, Ye ZJ, et al. Visible-light-driven hydrogen production from water in a noble- metal-free system catalyzed by zinc porphyrin sensitized MoS2/ZnO. Dyes Pigments. 2013;123:285–292.
- Ge R, Li X, Zhuang B, et al. Assembly mechanism and photoproduced electron transfer for a novel cubic Cu2O/tetrakis(4-hydroxyphenyl)porphyrin hybrid with visible photocatalytic activity for hydrogen evolution. Appl Cat B: Environ. 2017;211:296–304.10.1016/j.apcatb.2017.04.056
- Hara K, Wang ZS, Sato T, et al. Oligothiophene-containing coumarin dyes for efficient dye-sensitized solar cells. J Phys Chem B. 2005;109:15476–15482.10.1021/jp0518557
- Houlding VH, Grätzel M. Photochemical H2 generation by visible light. Sensitization of titanium dioxide particles by surface complexation with 8-hydroxyquinoline. J Am Chem Soc. 1983;105:5695–5696.10.1021/ja00355a032
- Shimizu T, Iyoda T, Koide Y. An advanced visible-light-induced water reduction with dye-sensitized semiconductor powder catalyst. J Am Chem Soc. 1985;107:35–41.10.1021/ja00287a007
- Abe R, Hara K, Sayama K, et al. Steady hydrogen evolution from water on Eosin Y-fixed TiO2 photocatalyst using a silane-coupling reagent under visible light irradiation. J Photochem Photobiol A: Chem. 2000;137:63–69.10.1016/S1010-6030(00)00351-8
- Moser J, Grätzel M. Photosensitized electron injection in colloidal semiconductors. J Am Chem Soc. 1984;106:6557–6564.10.1021/ja00334a017
- Chattrtjee D. Effect of excited state redox properties of dye sensitizers on hydrogen production through photo-splitting of water over TiO2 photocatalyst. Catal Commun. 2010;11:336–339.10.1016/j.catcom.2009.10.026
- Jin Z, Zhang X, Lu G, et al. Improved quantum yield for photocatalytic hydrogen generation under visible light irradiation over eosin sensitized TiO2 —Investigation of different noble metal loading. J Mol Cat A: Chem. 2006;259:275–280.10.1016/j.molcata.2006.06.035
- Jin Z, Zhang X, Li Y, et al. 5.1% Apparent quantum efficiency for stable hydrogen generation over eosin-sensitized CuO/TiO2 photocatalyst under visible light irradiation. Cat Comm. 2007;8: 1267–1273.10.1016/j.catcom.2006.11.019
- Sreethawong T, Junbua C, Chavadej S. Photocatalytic H2 production from water splitting under visible light irradiation using Eosin Y-sensitized mesoporous-assembled Pt/TiO2 nanocrystal photocatalyst. J Power Sources. 2009;190:513–524.10.1016/j.jpowsour.2009.01.054
- Rrungiaroentawon N, Onsuratoom S, Chavadej S. Hydrogen production from water splitting under visible light irradiation using sensitized mesoporous-assembled TiO2-SiO2 mixed oxide photocatalysts. Int J Hydro Energy. 2012;37:11061–11071.10.1016/j.ijhydene.2012.04.120
- Chowdhury R, Gomaa H, Ray AK. Sacrificial hydrogen generation from aqueous triethanolamine with Eosin Y-sensitized Pt/TiO2 photocatalyst in UV, visible and solar light irradiation. Chemosphere. 2015;121:54–61.10.1016/j.chemosphere.2014.10.076
- Abe R, Sayama K, Arakawa H. Efficient hydrogen evolution from aqueous mixture of I− and acetonitrile using a merocyanine dye-sensitized Pt/TiO2 photocatalyst under visible light irradiation. Chem Phys Lett. 2002;362:441–444.10.1016/S0009-2614(02)01140-5
- Abe R, Sayama K, Arakawa H. Significant influence of solvent on hydrogen production from aqueous I3−/I− redox solution using dye-sensitized Pt/TiO2 photocatalyst under visible light irradiation. Chem Phys Lett. 2003;379:230–235.10.1016/j.cplett.2003.07.026
- Ikeda S, Abe C, Torimoto T, et al. Photochemical hydrogen evolution from aqueous triethanolamine solutions sensitized by binaphthol-modified titanium(IV) oxide under visible-light irradiation. J Photocehm Photobiol A: Chem. 2003;160:61–67.10.1016/S1010-6030(03)00222-3
- Zhang G, Choi W. A low-cost sensitizer based on a phenolic resin for charge-transfer type photocatalysts working under visible light. Chem Commun. 2012;48:10621–10623.10.1039/c2cc35751 h
- Kamegawa T, Matsuura S, Seto H, et al. A visible-light-harvesting assembly with a sulfocalixarene linker between dyes and a Pt-TiO2 photocatalyst. Angew Chem Int Ed. 2013;52:916–919.10.1002/anie.201206839
- Zhang X, Peng T, Song S. Recent advances in dye-sensitized semiconductor systems for photocatalytic hydrogen production. J Mater Chem A. 2016;4:2365–2402.10.1039/C5TA08939E
- Choi SK, Yang HS, Kin JH, et al. Organic dye-sensitized TiO2 as a versatile photocatalyst for solar hydrogen and environmental remediation. App Cat B: Environ. 2012;121-122:206–213.10.1016/j.apcatb.2012.04.011
- Lee SH, Park Y, Wee KR, et al. Significance of hydrophilic characters of organic dyes in visible-light hydrogen generation based on TiO2. Org Lett. 2010;12:460–463.10.1021/ol9026182
- Han WS, Wee KR, Kim HY, et al. Hydrophilicity control of visible-light hydrogen evolution and dynamics of the charge-separated state in dye/TiO2/Pt hybrid systems. Chem Eur J. 2012;18:15368–15381.10.1002/chem.v18.48
- Watanabe M, Hagiwara H, Ogata Y, et al. Impact of alkoxy chain length on carbazole-based, visible light-driven, dye sensitized photocatalytic hydrogen production. J Mater Chem A. 2015;3:21713–21721.10.1039/C5TA04991A
- Lee J, Kwak J, Ko KC, et al. Phenothiazine-based organic dyes with two anchoring groups on TiO2 for highly efficient visible light-induced water splitting. Chem Commun. 2012;48:11431–11433.10.1039/c2cc36501d
- Manfredi N, Cecconi B, Calabrese V. Dye-sensitized photocatalytic hydrogen production: distinct activity in a glucose derivative of a phenothiazine dye. Chem Commun. 2016;52:6977–6980.10.1039/C6CC00390G
- Tiwari A, Mondal I, Pal U. Visible light induced hydrogen production over thiophenothiazine-based dye sensitized TiO2 photocatalyst in neutral water. RSC Adv. 2015;5:31415–31421.10.1039/C5RA03039 K
- Watanabe M, Hagiwara H, Iribe A, et al. Spacer effects in metal-free organic dyes for visible-light-driven dye-sensitized photocatalytic hydrogen production. J Mater Chem A. 2014;2:12952–12961.10.1039/C4TA02720E
- Tiwari A, Pal U. Effect of donor-donor-π-acceptor architecture of triphenylamine-based organic sensitizers over TiO2 photocatalysts for visible-light-driven hydrogen production. Int J Hydro Energy. 2015;40:9069–9079.10.1016/j.ijhydene.2015.05.101
- Narayanaswamy K, Tiwari A, Mondal I. Dithiafulvalene functionalized diketopyrrolopyrrole based sensitizers for efficient hydrogen production. Phys Chem Chem Phys. 2015;17:13710–13718.10.1039/C5CP01777G
- Yu F, Cui SC, Li X. Effect of anchoring groups on N-annulated perylene-based sensitizers for dye-sensitized solar cells and photocatalytic H2 evolution. Dyes Pigments. 2017;139:7–18.10.1016/j.dyepig.2016.12.013
- Wang X, Maeda K, Thomas A, et al. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat Mater. 2008;9:76–80.
- Sierra M, Borges E, Esparaza P, et al. Photocatalytic activities of coke carbon/g-C3N4 and Bi metal/Bi mixed oxides/g-C3N4 nanohybrids for the degradation of pollutants in wastewater. Sci Technol Adv Mater. 2016;17:659–668.10.1080/14686996.2016.1235962
- Takanabe K, Kamata K, Wang X, et al. Photocatalytic hydrogen evolution on dye-sensitized mesoporous carbon nitride photocatalyst with magnesium phthalocyanine. Phys Chem Chem Phys. 2010;12:13020–13025.10.1039/c0cp00611d
- Lijuan Yu, Zhang X, Zhuang C, et al. Syntheses of asymmetric zinc phthalocyanines as sensitizer of Pt-loaded graphitic carbon nitride for efficient visible/near-IR-light-driven H2 production. Phys Chem Chem Phys. 2014;16:4106–4114.
- Zhang X, Yu L, Peng T, et al. Highly asymmetric phthalocyanine as a sensitizer of graphitic carbon nitride for extremely efficient photocatalytic H2 production under near-infrared light. ACS Catal. 2014;4:162–170.10.1021/cs400863c
- Min S, Lu G. Enhanced electron transfer from the excited eosin Y to mpg-C3N4 for highly efficient hydrogen evolution under 550 nm irradiation. J Phys Chem C. 2012;116:19644–19652.10.1021/jp304022f
- Yan H, Huang Y. Polymer composites of carbon nitride and poly(3-hexylthiophene) to achieve enhanced hydrogen production from water under visible light. Chem Commun. 2011;47:4168–4170.10.1039/c1cc10250 h
- Zhang X, Peng B, Zhang S, et al. Robust wide visible-light-responsive photoactivity for H2 production over a polymer/polymer heterojunction photocatalyst: the significance of sacrificial regent. ACS Sustainable Chem Eng. 2015;3:1501–1509.10.1021/acssuschemeng.5b00211
- He F, Chen G, Yu Y, et al. Facile approach to synthesize g-PAN/g-C3N4 composites with enhanced photocatalytic H2 evolution activity. ACS Appl Mater Interfaces. 2014;6:7171–7179.10.1021/am500198y
- Yan SC, Lv SB, Li ZC, et al. Organic–inorganic composite photocatalyst of g-C3N4 and TaON with improved visible light photocatalytic activities. Dalton Trans. 2010;39:1488–1491.10.1039/B914110C
- Yan H, Yang H. TiO2–g-C3N4 composite materials for photocatalytic H2 evolution under visible light irradiation. J Alloys Compounds. 2011;509:L26–L29.10.1016/j.jallcom.2010.09.201
- Gao ZD, Qu YF, Zhou X, et al. Pt-decorated g- C3N4 / TiO2 nanotube arrays with enhanced visible-light photocatalytic activity for H2 evolution. ChemistryOpen. 2016;5:197–200.10.1002/open.201500219
- Yang J, Wang D, Han H, et al. Roles of cocatalysts in photocatalysis and photoelectrocatalysis. Acc Chem Res. 2013;46:1900–1909.10.1021/ar300227e
- Trasatti S. Work function, electronegativity, and electrochemical behaviour of metals III. Electrolytic hydrogen evolution in acid solutions. J Electroanal Chem. 1972;39:163–184.10.1016/S0022-0728(72)80485-6
- Trasatti S. Electrocatalysis by oxides — Attempt at a unifying approach. J Electroanal Chem. 1980;111:125–131.10.1016/S0022-0728(80)80084-2
- Zakharenko VS, Blatov AV, Parmon VN. Pt(arizarine)2 complex adsorbed on titanium, dioxide as a sensitizer in photocatalytic evolution of dihydrogen. React Kinet Catal Lett. 1988;36:295–300.10.1007/BF02063821
- Zhang J, Du P, Schneider J, et al. Photogeneration of hydrogen from water using an integrated system based on TiO2 and platinum(II) diimine dithiolate sensitizers. J Am Chem Soc. 2007;129:7726–7727.10.1021/ja071789 h
- Jarosz P, Du P, Schneider J, et al. Platinum(II) terpyridyl acetylide complexes on platinized TiO2: toward the photogeneration of H2 in aqueous media. Inorg Chem. 2009;48:9653–9663.10.1021/ic9001913
- Wilson AD, Newell RH, McNevin MJ, et al. Hydrogen oxidation and production using nickel-based molecular catalysts with positioned proton relays. J Am Chem Soc. 2006;128:358–366.10.1021/ja056442y
- Wilson A, Shoemaker RK, Miedaner A, et al. Nature of hydrogen interactions with Ni(II) complexes containing cyclic phosphine ligands with pendant nitrogen bases. Proc Natl Acad Sci USA. 2007;104:6951–6956.10.1073/pnas.0608928104
- Jacobsen GM, Yang JY, Twamley B, et al. Hydrogen production using cobalt-based molecular catalysts containing a proton relay in the second coordination sphere. Energy Environ Sci. 2008;1:167–174.10.1039/b805309j
- Wang D, Zhang Y, Chen W. A novel nickel–thiourea–triethylamine complex adsorbed on graphitic C3N4 for low-cost solar hydrogen production. Chem Commun. 2014;50:1754–1756.10.1039/c3cc48141 g
- Das A, Han Z, Brennessel WW, et al. Nickel complexes for robust light-driven and electrocatalytic hydrogen production from water. ACS Catal. 2015;5:1397–1406.10.1021/acscatal.5b00045
- Gross MA, Reynal A, Durrant JR, et al. Versatile photocatalytic systems for H2 generation in water based on an efficient DuBois-type nickel catalyst. J Am Chem Soc. 2014;136:356–366.10.1021/ja410592d
- Eckenhoff WT, McNamara WR, Du P, et al. Cobalt complexes as artificial hydrogenases for the reductive side of water splitting. Biochim Biophys Acta. 2013;1827:958–973.10.1016/j.bbabio.2013.05.003
- Lakadamyali F, Reisner E. Photocatalytic H2 evolution from neutral water with a molecular cobalt catalyst on a dye-sensitised TiO2 nanoparticle. Chem Commun. 2011;47:1695–1697.10.1039/c0cc04658b
- Lakadamyali F, Reynal A, Kato M, et al. Electron transfer in dye-sensitised semiconductors modified with molecular cobalt catalysts: photoreduction of aqueous protons. Chem Eur J. 2012;18:15464–15475.10.1002/chem.v18.48
- Reynal A, Lakadamyali F, Gross MA, et al. Parameters affecting electron transfer dynamics from semiconductors to molecular catalysts for the photochemical reduction of protons. Energy Environ Sci. 2013;6:3291–3300.10.1039/c3ee40961a
- Willkomm J, Muresan NM, Reisner E. Enhancing H2 evolution performance of an immobilised cobalt catalyst by rational ligand design. Chem Sci. 2015;6:2727–2736.10.1039/C4SC03946G
- Bard AJ. Photoelectrochemistry and heterogeneous photocatalysis at semiconductors. J Photochem. 1979;10:59–75.10.1016/0047-2670(79)80037-4
- Maeda K. Z-scheme water splitting using two difficient semiconductor photocatalysts. ACS Catal. 2013;3:1486–1503.10.1021/cs4002089
- Li H, Tu W, Zhou Y, et al. Z-scheme photocatalytic systems for promoting photocatalytic performance: recent progress and future challenges. Adv Sci. 2016;3:1500389.10.1002/advs.201500389
- Grätzel M. Photoelectrochemical cells. Nature. 2001;414:338–344.10.1038/35104607
- Abe R, Shinmei K, Hara K, et al. Robust dye-sensitized overall water splitting system with two-step photoexcitation of coumarin dyes and metal oxide semiconductors. Chem Commun. 2009; 3577–3579.10.1039/b905935 k
- Abe R, Shinmei K, Koumura N, et al. Visible-light-induced water splitting based on two-step photoexcitation between dye-sensitized layered niobate and tungsten oxide photocatalysts in the presence of a triiodide/iodide shuttle redox mediator. J Am Chem Soc. 2013;135:16872–16884.10.1021/ja4048637
- Iwase A, Ng YH, Ishiguro Y, et al. Reduced graphene oxide as a solid-state electron mediator in Z-scheme photocatalytic water splitting under visible light. J Am Chem Soc. 2011;133:11054–11057.10.1021/ja203296z
- Ma D, Wu J, Gao M, et al. Fabrication of Z-scheme g-C3N4/RGO/Bi2WO6 photocatalyst with enhanced visible-light photocatalytic activity. Chem Engineer J. 2016;290:136–146.10.1016/j.cej.2016.01.031
- Tian N, Huang H, He Y, et al. Mediator-free direct Z-scheme photocatalytic system: BiVO4/g-C3N4 organic – inorganic hybrid photocatalyst with highly efficient visible-light-induced photocatalytic activity. Dalton Trans. 2015; 44: 4297–4307.10.1039/C4DT03905 J
- Katsumata H, Sakai T, Suzuki T, et al. Highly efficient photocatalytic activity of g-C3N4/Ag3PO4 hybrid photocatalysts through Z-scheme photocatalytic mechanism under visible light. Ind Eng Chem Res. 2014;53:8018–8025.10.1021/ie5012036
- Katsumata H, Tachi Y, Suzuki T, et al. Z-scheme photocatalytic hydrogen production over WO3/g-C3N4 composite photocatalyst. RSC Adv. 2014;4:21405–21409.10.1039/C4RA02511C
- Martin DJ, Rerdon PJT, Moniz SJA, et al. Visible light-driven pure water splitting by a nature-inspired organic semiconductor-based system. J Am Chem Soc. 2014;136:12568–12571.10.1021/ja506386e
- Zhang Y, Mao F, Yan H, et al. A polymer – metal – polymer – metal heterostructure for enhanced photocatalytic hydrogen production. J Mater Chem A. 2015;3:109–115.10.1039/C4TA04636F
- Hagiwara H, Ono N, Inoue T, et al. Dye-sensitizer effects on a Pt/KTa(Zr)O3 catalyst for the photocatalytic splitting of water. Angew Chem Int Ed. 2006;45:1420–1422.10.1002/(ISSN)1521-3773
- Hagiwara H, Matsumoto H, Ishihara T. Improvement of photocatalytic activity of metal sulfide by organic dye for H2 formation from water. Electrochemistry. 2008;76:125–127.10.5796/electrochemistry.76.125
- Hidehisa H, Kumagae K, Ishihara T. Effects of nitrogen dopingon photocatalytic water-splitting activity of Pt KTa0.92Zr0.08O3 perovskite oxide catalyst. Chem Lett. 2010; 39: 498–499.
- Hidehisa H, Inoue T, Kaneko K, et al. Charge-transfer mechanism in Pt/KTa(Zr)O3 photocatalysts modified with porphyrinoids for water splitting. Chem Eur J. 2009;15:12862–12870.
- Hidehisa H, Ono N, Ishihara T. Effects of redox potential of metallophthalocyanine dye on photocatalytic activity of KTa(Zr)O3 for water splitting. Chem Lett. 2010;39(178):179.
- Nagatomo M, Hagiwara H, Ida S, et al. Modification of organic dyes on photocatalytic water splitting activity of KTa(Zr)O3. Electrochemisty. 2011;79:779–782.10.5796/electrochemistry.79.779
- Hagiwara H, Higashi K, Watanabe M, et al. Effect of porphyrin molecular structure on water splitting activity of a KTaO3 photocatalyst. Catalysis. 2016;6:42.
- Hagiwara H, Watanabe M, Daio T, et al. Modification effects of meso-hexakis(pentafluorophenyl) [26]hexaphyrin aggregates on the photocatalytic water splitting. Chem Commun. 2014;50:12515–12518.10.1039/C4CC05127 K