
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
We report the mechanistic investigation of catalytic H2 evolution from formic acid in water using a formate-bridged dinuclear Ru complex as a formate hydrogen lyase model. The mechanistic study is based on isotope-labeling experiments involving hydrogen isotope exchange reaction.
Graphical Abstract
Classification:
1. Introduction
Formate hydrogen lyase (FHL) is an enzyme complex that catalyzes the conversion of HCOOH to evolve CO2 and H2 (Eq. 1) [Citation1–3]. This enzyme complex is composed of a formate dehydrogenase (FDH) [Citation4,5] and a [NiFe]hydrogenase ([NiFe]H2ase) [Citation6–9], and catalyzes oxidation of HCOOH (Eq. 2) and evolution of H2, respectively (Eq. 3).(1)
(1)
(2)
(2)
(3)
(3)
We have previously reported a NiIIRuII model complex [NiII(X)(HCOO)(μ-H)RuII(C6Me6)] (X = N,N′-dimethyl-3,7-diazanonane-1,9-dithiolato) that can catalyze fast conversion of HCOOH to H2 and CO2 [Citation10] [turnover frequency {TOF = (mol of evolved H2/mol of catalyst) per hour} = 857 h−1]. This extremely fast reaction rate, however, prevented us from investigating the mechanism of H2 formation.
As part of our efforts to investigate the mechanism of H2/CO2 formation from HCOOH, we now report a dinuclear RuI complex [RuI2(CO)4(μ-HCOO)2(DMSO)2] (1) that catalyzes the above reaction at a significantly slower reaction rate (TOF = 13.1 h−1). Here we disclose the detailed mechanism of H2 formation from HCOOH based on isotope-labeling experiments.
2. Experimental details
2.1. Materials and methods
All experiments were carried out under N2 or Ar atmosphere by using standard Schlenk techniques and a glovebox. Tetrahydrofuran (THF) was distilled from Na/benzophenone under N2 atmosphere prior to use. HCOOH, DCOOD, 40% NaOD/D2O, and dimethylsulfoxide (DMSO) were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). HCOOD and DCOOH were purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). These materials were used without further purification. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a JEOL JNM-AL300 spectrometer (JEOL, Tokyo, Japan) at 25 °C, in which the chemical shifts were referenced to tetramethylsilane (TMS) in chloroform-d1 and DMSO in DMSO-d6. UV–vis absorption spectra were recorded on a JASCO V–670 UV–Visible–NIR spectrophotometer (the light pass length was 1.0 cm). An IR spectrum was recorded on a Thermo Nicolet NEXUS 870 Fourier transform infrared (FTIR) instrument (Thermo Fisher Scientific, Massachusetts, USA) at 25 °C. Gas chromatographic (GC) analyses were conducted by a Shimadzu GC–8A (He carrier) (Shimadzu, Kyoto, Japan) with a MnCl2–alumina column (model: Shinwa OGO–SP) at –196 °C (liquid N2) for quantitative analyses of H2, HD, and D2 and by a Shimadzu GC-2014 (Ar carrier) (Shimadzu, Kyoto, Japan) with activated charcoal at 100 °C for quantitative analyses for H2, HD, D2, and CO2 using a thermal conductivity detector. Elemental analysis data were obtained by a PerkinElmer 2400II series CHNS/O analyzer (PerkinElmer, Massachusetts, USA) using Ar as the carrier gas. Dynamic light scattering measurements were conducted with a Malvern Zetasizer Nano (Malvern, Worcestershire, UK). X-band electron spin resonance (ESR) spectra were measured by a JEOL JES-FA200 spectrometer (JEOL, Tokyo, Japan) at –150 °C.
pH Adjustment. The pH of the solution was adjusted by using HCOOH and NaOH/H2O (1.0–7.0). The pD of the solution was adjusted by using HCOOH, DCOOH, HCOOD, DCOOD, and 40% NaOD/D2O (1.0–7.0), in which H+ concentration of HCOOH and DCOOH is negligible quantity compared to D+ concentration of D2O. In a pH (or pD) range of 1.0–7.0, the pH (or pD) values of the solutions were determined by a pH meter (model: TOA HM20 J; DKK-TOA, Tokyo, Japan) equipped with a pH combination electrode (model: TOA GST-5725C; DKK-TOA, Tokyo, Japan). Values of pD were corrected by adding 0.4 to the observed values (pD = pH meter reading + 0.4) [Citation11,12].
[RuI2(CO)4(μ-HCOO)2(DMSO)2] (1). A THF solution of [Ru03(CO)12] (1.00 g, 1.56 mmol) and HCOOH (12 mL, 0.32 mol) was refluxed for 3.5 h. The resulting solution was cooled to room temperature, to which was added DMSO (335 μL, 4.72 mmol) followed by its stirring for 30 min. The solvent was removed under reduced pressure to yield a yellow powder of 1, which was collected and dried in vacuo {yield: 85% based on the amount of [Ru03(CO)12] added}. 1H NMR (300 MHz, in chloroform-d1, referenced to TMS): δ 8.23 (s, 2H, HCOO), 3.06 (s, 12H, (CH3)2SO). Anal. Calcd for 1 (C10H14O10Ru2S2): C, 21.43; H, 2.52%. Found: C, 21.45; H, 2.22%.
Typical procedure of H2 evolution from HCOOH catalyzed by 1. In a 3.0 mL vial capped with a septum, a solution of HCOOH (2.60 mmol) in H2O (1.0 mL) was added to 1 (1.25 μmol). The pH of the resulting solution was adjusted to 1.0–7.0 and the solution was heated at 80 °C for 1 h. The gas above the solution within the vial was sampled with a gas-tight syringe (500 μL) and analyzed for H2 and CO2 by GC. No CO was observed. No nanoparticles were formed in the catalytic reaction, which was confirmed by dynamic light scattering measurements.
Isotope-labeling experiments for catalytic H2, HD, and D2 evolution. In a 3.0 mL vial capped with a septum, DCOOH (2.60 mmol) in H2O (1.0 mL), HCOOD (2.60 mmol) in D2O (1.0 mL), or DCOOD (2.60 mmol) in D2O (1.0 mL) was added to 1 (1.25 μmol). The pH or pD of the resulting solution was adjusted to 3.5 by the addition of NaOH or NaOD, respectively, and it was stirred at 80 °C for 1 h. The gas above the solution within vial was sampled with a gas-tight syringe (500 μL) and analyzed for H2, HD, D2, and CO2 by GC. No CO was observed. No nanoparticles were formed in the catalytic reaction, which was confirmed by a dynamic light scattering measurements.
The initial rate of the catalytic H2 evolution against the catalyst concentration. In a 3.0 mL vial capped with a septum, a solution of HCOOH (2.60 M) in H2O was added into 1 (0.63, 1.25, 2.5, or 5.0 mM). The pH of the resulting solution was adjusted to 3.5 and the solution was heated at 80 °C for 300 s. The gas above the solution within the vial was sampled with a gas-tight syringe (500 μL) and analyzed for H2 by GC. The catalytic reaction is first-order against the catalyst concentration.
Reactivity of hydride species 2 toward proton of HCOOH. In a 3.0 mL vial capped with a septum, 10 equivalents of HCOOH (9.4 μL, 250 μmol) was added into a DMSO (2.0 mL) solution of 2 (vide infra), which was prepared from the reaction of 1 (14 mg, 25 μmol) with HCOONa (1.7 mg, 25 μmol) at 80 °C for 1 h. No H2 gas was formed, as confirmed by GC analysis. The same reaction was conducted using DMSO-d6 (450 μL) instead of DMSO (2.0 mL), which was monitored by 1H NMR spectroscopy. No decrease of the hydride-derived peak of 2 was observed.
H+/D+ exchange of hydride ligand of 2. In an NMR sample tube, a DMSO-d6 solution (400 μL) of 1 (14 mg, 25 μmol) with HCOONa (1.7 mg, 25 μmol) was heated at 80 °C for 1 h to form 2, which was confirmed by 1H NMR spectroscopy. Then, D2O (50 μL) was added into the resulting DMSO-d6 solution under N2 atmosphere. The H+/D+ exchange of hydride ligand of 2 was confirmed by 1H NMR spectroscopy with CH2Br2 as an internal standard to investigate the intensities of hydride- and formate-derived peaks.
X-ray crystallographic analysis of 1. A single crystal of 1 suitable for X–ray analysis was obtained from the diffusion of diethyl ether into its THF solution. Measurements were performed on a Rigaku/MSC Saturn CCD diffractometer (Rigaku, Tokyo, Japan) with confocal monochromated Mo-Kα radiation (λ = 0.7107 Å). Data were collected and processed using the CrystalClear program. All calculations were performed using the CrystalStructure crystallographic software package except for refinement, which was performed using SHELXL–97. Crystallographic data for 1 have been deposited at the Cambridge Crystallographic Data Centre as Supplementary Publication No. CCDC 1556459. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK {fax.: (+44)1223–336–033; e-mail: [email protected]}.
3. Results and discussion
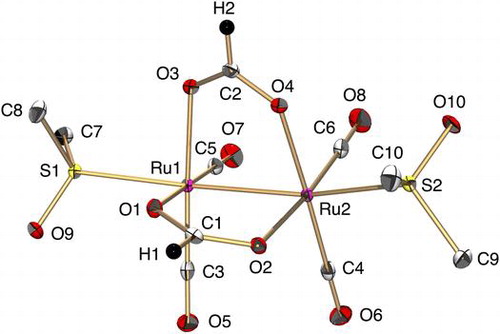
A bis(μ-formate) RuI2 complex, [RuI2(CO)4(μ-HCOO)2(DMSO)2] (1) was synthesized from the reaction of [Ru03(CO)12] with HCOOH and DMSO in THF under a N2 atmosphere, which was then characterized by X-ray analysis, and 1H NMR, UV-vis, and IR spectroscopies (Figures ). Both Ru atoms adopt a distorted octahedral geometry in which the Ru ion is ligated by two C(CO), an S(DMSO), two O(μ-formate), and an adjacent Ru atom (Figure ). Two Ru atoms are tethered by a Ru–Ru bond and two formate ligands. The bond distance of Ru1–Ru2 {2.6654(3) Å} is similar to those of other formate-bridged dinuclear Ru complexes (2.679 and 2.720 Å) [Citation13,14]. The C–O bond distances of each HCOO– ligand {C1–O1 = 1.258(2), C1–O2 = 1.262(2), C2–O3 = 1.263(2), and C2–O4 = 1.259(2) Å} are almost the same, which indicates that this could be explained by the delocalization of a double bond over two C–O bonds. The C–O bond distances of four CO ligands {1.142(2)–1.148(2) Å} are slightly longer than that of free CO (1.128 Å), suggestive of back donation of electron density from the t2 g orbital of low-valent RuI to the π*-antibonding orbital of CO.
Figure 1. An ORTEP drawing of 1 with thermal ellipsoids at 50% probability. The hydrogen atoms of DMSO are omitted for clarity. Selected interatomic distances (l/Å) and angles (ϕ/°): Ru1–Ru2 = 2.6654(3), Ru1–O1 = 2.1152(13), Ru1–O3 = 2.1290(13), Ru1–S1 = 2.4107(5), Ru1–C3 = 1.855(2), Ru1–C5 = 1.853(2), Ru2–O2 = 2.1374(13), Ru2–O4 = 2.1221(13), Ru2–S2 = 2.4134(5), Ru2–C4 = 1.855(2), Ru2–C6 = 1.849(2), C1–O1 = 1.258(2), C1–O2 = 1.262(2), C2–O3 = 1.263(2), C2–O4 = 1.259(2), C3–O5 = 1.145(2), C4–O6 = 1.142(2), C5–O7 = 1.148(2), C6–O8 = 1.146(2), O1–C1–O2 = 126.67(18), O3–C2–O4 = 126.76(17).
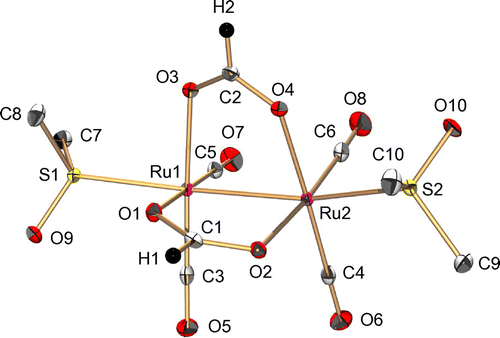
Figure 2. An IR spectrum of 1 in a KBr disk.
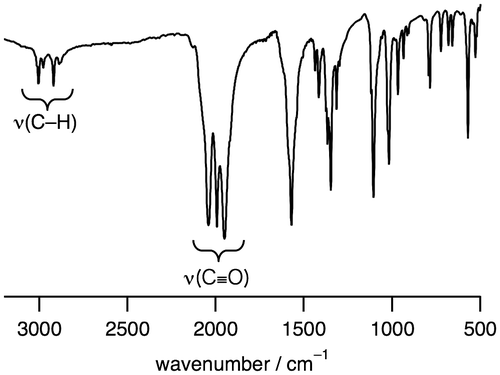
Figure 3. A 1H NMR spectrum of 1 in chloroform-d1.
Figure 4. A UV-vis absorption spectrum of 1 (0.060 mM) in H2O.
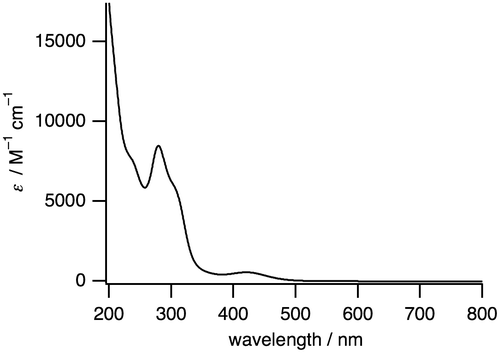
The IR spectrum of 1 shows the stretching frequencies of CO coordinated to RuI centers at 1950, 1994, and 2041 cm−1 (Figure ), which are lower than the free CO stretching frequency (2143 cm−1). This weakened CO bond is also caused by the back donation from the low-valent RuI to the CO ligand. The 1H NMR signals observed in the diamagnetic region revealed that 1 is diamagnetic, which originates from an antiferromagnetic exchange interaction between two RuI centers through metal–metal bonding (Figure ). The UV-vis absorption spectrum of 1 in H2O shows a sharp band at 280 nm (8480 M−1 cm−1) and a broad band at 420 nm (550 M−1 cm−1) (Figure ).
The bis(μ-formate) RuI2 complex 1 can convert to a (hydride)(formate) species 2 with evolution of CO2 in DMSO at 80 °C in the presence of 1 equivalent of HCOONa. β-Hydrogen elimination is expected to be assisted by the metal centers to release CO2 and form the hydride ligand [Citation15]. The 1H NMR spectrum of 2 in DMSO-d6 shows hydride- and formate-derived signals at –12.4 and 7.76 ppm, respectively, with the same intensities (Figure (a)). The hydride-derived peak is typical of those found in hydride-bridged Ru complexes [Citation16–19]. The dimer structure of 2 with an antiferromagnetic exchange interaction between two RuI centers, was suggested by the signals observed in the diamagnetic region of the 1H NMR spectrum and its ESR silent character.
Figure 5. 1H NMR spectra of 2 (a) before and (b) after addition of D2O, in which 2 was obtained from heating of 1 at 80 °C in DMSO-d6 for 1 h in the presence of 1 equivalent of HCOONa. †: the peak of CH2Br2 as an internal standard.
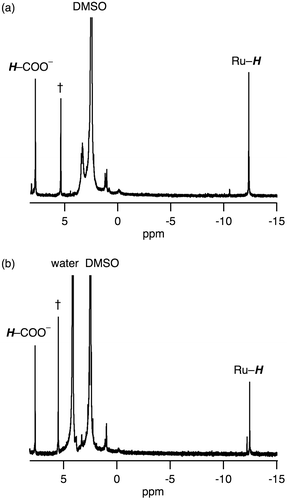
We have confirmed that the hydride ligand of 2 has a protic character rather than hydridic character based on the following investigations. We observed 2 being unreactive toward proton of HCOOH, i.e. dihydrogen gas was not formed via protonation of the hydride ligand, which was confirmed by 1H NMR spectroscopy and GC analysis. Then, we observed an H+/D+ exchange of the hydride ligand of 2, as confirmed by 1H NMR spectroscopy (Figure ). The intensity of hydride-derived peak of 2 only decreased by the addition of D2O into DMSO-d6 solution of 2 (Figure ), meaning that the hydride ligand underwent the H+/D+ exchange with D+.
Complex 1 is a precursor to catalyze the pH-dependent conversion of HCOOH to H2 and CO2 in water at pH 1.0–7.0 (Figures and Table ). H2 and CO2 gases were detected by GC. No nanoparticles were formed in the catalytic reaction, which was confirmed by dynamic light scattering measurements. Figure shows the time-dependent profile of turnover numbers (TONs, mol of H2 evolved/mol of 1) of H2 evolution in the reaction of 1 with an excess amount of HCOOH in water at 80 °C. The pH-dependent TON shows a maximum around pH 3.5 (Figure ). We investigated the dependence of initial rate for H2 production against the concentration of 1 (0.63–5.0 mM). This linear correlation clearly indicates that the catalytic reaction is first-order against the catalyst concentration (Figure ).
Figure 6. A plot of turnover numbers (TONs) vs. time for the H2 evolution from HCOOH catalyzed by 1 (1.25 mM) with HCOOH (2.60 M) in H2O at 80 °C and pH 3.5.
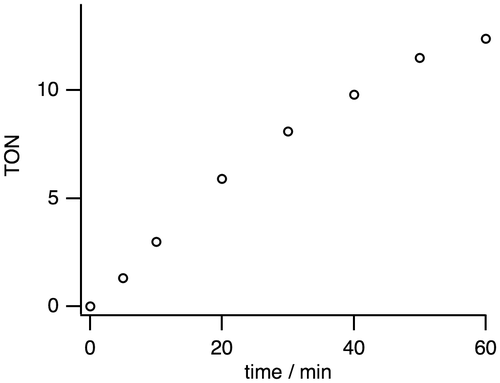
Figure 7. A pH-dependent H2 evolution from HCOOH catalyzed by 1 (1.25 mM) with HCOOH (2.60 M) in H2O for 1 h at 80 °C. The maximum turnover number (TON) is 13.1 at pH 3.5.
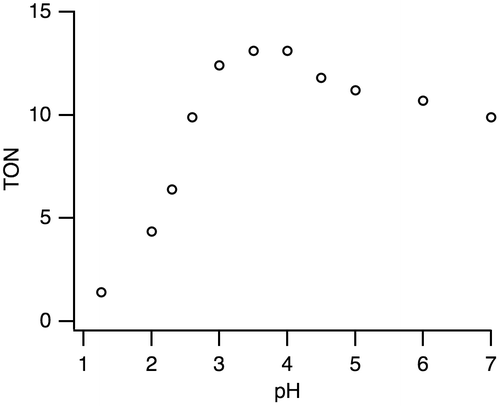
Figure 8. A plot of initial rate of H2 production against the concentration of 1 (0.63–5.0 mM) in the reaction of 1 with HCOOH (2.60 M) at 80 °C and pH 3.5. The initial rate of H2 production was determined for 300 s.
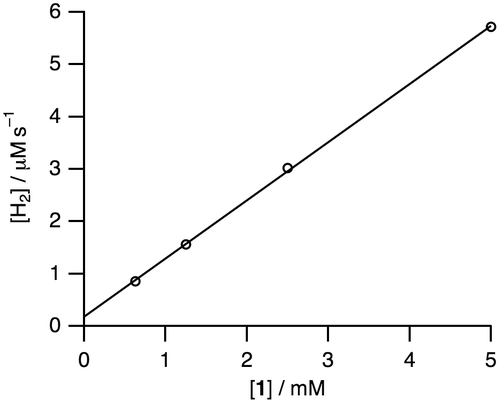
Figure 9. A proposed reaction mechanism for the conversion of HCOOH to H2 and CO2 catalyzed by dinuclear Ru complexes in H2O.
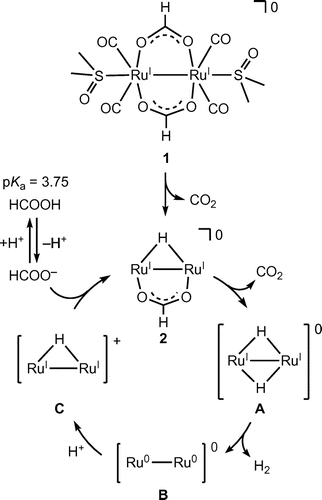
Figure 10. A proposed reaction mechanism for the hydrogen isotope exchange reaction in the conversion of HCOOD to H2, HD, D2, and CO2 in D2O catalyzed by dinuclear Ru complexes (entry 3 of Table 1).
Table 1. Dehydrogenation of HCOOH, DCOOH, HCOOD, or DCOOD in H2O or D2O by using 1 as a precursorTable Footnotea
In order to investigate the reaction mechanism of the dehydrogenation of HCOOH, deuterium isotope-labeling experiments were conducted as shown in Table . Entries 2 (DCOOH in H2O at pH 3.5) and 3 (HCOOD in D2O at pD 3.5) show the TONs of 5.2 and 6.7, respectively, which are lower than that of 13.1 in the entry 1 (HCOOH in H2O at pH 3.5). These results suggest that the cleavage of C–H bond in HCOOH and O–H bond in H2O should be involved in the rate-determining step. The lowest TON value of 2.4 in entry 4 (DCOOD in D2O at pD 3.5) can be expected by the results of entries 2 and 3. According to the results of entries 1–4, we propose the reaction mechanism shown in Figure . The bis(μ-formate) complex 1 is converted to the (hydride)(formate) species 2 with the release of CO2. The subsequent decarboxylation of 2 is expected to result in formation of a dihydride species A, which can be proposed owing to the previously reported analogous structures [Citation20–24]. Then reductive elimination of H2 can yield a low-valent species B. Protonation of B would provide a monohydride species C, which can bind HCOO– to return to 2. This proposed mechanism involves protonation step of B and binding step of HCOO–, with a pKa of 3.75, to C, which causes a maximum TON around pH 3.5 because acidic conditions facilitate the protonation step and basic conditions deprotonating HCOOH facilitate the binding step of HCOO– to C.
We also investigated the hydrogen isotope exchange reaction by observation of evolved dihydrogen gases in Table . The entry 2 shows H2 (7%) and HD (93%) are evolved in the reaction of 1 with DCOOH in H2O. This result clearly indicates that dihydrogen gases (H2 and HD) originate from H of H2O and D of DCOOH. The evolved H2 is a key product to elucidate the reaction mechanism since H2 comes from only H+ in H2O, which enables us to exclude the possibility of protonation of the hydride ligand to evolve dihydrogen gas. The same is true of entry 3. In this context, we can propose the reaction mechanism of the hydrogen isotope exchange reaction, involving the H+/D+ exchange of the protic hydride ligand, causing reductive elimination of dihydrogen gas, as shown in Figure [Citation25].
4. Conclusions
Isotope-labeling experiments suggest that the formation of dihydrogen gas from formic acid takes place via H+/D+ exchange of the protic hydride ligands and reductive elimination from the protic hydride ligands of the dinuclear Ru complex in H2O/D2O.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by Japan Society for the Promotion of Science (JSPS), [grant numbers JP26000008 (Specially Promoted Research); JP16K05727; JP15K05566]; World Premier International Research Centre Initiative (WPI), Japan.
Supplemental data
The supplemental material for this paper is available online at https://doi.org/10.1080/14686996.2017.1379857
suppl.zip
Download Zip (240.8 KB)Related Research Data
References
- Prinske C, Jaroschinsky M, Sawers RG. Levels of control exerted by the Isc iron-sulfur cluster system on biosynthesis of the formate hydrogenlyase complex. Microbiology. 2013;159:1179–1189. 10.1099/mic.0.066142-0
- Forzi L, Sawers RG. Maturation of [NiFe]-hydrogenases in Escherichia coli. Biometals. 2007;20:565–578. 10.1007/s10534-006-9048-5
- Bagramyan K, Trchounian A. Structural and functional features of formate hydrogen lyase, an enzyme of mixed-acid fermentation from Escherichia coli. Biochemistry (Moscow). 2003;68:1159–1170. 10.1023/B:BIRY.0000009129.18714.a4
- Hille R, Hall J, Basu P. The mononuclear molybdenum enzymes. Chem Rev. 2014;114:3963–4038.10.1021/cr400443z
- Gonzalez PJ, Rivas MG, Mota CS, et al. Periplasmic nitrate reductases and formate dehydrogenases: Biological control of the chemical properties of Mo and W for fine tuning of reactivity, substrate specificity and metabolic role. Coord Chem Rev. 2013;257:315–331.10.1016/j.ccr.2012.05.020
- Special issue on Hydrogenases. Eur J Inorg Chem. 2011;2011:915–1171.
- Special issue on Renewable energy. Chem Soc Rev. 2009;38:1–300.
- Tard C, Pickett CJ. Structural and functional analogues of the active sites of the [Fe]-, [NiFe]-, and [FeFe]-hydrogenases. Chem Rev. 2009;109:2245–2274. 10.1021/cr800542q
- Special issue on Hydrogen. Chem Rev. 2007;107:3900–4435.
- Nguyen NT, Mori Y, Matsumoto T, et al. A [NiFe]hydrogenase model that catalyses the release of hydrogen from formic acid. Chem Commun. 2014;50:13385–13387.10.1039/C4CC05911E
- Glasoe PK, Long FA. Use of glass electrodes to measure acidities in deuterium oxide. J Phys Chem. 1960;64:188–190. 10.1021/j100830a521
- Mikkelsen K, Nielsen SO. Acidity measurements with the glass electrode in H2O-D2O mixtures. J Phys Chem. 1960;64:632–637. 10.1021/j100834a026
- Morris DJ, Clarkson GJ, Wills M. Insights into hydrogen generation from formic acid using ruthenium complexes. Organometallics. 2009;28:4133–4140. 10.1021/om900099u
- Czaun M, Goeppert A, Kothandarman J, et al. Formic acid as a hydrogen storage medium: ruthenium-catalyzed generation of hydrogen from formic acid in emulsions. ACS Catal. 2014;4:311–320. 10.1021/cs4007974
- Robertson A, Matsumoto T, Ogo S. The development of aqueous transfer hydrogenation catalysts. Dalton Trans. 2011;40:10304–10310. 10.1039/c1dt10544b
- Churchill MR, Janik TS, Duggan TP, et al. Synthesis, characterization, and crystal structure of (μ-H)2Ru3(μ3-η2-CHC(O)OCH3)(CO)9, a stabilized intermediate in the reductive elimination of hydrocarbons from trimetallic clusters. Organometallics. 1987;6:799–805. 10.1021/om00147a019
- Gao Y, Kuncheria JK, Jenkins HA, et al. The interconversion of formic acid and hydrogen/carbon dioxide using a binuclear ruthenium complex catalyst. J Chem Soc Dalton Trans. 2000;3212–3217.
- Pelayo-Vázquez JB, González FJ, Leyva MA, et al. A ruthenium carbonyl cluster containing a hydroquinone ligand: A layered structure with a polymetallic species. Structure and electrochemical characterization. J Organomet Chem. 2012;716:289–293. 10.1016/j.jorganchem.2012.06.030
- Hossain MJ, Rajbangshi S, Khan MMM, et al. Experimental and computational studies on the reaction of silanes with the diphosphine-bridged triruthenium clusters Ru3(CO)10(μ-dppf), Ru3(CO)10(μ-dppm) and Ru3(CO)9{μ3-PPhCH2PPh(C6H4)}. J Organomet Chem. 2014;767:185–195. 10.1016/j.jorganchem.2014.05.040
- Mayer T, Böttcher H-C. Protonation of metal–metal bonds in coordinatively unsaturated diruthenium cores. Polyhedron. 2013;50:507–511. 10.1016/j.poly.2012.11.049
- Siedle AR, Newmark RA, Pignolet LH. Organometallic chemistry of fluorocarbon acids. synthesis, structure, and solvolysis of a sulfinate-bridged diruthenium dihydride cluster, [(Ph3P)4Ru2(μ-H)2(μ-CF3SO2)(CO)2]HC(SO2CF3)2. Inorg Chem. 1986;25:1345–1351. 10.1021/ic00229a010
- Geetharani K, Bose SK, Sahoo S, et al. Cluster expansion reactions of group 6 and 8 metallaboranes using transition metal carbonyl compounds of groups 7–9. Inorg Chem. 2011;50:5824–5832. 10.1021/ic200802c
- Nagashima H, Fukahori T, Aoki K, et al. Partial hydrogenation of acenaphthylene on the face of a triruthenium cluster: formation of (μ2:η1:η5-C12H10)Ru3H2(CO)7 from (μ3:η2:η3:η5-C12H8)Ru3(CO)7. J Am Chem Soc. 1993;115:10430–10431. 10.1021/ja00075a097
- van Buijtenen J, Meuldijk J, Vekemans JAJM, et al. Dinuclear ruthenium complexes bearing dicarboxylate and phosphine ligands. Acceptorless catalytic dehydrogenation of 1-phenylethanol. Organometallics. 2006;25:873–881. 10.1021/om050789s
- Ogo S. Electrons from hydrogen. Chem Commun. 2009;3317–3325.