
Abstract
Sterically large substituents can provide kinetic stabilization to various types of low-coordinate compounds. For example, regarding the chemistry of the group 14 elements, since West et al. introduced the concept of kinetic protection of the otherwise highly reactive Si=Si double bond by bulky mesityl (2,4,6-trimethylphenyl) groups in 1981, a number of unsaturated compounds of silicon and its group homologs have been successfully isolated by steric effects using the appropriate large substituents. However, the functions and applications of the Si–Si π-bonds consisting of the 3pπ electrons on the formally sp 2-hybridized silicon atoms have rarely been explored until 10 years ago, when Scheschkewitz and Tamao independently reported the model systems of the oligo(p-phenylenedisilenylene)s (Si–OPVs) in 2007. This review focuses on the recent advances in the chemistry of π-electron systems containing Si=Si double bonds, mainly published in the last decade. The synthesis, characterization, and potential application of a variety of donor-free π-conjugated disilene compounds are described.
1. Introduction
In the periodic table, silicon is located just below carbon in the Group 14 elements, but carbon and silicon have different roles and functions in nature. While carbon is a central element in the organic substances that constitute the body of all living things, silicon is a key element of inorganic substances that constitute the earth’ s crust and is widely used in glass, semiconductors, concrete, ceramics, etc. Strangely, there is no organosilicon material containing C–Si bonds in nature except for silicon carbide in meteorites. Therefore, every organosilicon compound is an artificial substance created by human technology.
While a variety of allotropes of carbon, such as diamond, graphite, fullerenes, and carbon nanotubes, are known as a stable material mainly consisting of the sp 3- or sp 2-hybridized carbon atoms, the stable form of silicon only possesses a diamond-type structure based on the formally sp 3-hybridized silicon atoms. Can sp 2-hybridized silicon atoms exist as a stable substance? In recent years, silicene, the silicon analog of graphene, has attracted much attention, from both experimentalists and theoreticians, as a new two-dimensional allotrope of silicon [Citation1–3]. Theoretical studies predict that repeating units in pure silicon nanosheets do not exhibit a planar hexagonal geometry like graphene but a non-classical propellane motif, presumably due to the instability associated with sp 2-hybridized silicon atoms [Citation4].
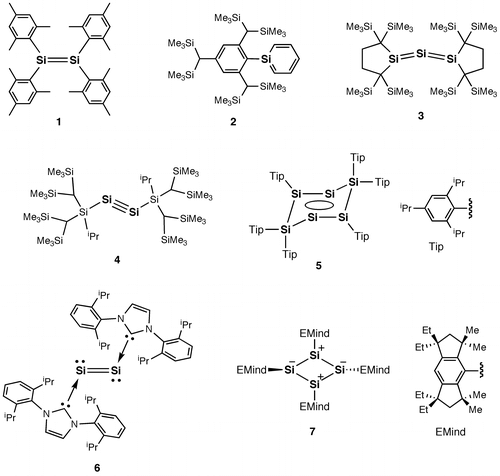
In general, the Si–Si π-bond is much weaker than the C–C π-bond essentially due to the less effective overlap of the two adjacent 3p orbitals relative to that of the 2p orbitals, corresponding to the greater covalent atomic radius of silicon (1.11(2) Å) than that of carbon (sp 3-C; 0.76(1), sp 2-C; 0.73(2), and sp-C; 0.69(1) Å) [Citation5]. In 1981, West, Fink, and Michl demonstrated for the first time that disilene (R2Si=SiR2) (1), the silicon analog of alkene, can be created based on the concept of kinetic stabilization using sterically demanding substituents, protecting the highly reactive Si=Si double bond, as shown in Figure , which produced a significant change in the main group chemistry [Citation6–11]. In fact, after the finding of this isolatable disilene 1, many kinds of unsaturated compounds of silicon have been successfully obtained by virtue of the steric effects of the bulky protecting groups [Citation12–22]. For recent representative examples, since 2000, silaaromatics (2) [Citation23–25], trisilaallenes (R2Si=Si=SiR2) (3) [Citation26,27], and disilynes (RSi≡SiR) (4) [Citation28–31] have been isolated using the appropriately designed bulky aryl, alkyl, and silyl substituents, respectively. Also, a tricyclic aromatic isomer of hexasilabenzene (5) [Citation32] and a disilicon(0) fragment coordinated by the N-heterocyclic carbenes (NHCs) (6) [Citation33] were synthesized as stable crystalline compounds. In 2011, we reported the synthesis of a cyclobutadiene (CBD) silicon analog, i.e. tetrasilacyclobutadiene (7), with a planar rhombic charge-separated structure originating from the polar Jahn–Teller distortion [Citation34]. This is the first persila[n]annulene compound, (SiR) n (n is an even number equal to or greater than 4), with a cyclic structure consisting of formally sp 2-hybridized silicon atoms, which will open a new facet of silicon π-science.
Figure 1. Examples of stable unsaturated silicon compounds.
Although the fundamental chemistry of the low-coordinated compounds of silicon has been steadily established year-by-year, the functions and applications of the Si–Si π-bonds consisting of the 3pπ electrons on the formally sp 2-hybridized silicon atoms have rarely been explored. In main group element chemistry, recent synthetic efforts have been directed toward investigating the combination of the multiple bonds of the heavier main group elements and the carbon π-electron systems due to their unique electronic properties and potentially useful technological applications for organic electronics, which would offer a new avenue to functional organoelement materials [Citation35–45]. However, this chemistry always faces a formidable challenge. While the sufficient steric effects of the bulky substituents are crucial in protecting the highly reactive heavier multiple bonds, it may cause twisting of the π-framework, which prevents the preferred extension of the π-conjugation over the skeleton.
In order to further develop this chemistry toward advanced materials science and technology, we have designed a series of fused-ring bulky 1,1,3,3,5,5,7,7-octa-R-substituted s-hydrindacen-4-yl groups, called the ‘Rind’ groups, as shown in Figure , where R denotes the initial of the substituents on the benzylic positions of the hydrindacene skeleton [Citation46]. The Rind groups are actually giant aryl hydrocarbon substituents. Nevertheless, they can be easily prepared by organic synthetic methods, including the intramolecular Friedel-Crafts double cyclization [Citation47]. In addition, various R groups (R1, R2, R3, and R4 groups) can be introduced at the four benzylic positions of the hydrindacene skeleton. While the peripheral R1 and R2 groups can control their physical properties such as crystallinity and solubility, the proximal R3 and R4 groups can directly change the steric (size and shape) effects of the Rind groups. Also, the Rind groups have a rigid structure based on the fused-ring system and show a high chemical stability due to the full substitution at all the benzylic positions, whose C–H bonds are generally more reactive than other C–H bonds. The term ‘Rind’ in English describes the thick outer skin of some types of fruits such as an orange and melon, which is fully in accordance with our research idea, i.e. Rind can keep the inside fresh. Actually, the Rind groups provide us great opportunities to study a variety of low-coordinate compounds of the main group elements [Citation34,48–62] and coordinatively unsaturated transition metal complexes [Citation63–71].
Figure 2. Rind groups.
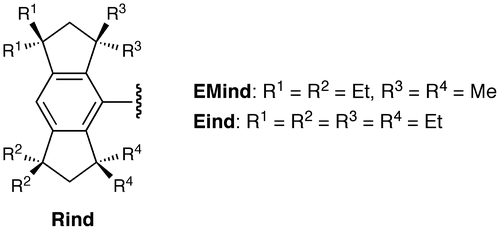
Figure 3. Disilenes 8 and 15.
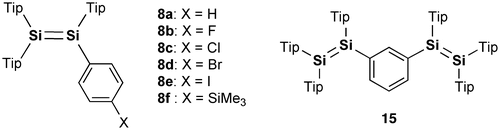
Figure 4. Molecular structures of 16 (a) and 17 (b) determined by X-ray crystallography.
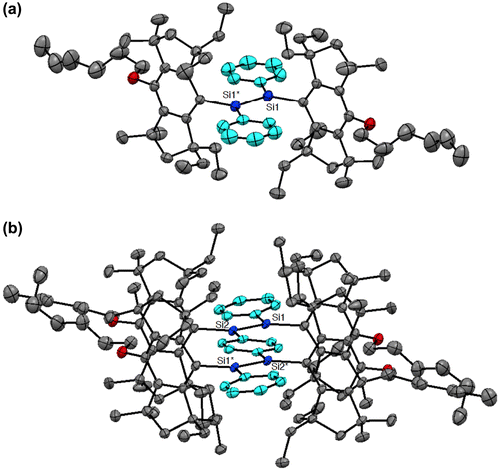
Figure 5. Photographs of the THF solutions of 16–19 at room temperature: (a) under room light; (b) under 360 nm UV light.

Figure 6. Disilenes 22 and 23.
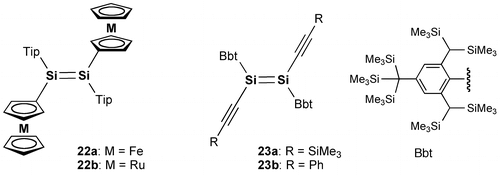
Figure 7. Molecular structures of 24 (a), 25a (b), and 25b (c) determined by X-ray crystallography.
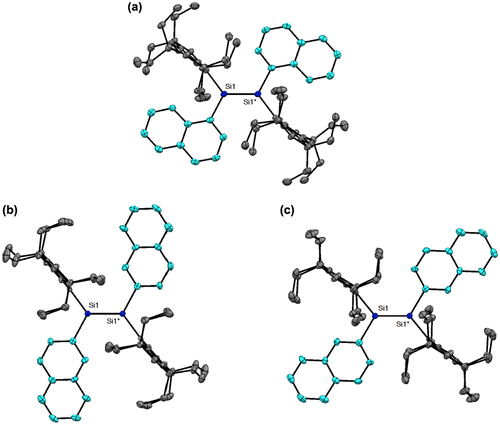
Figure 8. Photographs of the THF solutions (left) and solid in the air (right) of 24 and 25: (a) under room light; (b) under 365 nm UV light.
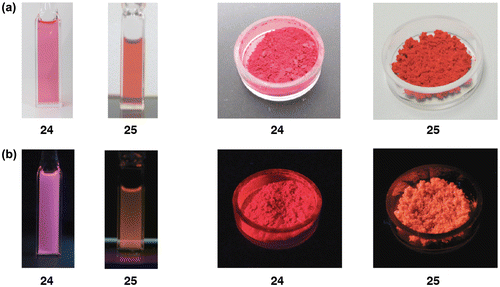
Figure 9. Frontier molecular orbitals of 24, 25a, and 25b together with the energy levels.
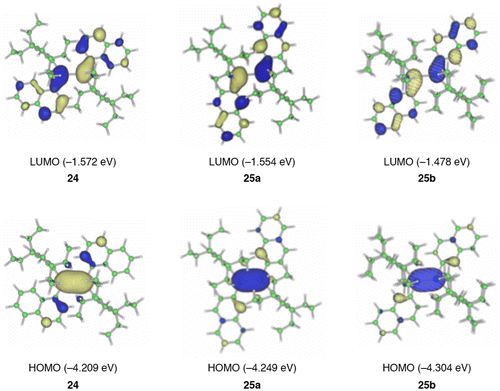
Figure 10. (a) The configuration of the EL device; (b) EL from the device at a 5 V applied voltage.
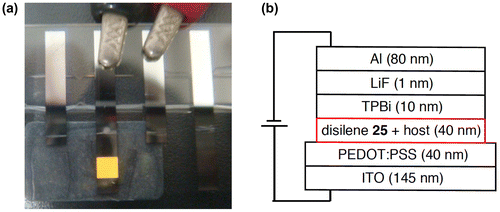
Figure 11. Molecular structure of 29 determined by X-ray crystallography.
Figure 12. Photographs of the solutions of 29 under 365 nm UV light: (a) hexane; (b) THF; (c) acetone.

Figure 13. Selected molecular orbitals of 29 together with the energy levels.
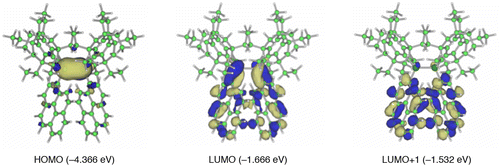
Figure 14. Disilenes 31–33.
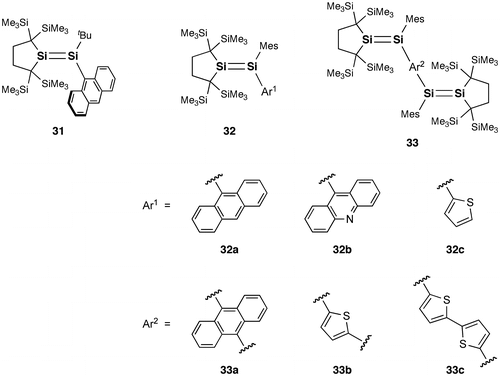
Figure 15. Disilenes 34 and 35.
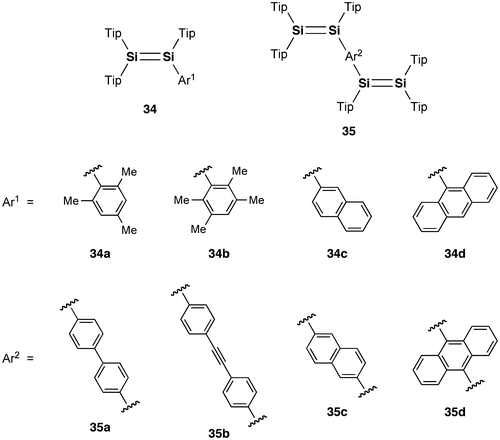
Figure 16. Photographs of the THF solutions of 29: (a) before irradiation; (b) after irradiation at 530 nm.

Figure 17. Oligothiophenes 36.
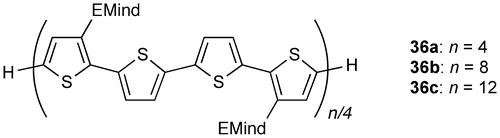
Figure 18. Molecular structures of (a) 37a and (b) 38a determined by X-ray crystallography.
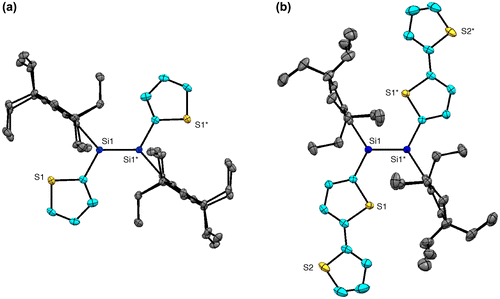
Figure 19. Rotational isomers of 37 and 38.
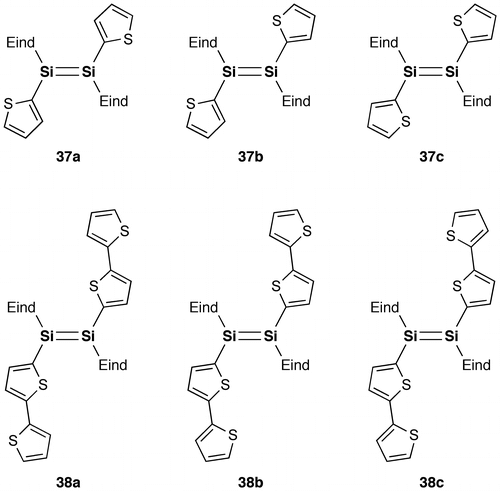
Figure 20. Photographs of the THF solutions: (a) 37 under room light; (b) 38 under room light; (c) 38 under 365 nm UV light.

Figure 21. Frontier molecular orbitals of 37a and 38a together with the energy levels.
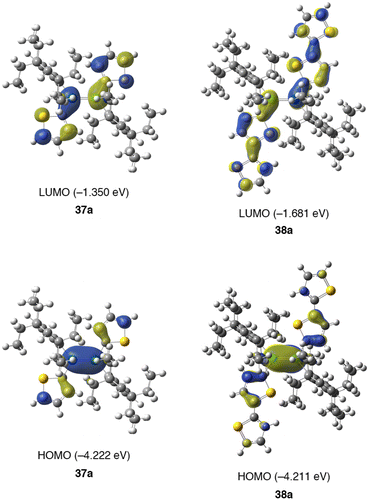
Figure 22. Compounds 5, 7, and 41–43.
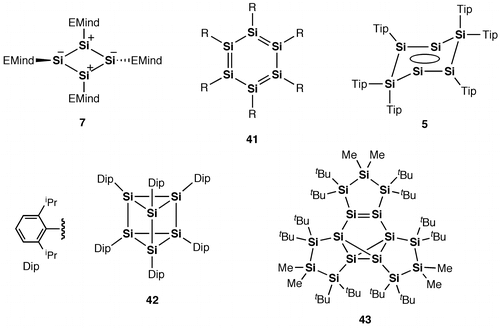
Figure 23. Compounds 44–47.
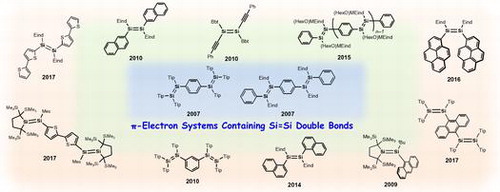
In this review, we describe the recent progress in developing organic π-electron architectures featuring Si=Si double bonds, mainly focusing on the following four topics: (1) oligo(p-phenylenedisilenylene)s; π-conjugation between benzene rings and Si=Si units, (2) air-stable emissive disilenes with naphthyl groups; potential applicability in electroluminescence devices, (3) disilene π-system with pyrenyl groups; evidence for intramolecular charge-transfer emission, and (4) disilene–thiophene π-systems; future organosilicon chemistry for developing advanced materials.
2. Oligo(p-phenylenedisilenylene)s; π-conjugation between benzene rings and Si=Si units
Poly(p-phenylenevinylene)s (PPVs) with alternating benzene rings and C=C double bonds are some of the most attractive conducting polymers due to their excellent stability and processability and unique electronic and optical properties, which can be used for various applications in modern electrochemistry [Citation72,73]. Oligo(p-phenylenevinylene)s (OPVs) have also received extensive attention as linear monodispersed π-conjugated oligomers with well-defined molecular structures and tunable optoelectronic properties [Citation74–76]. Since the Si=Si units possess a narrower energy gap between the highest occupied molecular orbital and the lowest unoccupied molecular orbital (HOMO–LUMO) relative to the C=C units [Citation12–22], the disilene analogs of OPVs, oligo(p-phenylenedisilenylene)s (Si–OPVs), would provide new opportunities for application to a range of organic electronic devices.
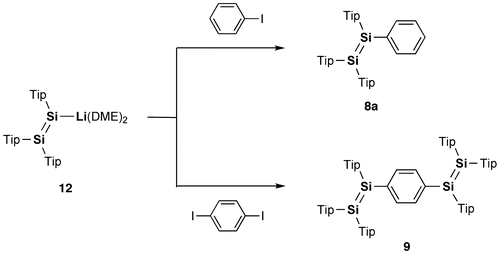
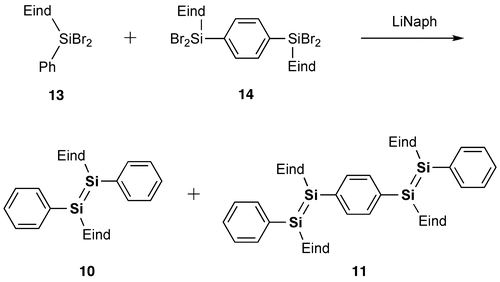
In 2007, model systems of the Si–OPVs (8a and 9–11) were synthesized by the groups of Scheschkewitz and Tamao employing the bulky Tip (2,4,6-triisopropylphenyl) and Eind (R1 = R2 = R3 = R4 = Et) groups [Citation77,78]. As shown in Scheme , the phenyl-substituted disilene, (Tip)2Si=Si(Tip)Ph (8a), and the para-phenylene-bridged tetrasiladiene 9 can be obtained as yellow and red crystals in 58% and 72% yields by the reaction of the isolatable disilenyllithium, (Tip)2Si=Si(Tip)Li(DME)2 (DME = 1,2-dimethoxyethane) (12) [Citation79], which is a silicon analog of vinyllithium, with iodobenzene and 1,4-diiodobenzene. In contrast, compounds 10 and 11 have been prepared by the reductive co-condensation of the two kinds of dibromosilanes, (Eind)PhSiBr2 (13) and [(Eind)SiBr2]2(1,4-C6H4) (14), with a 5:1 molar ratio using lithium naphthalenide (LiNaph) as a homogeneous reducing agent (Scheme ), in which 13 and 14 serve as the end-capping unit and central building unit for the Si–OPVs, respectively. The resulting monomer 10 and dimer 11 can be separated by silica gel column chromatography in a glove box using hexane and toluene as the eluents, leading to the isolation of yellow-orange crystals of 10 in 35% yield and red crystals of 11 in 15% yield. However, the higher oligomers, such as the trimer and tetramer, could not be obtained in a pure form mainly due to their poor solubility in common organic solvents.
Scheme 1. Synthesis of compounds 8a and 9.
Scheme 2. Synthesis of compounds 10 and 11.
In addition to the initial achievements, Scheschkewitz et al. prepared a series of para-functionalized-phenyl-substituted disilenes, (Tip)2Si=Si(Tip)(4–X–Ph) (X = F (8b), Cl (8c), Br (8d), I (8e), SiMe3 (8f)) [Citation80], which are shown in Figure . The laterally functionalized disilenes 8b–e can be synthesized by a similar reaction between the disilenyllithium 12 and the para-functionalized-phenyl iodides (4–X–PhI). In addition, the para-trimethylsilylphenyldisilene 8f has been obtained as a major product by the reaction of the para-bromophenyldisilene 8d with 2 equiv of tert-butyllithium ( t BuLi) followed by the addition of trimethylchlorosilane (Me3SiCl). The disilenes 8a–d exhibit a good correlation of the ultraviolet–visible (UV–vis) absorptions with the electronic Hammett parameters. As shown in Figure , the meta-phenylene-bridged tetrasiladiene (15), which is a regioisomer of 9, has also been isolated as orange crystals in 85% yield by the reaction of 12 with 1,3-diiodobenzene [Citation80].
As shown in Scheme , we have recently obtained the new Si–OPVs (16–19) having the modified Rind group, (HexO)MEind group, with a hexyloxy chain at the para position of the MEind (R1 = R2 = Me, R3 = R4 = Et) group for improving their solubility [Citation81]. A similar one-pot reductive co-condensation reaction of [(HexO)MEind]-PhSiBr2 (20) and [{(HexO)MEind}SiBr2]2(1,4-C6H4) (21) in a 2:1 molar ratio has led to the successful isolation of the yellow crystals of the monomer 16 in 10% yield, deep red crystals of the dimer 17 in 39%, a purple powder of the trimer 18 in 7% yield, and a deep purple powder of the tetramer 19 in 5% yield, which can be separated by silica gel column chromatography in a glove box using toluene and tetrahydrofuran (THF) as the eluents.
Scheme 3. Synthesis of compounds 16–19.
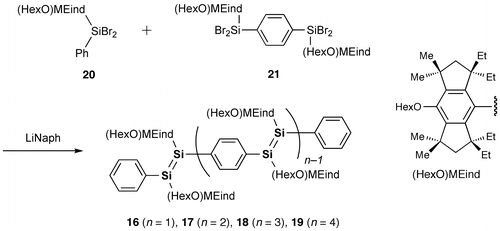
Figure shows the molecular structures of 16 and 17 based on the single-crystal X-ray diffraction analysis. The bulky (HexO)MEind groups effectively encapsulate the reactive Si=Si units and produce the highly coplanar Si–OPVs π-frameworks. The selected structural parameters of 16 and 17 are summarized in Table , together with those of 8a–d, 9–11, and 15 for comparison. The disilene cores of 16 and 17 display an almost planar geometry. The trans-bent angles (θ) between the Si–Si vector and the C–Si–C plane are estimated to be 1.65(8)° for 16 and 0.62(12) and 3.38(13)° for 17, which are comparable to those of 10 (θ = 2.72(14)°) and 11 (θ = 0.7(3) and 2.7(3)°) and much smaller than those of 8a (θ = 22.8 and 22.0°), 9 (θ = 16.45(10) and 19.31(10)°), and 15 (θ = 20.3 and 26.7°). These X-ray data show the excellent structural controllability of the Rind groups, where the proximate ethyl side chains on the s-hydrindacene skeletons can interlock with one another above and below the Si=Si moieties to enforce the planar structure.
Table 1. X-ray structural parameters of disilenes.
The photophysical data of the Si–OPVs (16–19) are summarized in Table . The absorption color gradually changes from yellow for 16 to blue for 19, as shown in Figure (a). The UV–vis spectra in THF exhibit absorption maxima (λ max(abs)) at 465 nm for 16, 546 nm for 17, 581 nm for 18, and 610 nm for 19, some of which are comparable to those of the Eind-based Si–OPVs (461 nm for 10 and 543 nm for 11 in hexane). The high molar extinction coefficients of 16–19 (ε = 2.8–7.1 × 104 cm–1 M–1) are assignable to the allowed HOMO → LUMO (π → π*) transitions, which are supported by the theoretical calculations using time-dependent density functional theory (TD-DFT) [Citation82]. It should be noted that the λ max(abs) values of 10 (461 nm) and 16 (465 nm) are more than 40 nm red-shifted from that of tetramesityldisilene 1 (420 nm) [Citation6], indicating the π-conjugation over the 1,2-diphenyldisilene (disilastilbene) skeleton. In addition, the considerable bathochromic shifts with the increasing Si=Si units are most likely interpreted in terms of the extended π-conjugation over the entire Si–OPVs skeleton, thus providing clear evidence for the efficient π-conjugation between the benzene rings and Si=Si double bonds. The λ max(abs) values provide a good fit to Meier’s equation [Citation83], which enables the estimation of an effective conjugation length (ECL) of nine repeat units (n ECL = 9) and the absorption maximum of 635 nm for the infinite chain (λ ∞(abs) = 635 nm). These estimated values (n ECL = 9 and λ ∞(abs) = 635 nm) for the Si–OPVs are lower and longer than those of the carbon counterpart, OPVs (n ECL = 11 and λ ∞(abs) = 481 nm), respectively [Citation84]. Thus, the inclusion of the Si=Si double bonds into the carbon π-conjugated systems may cause a significant change in the electronic and absorption properties.
Table 2. Photophysical data for disilenes.
While the monomer 16 does not show any emission like 10, the dimer 17, trimer 18, and tetramer 19 exhibit an intense fluorescence in THF at room temperature (Figure (b)). The emission maxima (λ max(ex)) are observed at 613 nm for 17, 643 nm for 18, and 668 nm for 19, one of which is similar to that of 11 (612 nm). The quantum yields (ΦF) increased from 0.11 to 0.48 with the increasing number of repeated units. In contrast, the Stokes shifts (Δν Stokes) decreased from 2000 cm–1 for 17 to 1420 cm–1 for 19, which are lower than those of the flexible carbon-based OPVs (3199–3029 cm–1) [Citation83] and higher than those of the rigid carbon-bridged OPVs (772–583 cm–1) [Citation85], suggesting the moderate rigidity of the Si–OPVs frameworks supported by the perpendicularly-fixed (HexO)MEind groups.
These studies reveal the possibility of constructing the π-conjugated disilene systems alternating the carbon-based 2pπ and silicon-based 3pπ-electrons, where the Si=Si double bonds would be promising building blocks. We hope that the present studies would provide a further challenge for the pure and applied chemistry of disilene copolymers containing various carbon π-electron systems. Actually, we very recently succeeded in obtaining some disilene–thiophene π-systems as model compounds for the disilene–thiophene copolymers [Citation86], which are described in the latter part of this review.
3. Air-stable emissive disilenes with naphthyl groups; potential applicability in electroluminescence devices
After the initial findings of the model compounds of the Si–OPVs, the π-electron systems containing a Si=Si chromophore attracted much attention from the viewpoint of their potentially useful properties and unique functions. For example, as shown in Figure , a new type of disilene bearing metallocenyl groups has been synthesized by Sasamori, Tokitoh, and co-workers [Citation87,88]. The 1,2-bis(metallocenyl)disilenes (22) exhibit a multistep redox process in the cyclic voltammograms, thus indicating the potential application of the disilene π-system as electrochemical materials. Tokitoh et al. have also shown that the 1,2-dialkynyldisilenes (23) can be obtained as a stable crystalline compound by taking advantage of the steric protection using the bulky Bbt groups (Bbt = 2,6-bis[bis(trimethylsilyl)methyl]-4-[tris(trimethylsilyl)methyl]phenyl) (Figure ) [Citation89]. The UV–vis spectrum of 23b in hexane exhibits an absorption maximum (λ max(abs)) at 469 nm, which is 32 nm red-shifted compared with that of 23a (437 nm), indicating the effective π-conjugation between the central Si=Si unit and two terminal phenyl groups via the C≡C triple bonds.
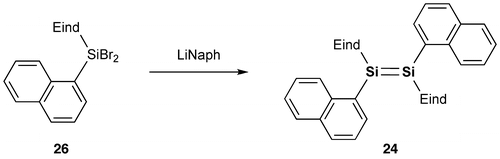
We have set out to investigate the other types of disilene π-systems with two polycyclic aromatic groups on the Si=Si core incorporating the two bulky Rind groups. As the first target molecules, we designed two kinds of 1,2-dinaphthyldisilene regioisomers, (E)-1,2-di(1-naphthyl)disilene (24) and (E)-1,2-di(2-naphthyl)-disilene (25), as shown in Schemes and [Citation90,91]. The disilenes 24 and 25 were obtained as red crystals in 59% and 57% yields, respectively, by the reductive coupling of (Eind)(1-naphthyl)SiBr2 (26) and (Eind)(2-naphthyl)SiBr2 (27). The disilenes 24 and 25 are extraordinarily air-stable in the solid state of more than several years with no detectable change as confirmed by the proton nuclear magnetic resonance (1H NMR) spectra, which indicates the excellent protection abilities of the Eind group. The disilenes 24 and 25 decompose in a dilute solution (ca. 10–5 mol L–1) upon exposure to air with a half-lifetime of 2–4 h, which is rather longer than that previously reported for (Tip)2Si=Si(Tip)2 (17 min) [Citation92], as monitored by the UV–vis absorption spectroscopy.
Scheme 4. Synthesis of compound 24.
Scheme 5. Synthesis of compound 25.
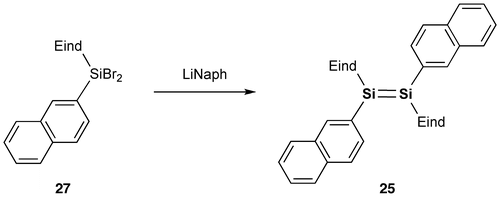
Figure shows the molecular structures of 24 and 25 confirmed by X-ray crystallography. Both molecules have an inversion center at the midpoint of the Si=Si double bond with an E-configuration. In the crystal of 24, the hydrogen atoms at the peri-position on the 1-naphthyl groups participate in the CH–π interaction with the benzene ring of the perpendicularly-oriented Eind groups, producing the highly coplanar di(1-naphthyl)disilene skeleton, favorable for the efficient π-conjugation involving the Si=Si unit. In the crystal of 25, the Si atoms and the 2-naphthyl groups are disordered over the two positions, which corresponds to a mixture of two rotational isomers, s-cis, s-cis (25a) and s-trans, s-trans (25b), with the occupancy factors of ca. 0.40/0.60. Each rotational isomer has an essentially coplanar di(2-naphthyl)disilene framework. The selected structural parameters of 24, 25a, and 25b are summarized in Table . The trans-bent angles (θ) are estimated to be 4.93(12)° for 24, 2.25(14)° for 25a, and 9.57(11)° for 25b. The Si=Si bond distance is 2.1688(7) Å for 24, 2.1623(18) Å for 25a, and 2.1667(12) Å for 25b, which are comparable to those of 10 (2.1593(16) Å), 11 (2.156(2) Å), 16 (2.1626(8) Å), and 17 (2.1642(8) Å) and in the standard range of those reported for disilenes [Citation15].
As shown in Figure , the disilenes 24 and 25 exhibit a strong absorption and emission at room temperature both in solution and in the solid state mainly due to the highly coplanar 1,2-dinaphthyldisilene π-frameworks. The photophysical data of 24 and 25 are summarized in Table . In the UV–vis spectra in THF, the absorption maxima (λ max(abs)) appear at 521 nm for 24 and 504 nm for 25, which are red-shifted from that of 10 (461 nm), indicative of the effective π-conjugation over the dinaphthyldisilene skeletons. The emission maxima (λ max(ex)) are found at 614 nm for 24 and at 586 nm for 25. The Stokes shift (Δν Stokes) values are estimated to be 2910 cm–1 for 24 and 2780 cm–1 for 25, which are higher than that of 11 (2080 cm–1) but much lower than those of the tetramesityldisilene 1 (4000 cm–1) [Citation8] and tetraneopentyldisilene (7300 cm–1) [Citation93], thus indicating the structural rigidity of the dinaphthyldisilene skeletons. Each of the disilenes 24 and 25 shows a weaker emission in solution relative to that in the solid state mainly ascribed to the free-rotation of the naphthyl groups around the Si–C bonds in solution.
Figure shows the frontier molecular orbitals of 24, 25a, and 25b afforded by the DFT computations at the B3LYP/6-31G** level [Citation82]. While the HOMOs mainly consist of the π(Si–Si) orbital, the LUMOs involve the appreciable contribution of the π*(Si–Si)–π*(naphthalene) conjugation. The HOMO and LUMO energy levels of 24 (–4.209 and –1.572 eV) are somewhat higher and lower than those of 25a (–4.249 and –1.554 eV) and 25b (–4.304 and –1.478 eV). Accordingly, the HOMO–LUMO energy gap for 24 (2.637 eV) is slightly smaller than those of 25a (2.695 eV) and 25b (2.826 eV). These calculations are in good qualitative agreement with the experimental data, a slightly longer absorption maximum (λ max(abs)) for 24 (521 nm) relative to 25 (504 nm), which are based on the fact that the larger HOMO and LUMO lobes are at the 1-position than at the 2-position of the naphthalene ring.
The resulting 1,2-dinaphthyldisilenes 24 and 25 also exhibit a high thermal stability with a decomposition point of 245–248 °C for 24 and 282–285 °C for 25 under an argon atmosphere. The exceptional air and thermal stabilities would open up new opportunities for application in a range of organic electronic devices, since the Si=Si unit possesses a narrower HOMO–LUMO energy gap than the C=C unit [Citation12–22].
Actually, as shown in Figure , we have found that the disilene 25 can emit light in an organic light-emitting diode (OLED) [Citation94]. To the best of our knowledge, this is the first demonstration of electroluminescence (EL) from a disilene compound in OLEDs. A typical multi-thin-layer pattern has been used for the OLEDs, in which three organic molecular layers, i.e. poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT-PSS) [Citation95] as the hole-injecting layer, the disilene 25 with a host molecule of poly(9,9-dioctylfluorene) (PFO) in the weight ratio of 1:1 as the light-emitting and hole transporting layer, and 2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole) (TPBi) as the hole-blocking and electron-transporting layer, are sandwiched between a transparent ITO (indium tin oxide) anode and metallic LiF/Al cathode on a glass substrate. This device emits a bright orange light from the disilene 25 at the applied voltage of 5 V.
Although the total performance is rather low and still far from a practical application (the maximum brightness of L max = 119 cd m–2 at the driving voltage of 8.5 V, the luminance efficiency η 100 and the current efficiency L/J 100 at a luminance of 100 cd m–2 of 0.013 lm W–1 and 0.035 cd A–1, the external quantum efficiency (EQE) of 0.014%, and the half-life of 76.5 min of the orange light emission at a 25 mA cm–2 current density with the initial light output of 14.6 cd m–2), this study provides a new avenue for investigations to explore the applied chemistry of unsaturated compounds of the heavier main group elements. The air-stable, emissive 1,2-dinaphthyldisilenes 24 and 25 are now commercially available [Citation96].
4. Disilene π-system with pyrenyl groups; evidence for intramolecular charge-transfer emission
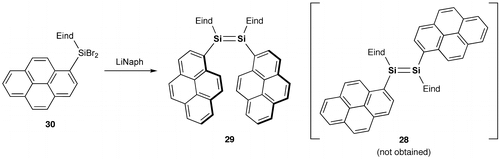
Following the successful achievements of the (E)-1,2-dinaphthyldisilenes 24 and 25, we focused on the development of further π-extended disilene molecules. For example, (E)-1,2-di(1-pyrenyl)disilene (28) can be considered as a fascinating π-system with two π-extended pyrenyl groups consisting of four fused benzene rings. As shown in Scheme , we have examined the reductive treatment of the Eind- and 1-pyrenyl-substituted dibromosilane, (Eind)(1-pyrenyl)SiBr2 (30), with a sufficient amount of lithium naphthalenide (LiNaph) in THF. However, unexpectedly, we obtained the Z isomer, (Z)-1,2-di(1-pyrenyl)disilene (29), as purple crystals in 43% yield [Citation97]. The disilene 29 is not very stable in the air and even in the solid state, which is in sharp contrast to the fact that the red crystals of (E)-1,2-dinaphthyldisilenes 24 and 25 can survive in the air for more than several years. Although the formation mechanism of 29 is not yet clear, this is the first selective formation of the Z isomer of the acyclic disilene by the reductive coupling of monosilane precursors. The attractive π–π interaction between the pyrenyl groups may play a role in determining the stereochemistry during in the Si–Si bond-forming processes.
Scheme 6. Synthesis of compound 29.
As shown in Figure , the molecular structure of 29 has been unambiguously characterized by X-ray crystallography to adopt a Z-configuration. The two Eind groups and the two 1-pyrenyl groups mesh in a gear-like fashion centering around the disilene core with the Si=Si bond length of 2.1718(6) Å and the trans-bent angles (θ) of 8.22(8) and 2.96(8)° (Table ). The two pyrene rings are twisted about the Si=Si unit with the Si–Si–C–C torsion angles of 52.66(13)° and 48.73(14)°, which intramolecularly interact with each other to have a π–π stacking with a distance between the centers of the two pyrene rings of 3.635 Å.
The photophysical data of 29 are summarized in Table . In the UV–vis spectrum of 29 in THF, two broad absorption bands are observed with the absorption maxima (λ max(abs)) at 519 and 575 nm, together with a strong absorption around 350 nm due to the pyrene ring itself. The λ max(abs) values are found to be not sensitive to the solvent polarity. In contrast, as shown in Figure , the emission maximum (λ max(ex)) of 29 is dependent on the solvent polarity and red-shifted from 661 nm in hexane to 676 nm in THF and to 694 nm in acetone (dielectric constant: hexane 1.88, THF 7.58, acetone 20.56) [Citation98]. These data indicate the intramolecular charge-transfer (ICT) emission at room temperature, which may originate from the arrangement of the two 1-pyrenyl groups twisted from the Si=Si double bond.
The photophysical properties of 29 are theoretically supported by DFT studies including excited-state calculations [Citation97]. Figure shows the three pertinent molecular orbitals of 29. Although the HOMO is represented by the π(Si–Si) orbital, the LUMO involves a substantial π*(Si–Si)–π*(pyrene) conjugation. The LUMO+1 corresponds to the π*(pyrene) orbital. The natural population analysis (NPA) charge distribution [Citation99] exhibits a more charge-separated character for the (Z)-1,2-di(1-pyrenyl)disilene skeleton in the excited state compared to the ground state, which is consistent with the π(Si–Si) → π*(pyrene) ICT excited state based on the electron transfer from the disilene π-donor toward the pyrene π-acceptor.
As shown in Figure , in regard to the ICT behavior of the disilenes, a unique ICT absorption by 9-anthryl-substituted trialkyldisilene (31) has been found by Iwamoto, Kira, and co-workers, in which the anthracene ring is orthogonal to the Si=Si unit [Citation100]. The disilene 31 can be obtained as blue-purple crystals by the reaction of the corresponding trialkyldisilenide with 9-bromoanthracene. Iwamoto et al. also very recently reported a series of heteroaryl-substituted disilenes (32a–c and 33a–c), where heteroaryl groups serve as electron acceptors for Si=Si double bonds in ICT transitions [Citation101]. These disilenes can also be prepared by the treatment of the corresponding dialkylaryldisilenide with heteroaryl halides and dihalides. In the crystal structures of 32a, 32b, 33b, and 33c, the heteroaryl groups are almost perpendicular to the Si=Si double bond mainly due to the steric repulsion between the bulky cyclic alkyl substituents, mesityl group, and the heteroaryl groups.
Very recently, Scheschkewitz et al. also reported the photophysical properties of some related (oligo)aromatic species having one or two Si=Si double bonds (34a–d and 35a–d), which have been prepared by the reaction of the triaryldisilenide 12 with aryl halides and dihalides [Citation102]. The tetrasiladienes 35b–d exhibit a fluorescence at room temperature. DFT calculations suggest the partial CT character of the excited state. It is important to note that the 9,10-anthracene-bridged tetrasiladiene 35d is the first example of a near-infrared emissive disilene compound ((λ max(ex)) = 816 nm).
In order to obtain the initial target molecule, (E)-1,2-di(1-pyrenyl)disilene 28, we examined the photoreaction of 29 [Citation103]. As shown in Figure , after the photolysis (λ = 530 nm) of 29 in THF at room temperature, the solution color has changed from purple to blue. In the UV–vis absorption spectrum, a relatively new sharp peak appeared around at 590 nm, which is 15 nm red-shifted from that of 29 (575 nm), with a higher molar extinction coefficient (ε = 1.3 × 104 cm–1 M–1) relative to that of 29 (ε = 7.2 × 103 cm–1 M–1), thus indicating the formation of a more π-extended system. The absorption spectral change with an isosbestic point at 540 nm suggests a clean photoisomerization process from (Z)-29 to (E)-28. Unfortunately, the blue 28 has not yet been isolated in a pure form due to its labile nature, but the DFT calculations indicate the highly coplanar (E)-1,2-di(1-pyrenyl)disilene skeleton induced by the orthogonal arrangement of the Eind groups [Citation90]. In order to confirm the formation of 28, we are now investigating on alternative synthetic route using the Eind-substituted (E)-1,2-dibromosilane, (Eind)BrSi=SiBr(Eind), as a precursor [Citation54].
5. Disilene–thiophene π-systems; future organosilicon chemistry for developing advanced materials
As already described, the construction of novel π-conjugated disilene systems consisting of the carbon-based 2pπ and silicon-based 3pπ-electrons would provide exciting opportunities to explore new organosilicon chemistry for organic–inorganic hybrid materials, since the Si=Si units have a narrower HOMO–LUMO gap compared to the C=C counterparts [Citation12–22]. Very recently, we studied a new type of π-conjugation between the Si=Si double bond and aromatic heterocycles. As shown in Figure , we previously examined the synthesis and electronic properties of a series of oligothiophenes (36a–c) with the bulky EMind (R1 = R2 = Et, R3 = R4 = Me) groups [Citation50]. The orthogonal orientation of the EMind groups has proven to be useful to produce a coplanar arrangement of oligothiophene backbones. Based on these fundamental investigations, we have set out to examine the possibility to construct disilene–thiophene π-conjugated systems by the introduction of the Rind groups.
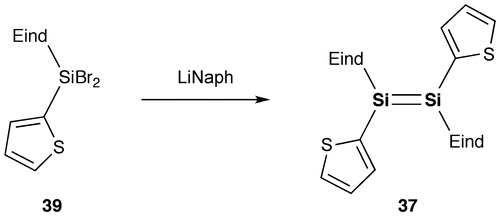
As shown in Schemes and , we have designed and synthesized two new disilenes, 1,2-bis(thiophen-2-yl)disilene (37) and 1,2-bis(2,2′-bithiophen-5-yl)disilene (38), as model compounds of the disilene–thiophene copolymers [Citation86]. The disilenes 37 and 38 can be isolated as orange and purple crystals, respectively, by the reduction of the corresponding thienyl- and bithienyl-substituted dibromosilanes (39 and 40).
Scheme 7. Synthesis of compound 37.
Scheme 8. Synthesis of compound 38.
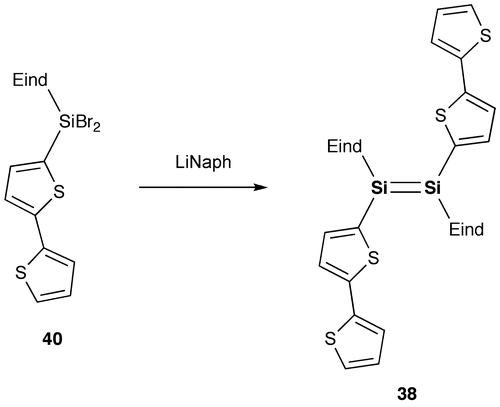
Figure shows the X-ray molecular structures of 37 and 38. Each molecule has an inversion center at the middle of the Si=Si double bond with an E configuration. As shown in Figure , there were found several rotational isomers of 37 and 38 in the crystals. For 37, the thiophene units are disordered over the two positions with the occupancy factors of ca. 0.90/0.10, which is consistent with the existence of a mixture of three rotational isomers, s-cis, s-cis (37a), s-cis, s-trans (37b), and s-trans, s-trans (37c), with the occupancy factors of ca. 0.81/0.18/0.01. For 38, while the inner thiophene rings are ordered in the crystal with an s-trans, s-trans conformation, the outer thiophene rings are disordered over the two orientations with the occupancy factors of ca. 0.76/0.24. Thus, the three rotational isomers, anti-(s-trans, s-trans)-anti (38a), syn-(s-trans, s-trans)-anti (38b), and syn-(s-trans, s-trans)-syn (38c), exist in the crystal with the ratio of ca. 0.58/0.36/0.06. It is worth mentioning that all the NMR data for 37 and 38 indicate the free-rotation around the Si–C bonds and the exocyclic C–C bonds in solution at room temperature on the NMR time scale.
The major structural parameters of 37 and 38 are summarized in Table . The disilene 37 has a trans-bent structure with the trans-bent angles (θ) of 19.12(12)° for 37a and 37b and 13.5(6)° for 37b and 37c. The Si atoms assume a somewhat pyramidal geometry with the sum of the bond angles around the Si atom (ΣSi) of ca. 355.5–357.5°. The disilene core of 38 exhibits a more coplanar arrangement relative to 37 with the trans-bent angle (θ) of 5.44(10)°. The Si atoms have an almost planar geometry; the sum of the bond angles around the Si atom (ΣSi) is 359.7°. The bithiophene moieties in 38 are slightly twisted with the dihedral angles between the inner and outer thiophene rings of 21.0(3)° for 38a and 38b and 28.6(10)° for 38b and 38c. The Si=Si double bond lengths of 2.1712(11) Å for 37 and 2.1584(9) Å for 38 are in the range of those for typical disilenes [Citation16].
The photophysical data of 37 and 38 are summarized in Table . As shown in Figure , the absorption color clearly changes from the yellow of 37 to the red-purple of 38. In the UV–vis spectrum of 37 in THF, the absorption maximum (λ max(abs)) appears at 459 nm, comparable to that of 10 (461 nm), which indicates the efficient π-conjugation between the Si=Si double bond and the two thiophene moieties. For 38, the absorption peak is observed at 530 nm, which is 71 nm red-shifted from that of 37 (459 nm) and similar to that of 11 (543 nm). This large bathochromic shift is most likely interpreted in terms of the extension of the π-conjugation with the increasing number of thiophene units. In addition, the λ max(abs) value of 38 is 116 nm longer than that of the EMind-substituted quaterthiophene 36a (414 nm in CH2Cl2) [Citation50]. Thus, the insertion of the Si=Si double bond into the quaterthiophene skeleton causes a considerable narrowing of the HOMO–LUMO gap.
As shown in Figure , the π-extended disilene 38 displays a weak but distinct emission both in solution and in the solid state, while the disilene 37 does not show any emission at room temperature. The emission maximum (λ max(ex)) of 38 appears at 688 nm in THF with the quantum yield (ΦF) of 0.01. The Stokes shift (Δν Stokes) of 38 (4330 cm–1) is more than twice as high as that of 11 (2080 cm–1). These emission properties in solution may be explained by the structural flexibility of the 1,2-bis(bithienyl)disilene skeleton compared to the 1,4-bis(disilenyl)benzene skeleton. The disilene 38 exhibits a relatively strong emission at 691 nm in the solid state with the quantum yield (ΦF) of 0.11, which is about 10 times stronger than that in solution. The weaker emission in solution is attributable to the free-rotation of the bithienyl groups around the Si–C bonds and the exocyclic C–C bonds as observed in the 1H NMR spectrum.
In order to further clarify the structural and electronic properties of the disilenes 37 and 38, we performed DFT calculations at the B3LYP-D3/6-31G(d,p) level [Citation82]. The DFT studies indicated a rather flexible geometry around the disilene core in 37. Thus, the optimized structures of 37a (C i symmetry), 37b (C s symmetry), and 37c (C 1 symmetry) exhibit an entirely coplanar 1,2-dithienyldisilene skeleton (θ = 0.0–0.1°), which are different from the X-ray structures (θ = 19.12(12)° and 13.5(6)°) found in the crystal. These rotational isomers have almost the same energies with the relative energies of 0.00 (37a), 1.12 (37b), and 2.48 (37c) kcal mol–1. The optimized structure of 38a (C 1 symmetry) shows a slightly more trans-bent configuration (θ = 10.8°) relative to that found in the crystal (θ = 5.44(10)°). The dihedral angles between the inner and outer thiophene rings are estimated to be 17.8°, which are somewhat smaller than those of the experimental X-ray values (21.0(3)° and 28.6(10)°). The differences between the X-ray and DFT structures are mainly due to the flexibility of the main chains consisting of the Si=Si unit and the thiophene rings, which would be easily affected by the crystal packing forces.
The molecular orbitals of 37a and 38a are depicted in Figure in which the HOMOs mainly consist of the π(Si–Si) orbital along with a small contribution of the π(thiophene) and π(bithiophene) orbitals, while the LUMOs delocalize over the entire 1,2-dithienyldisilene and 1,2-bis(bithienyl)disilene frameworks. The HOMO level of 38a (–4.211 eV) is comparable to that of 37a (–4.222 eV), while the LUMO level of 38a (–1.681 eV) is much lower than that of 37a (–1.350 eV) due to the extended π*(Si–Si)–π*(bithiophene) conjugation. The HOMO–LUMO energy gap for 38a (2.530 eV) is smaller than that for 37a (2.871 eV), which are in good agreement with the UV–vis absorption data. The TD-DFT calculations almost reproduce the absorption spectra with the absorption wavelengths at 466 nm for 37a and 558 nm for 38a, which are comparable to those observed for 37 (459 nm) and 38 (530 nm), assignable to the HOMO → LUMO (π–π*) transitions.
In this study, we have demonstrated for the first time that the Si=Si double bond can fully conjugate with aromatic heterocycles using the appropriate steric effects due to the bulky Eind groups. The experimental and theoretical studies provide clear evidence for the effective π-conjugation between the Si=Si chromophore and thiophene units, originating from the essentially coplanar (bi)thiophene–Si=Si–(bi)thiophene skeletons. Further studies to develop promising disilene–thiophene copolymers for future application in a range of organic electronic devices are now in progress.
6. Concluding remarks
In this review, we have addressed the recent progress related to the chemistry of π-electron systems containing Si=Si double bonds. Especially, the unique steric effects of the fused-ring bulky Rind groups enabled us to isolate a series of structurally well-defined discrete π-conjugated disilene molecules, which exhibit an efficient delocalization of 2pπ- and 3pπ-electrons over the skeletons. In other words, the properly designed bulky protecting groups play pivotal roles in the structural determination and in the control of the electronic properties in the π-electron systems consisting of the carbon π-systems and Si=Si double bond(s). We hope that the present studies will provide a future challenge of pure and applied organoelement chemistry toward advanced materials science and technology.
As shown in Figure , after the achievement of the first persila[n]annulene compound, i.e. tetrasilacyclobutadiene (n = 4) (7) [Citation34], extensive efforts have been devoted to the design and synthesis of hexasilabenzene (n = 6) (41) with a cyclic system consisting of the six formally sp 2-hybridized silicon atoms. The hexasilabenzene 41 can be regarded as the smallest fragment for silicene [Citation1–3], the silicon analog of graphene with a two-dimensional honeycomb structure. Although some related hexasilabenzene isomers (5, 42, and 43) were synthesized as a stable crystalline compound [Citation32,104,105], the long-considered hexasilabenzene 41 has not been isolated and remains elusive. In particular, novel electronic and optical properties as well as exotic silicon aromaticity arising from the six 3pπ-electrons in 41 have attracted much attention from both experimentalists and theoreticians [Citation106].
Also, as shown in Figure , a silicon analog of the linear polyacetylene, i.e. polysilyne (44), with repeating disilyne (RSi≡SiR) units, has yet to be achieved and remains as a dream compound for chemists, which would provide new opportunities for application in a range of electronic devices, because the Si=Si units have a smaller HOMO–LUMO energy gap compared to the C=C units. Some silicon analogs of 1,3-butadiene, i.e. the tetrasila-1,3-butadienes (45–47), have been obtained employing the bulky substituents [Citation107–109]. We hope that further progress will be made in the construction and functions of the cyclic and linear π-electron systems involving the Si=Si double bonds.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This research is partially supported by the Precursory Research for Embryonic Science and Technology (PRESTO) from Japan Science and Technology Agency (JST); Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan for Scientific Research on Innovative Areas, ‘Stimuli-responsive Chemical Species for the Creation of Functional Molecules’ [#2408] [grant number 24109003]; Scientific Research (B) [grant numbers 24350031, 15H03788]; MEXT-Supported Program for the Strategic Research Foundation at Private Universities 2014–2018 subsidy from MEXT and Kindai University. N.H. acknowledges the support by a Grant-in-Aid for JSPS Fellows from the Japan Society for the Promotion of Science (JSPS) [grant number JP16J01036].
Acknowledgments
The calculations and experiments described in this article have been performed with many collaborators, whose names appear in the references. The authors wish to express their sincere thanks to all of them for their kind collaboration, in particular, Prof Kohei Tamao, for his continuous discussions.
References
- Aufray B , Kara A , Vizzini S , et al . Graphene-like silicon nanoribbons on Ag(110): a possible formation of silicene. Appl Phys Lett. 2010;96:183102-(1–3).
- Lalmi B , Oughaddou H , Enriquez H , et al . Epitaxial growth of a silicene sheet. Appl Phys Lett. 2010;97:223109-(1–2).
- Spencer MJS , Morishita T . Silicene: structure, properties and applications. Cham: Springer; 2016.10.1007/978-3-319-28344-9
- Marutheeswaran S , Pancharatna PD , Balakrishnarajan MM . Preference for a propellane motif in pure silicon nanosheets. Phys Chem Chem Phys. 2014;16(23):11186–11190.10.1039/C4CP01286K
- Cordero B , Gómez V , Platero-Prats AE , et al . Covalent radii revisited. Dalton Trans. 2008;40:2832–2838.10.1039/b801115j
- West R , Fink MJ , Michl J . Tetramesityldisilene, a stable compound containing a silicon–silicon double bond. Science. 1981;214(4527):1343–1344.10.1126/science.214.4527.1343
- Fink MJ , Michalczyk MJ , Haller KJ , et al . The X-ray crystal structure of tetramesityldisilene. J Chem Soc, Chem Commun. 1983;18:1010–1011.10.1039/c39830001010
- Fink MJ , Michalczyk MJ , Haller KJ , et al . X-ray crystal structures for two disilenes. Organometallics. 1984;3(5):793–800.10.1021/om00083a025
- West R . The disilenes: chemistry of silicon-silicon double bonds. Pure Appl Chem. 1984;56(1):163–173.
- Shepherd BD , Campana CF , West R . Crystallographic analysis of thermochromic, unsolvated tetramesityldisilene at 173 K and 295 K. Heteroat Chem. 1990;1(1):1–7.10.1002/(ISSN)1098-1071
- Wind M , Powell DR , West R . Structure of the THF solvate of tetramesityldisilene. Organometallics. 1996;15(26):5772–5773.10.1021/om960753n
- Okazaki R , West R . Chemistry of stable disilenes. Adv Organomet Chem. 1996;39:231–273.
- Power PP . π-Bonding and the lone pair effect in multiple bonds between heavier main group elements. Chem Rev. 1999;99(12):3463–3504.10.1021/cr9408989
- Lee VY , Sekiguchi A . Heteronuclear heavy alkenes E=E′ (E, E′ = group 14 elements): germasilenes, silastannenes, germastannenes … next stop? Organometallics. 2004;23(12):2822–2834.10.1021/om040027 h
- Iwamoto T . Novel stable silicon-based π-electron systems: synthesis, unique structure, and properties of cyclic, spiroconjugated, and cumulative silicon–silicon doubly-bonded compounds. Bull Chem Soc Jpn. 2005;78(3):393–404.10.1246/bcsj.78.393
- Kira M , Iwamoto T . Progress in the chemistry of stable disilenes. Adv Organomet Chem. 2006;54:73–148.10.1016/S0065-3055(05)54003-6
- Wang Y , Robinson GH . Unique homonuclear multiple bonding in main group compounds. Chem Commun. 2009;32:5201–5213.10.1039/b908048a
- Fischer RC , Power PP . π-Bonding and the lone pair effect in multiple bonds involving heavier main group elements: developments in the new millennium. Chem Rev. 2010;110(7):3877–3923.10.1021/cr100133q
- Lee VY , Sekiguchi A . Organometallic compounds of low-coordinate Si, Ge, Sn and Pb. West Sussex: Wiley; 2010.10.1002/9780470669266
- Scheschkewitz D . The versatile chemistry of disilenides: disila analogues of vinyl anions as synthons in low-valent silicon chemistry. Chem Lett. 2011;40(1):2–11.10.1246/cl.2011.2
- Asay M , Sekiguchi A . Recent developments in the reactivity of stable disilynes. Bull Chem Soc Jpn. 2012;85(12):1245–1261.10.1246/bcsj.20120212
- Sasamori T , Tokitoh N . A new family of multiple-bond compounds between heavier group 14 elements. Bull Chem Soc Jpn. 2013;86(9):1005–1021.10.1246/bcsj.20130134
- Wakita K , Tokitoh N , Okazaki R , et al . Synthesis and properties of an overcrowded silabenzene stable at ambient temperature. Angew Chem Int Ed. 2000;39(3):364–636.
- Kinjo R , Ichinohe M , Sekiguchi A , et al . Reactivity of a disilyne RSi≡SiR (R = SiiPr[CH(SiMe3)2]2) toward π-bonds: stereospecific addition and a new route to an isolable 1,2-disilabenzene. J Am Chem Soc. 2007;129(25):7766–7767.10.1021/ja072759 h
- Han JS , Sasamori T , Mizuhata Y , et al . Reactivity of an aryl-substituted silicon–silicon triple bond: 1,2-disilabenzenes from the reactions of a 1,2-diaryldisilyne with alkynes. Dalton Trans. 2010;39(39):9238–9240.10.1039/c0dt00115e
- Ishida S , Iwamoto T , Kabuto C , et al . A stable silicon-based allene analogue with a formally sp-hybridized silicon atom. Nature. 2003;421(6924):725–727.10.1038/nature01380
- Tanaka H , Inoue S , Ichinohe M , et al . Synthesis and striking reactivity of an isolable tetrasilyl-substituted trisilaallene. Organometallics. 2011;30(13):3475–3478.10.1021/om200405e
- Sekiguchi A , Kinjo R , Ichinohe M . A stable compound containing a silicon–silicon triple bond. Science. 2004;305(5691):1755–1757.10.1126/science.1102209
- Wiberg N , Niedermayer W , Fischer G , et al . Synthesis, structure and dehalogenation of the disilene RClSi=SiClR [R = (tBu3Si)2MeSi]. Eur J Inorg Chem. 2002;5:1066–1070.10.1002/(ISSN)1099-0682
- Sasamori T , Hironaka K , Sugiyama Y , et al . Synthesis and reactions of a stable 1,2-diaryl-1,2-dibromodisilene: a precursor for substituted disilenes and a 1,2-diaryldisilyne. J Am Chem Soc. 2008;130(42):13856–13857.10.1021/ja8061002
- Ishida S , Sugawara R , Misawa Y , et al . Palladium and platinum η2-disilyne complexes bearing an isolable dialkyldisilyne as a ligand. Angew Chem Int Ed. 2013;52(49):12869–12873.10.1002/anie.201308517
- Abersfelder K , White AJP , Rzepa HS , et al . A tricyclic aromatic isomer of hexasilabenzene. Science. 2010;327(5965):564–566.10.1126/science.1181771
- Wang Y , Xie Y , Wei P , et al . A stable silicon(0) compound with a Si=Si double bond. Science. 2008;321(5892):1067–1071.
- Suzuki K , Matsuo T , Hashizume D , et al . A planar rhombic charge-separated tetrasilacyclobutadiene. Science. 2011;331(6022):1306–1309.10.1126/science.1199906
- Hissler M , Dyer PW , Réau R . Linear organic π-conjugated systems featuring the heavy Group 14 and 15 elements. Coord Chem Rev. 2003;244(1–2):1–44.10.1016/S0010-8545(03)00098-5
- Gates DP . Expanding the analogy between P=C and C=C bonds to polymer science. Top Curr Chem. 2005;250:107–126.10.1007/b98358
- Hissler M , Dyer PW , Reau R . The rise of organophosphorus derivatives in π-conjugated materials chemistry. Top Curr Chem. 2005;250:127–163.10.1007/b98358
- Baumgartner T , Réau R . Organophosphorus π-conjugated materials. Chem Rev. 2006;106(11):4681–4727.10.1021/cr040179 m
- Grimsdale AC , Müllen K . Oligomers and polymers based on bridged phenylenes as electronic materials. Macromol Rapid Commun. 2007;28(17):1676–1702.10.1002/(ISSN)1521-3927
- Scheschkewitz D . Anionic reagents with silicon-containing double bonds. Chem A Eur J. 2009;15(11):2476–2485.10.1002/chem.v15:11
- Matsuo T , Li B , Tamao K . π-Conjugated phosphasilenes stabilized by fused-ring bulky ‘Rind’ groups. C R Chim. 2010;13(8–9):1104–1110.10.1016/j.crci.2010.07.005
- Bates JI , Dugal-Tessier J , Gates DP . Phospha-organic chemistry: from molecules to polymers. Dalton Trans. 2010;39(13):3151–3159.10.1039/B918938F
- Matsuo T , Kobayashi M , Tamao K . π-Conjugated disilenes stabilized by fused-ring bulky ‘Rind’ groups. Dalton Trans. 2010;39(39):9203–9208.10.1039/c0dt00287a
- Simpson MC , Protasiewicz JD . Phosphorus as a carbon copy and as a photocopy: new conjugated materials featuring multiply bonded phosphorus. Pure Appl Chem. 2013;85(4):801–815.
- Präsang C , Scheschkewitz D . Reactivity in the periphery of functionalised multiple bonds of heavier group 14 elements. Chem Soc Rev. 2016;45(4):900–921.10.1039/C5CS00720H
- Matsuo T , Suzuki K , Fukawa T , et al . Synthesis and structures of a series of bulky ‘Rind-Br’ based on a rigid fused-ring s-hydrindacene skeleton. Bull Chem Soc Jpn. 2011;84(11):1178–1191.10.1246/bcsj.20110090
- Chang VSC , Kennedy JP . New telechelic polymers and sequential copolymers by polyfunctional initiator-transfer agents (inifers). Polym Bull. 1981;4(9):513–520.
- Li B , Matsuo T , Hashizume D , et al . π-Conjugated phosphasilenes stabilized by fused-ring bulky groups. J Am Chem Soc. 2009;131(37):13222–13223.10.1021/ja9051153
- Shoji Y , Matsuo T , Hashizume D , et al . A stable doubly hydrogen-bridged butterfly-shaped diborane(4) compound. J Am Chem Soc. 2010;132(24):8258–8260.10.1021/ja102913 g
- Otani T , Hachiya M , Hashizume D , et al . The synthesis of highly coplanar oligothiophenes induced by bulky Rind groups. Chemistry. 2010;6(2):350–354.
- Hachiya M , Ito M , Matsuo T , et al . Air- and moisture-stable p-monothiobenzoquinones incorporated in an octaalkyl-s-hydrindacene Skeleton. Org Lett. 2011;13(10):2666–2669.10.1021/ol200768b
- Li B , Matsuo T , Fukunaga T , et al . Neutral and cationic gold(I) complexes with π-conjugated phosphasilene ligands. Organometallics. 2011;30(13):3453–3456.10.1021/om2003442
- Shoji Y , Matsuo T , Hashizume D , et al . Boron–boron σ-bond formation by two-electron reduction of a H-bridged dimer of monoborane. J Am Chem Soc. 2011;133(29):11058–11061.10.1021/ja203333j
- Suzuki K , Matsuo T , Hashizume D , et al . Room-temperature dissociation of 1,2-dibromodisilenes to bromosilylenes. J Am Chem Soc. 2011;133(49):19710–19713.10.1021/ja209736d
- Li L , Fukawa T , Matsuo T , et al . A stable germanone as the first isolated heavy ketone with a terminal oxygen atom. Nat Chem. 2012;4(5):361–365.10.1038/nchem.1305
- Li B , Tsujimoto S , Li Y , et al . Synthesis and characterization of diphosphenes bearing fused-ring bulky rind groups. Heteroat Chem. 2014;25(6):612–618.10.1002/hc.2014.25.issue-6
- Shoji Y , Kaneda S , Fueno H , et al . An isolable diborane(4) compound with terminal B–H bonds: structural characteristics and electronic properties. Chem Lett. 2014;43(10):1587–1589.10.1246/cl.140507
- Agou T , Hayakawa N , Sasamori T , et al . Reactions of diaryldibromodisilenes with N-heterocyclic carbenes: formation of formal bis-NHC adducts of silyliumylidene cations. Chemistry. 2014;20(30):9246–9249.10.1002/chem.201403083
- Nagata K , Murosaki T , Agou T , et al . Activation of dihydrogen by masked doubly bonded aluminum species. Angew Chem Int Ed. 2016;55(41):12877–12880.10.1002/anie.201606684
- Murosaki T , Kaneda S , Maruhashi R , et al . Synthesis and structural characteristics of discrete organoboron and organoaluminum hydrides incorporating bulky Eind groups. Organometallics. 2016;35(19):3397–3405.10.1021/acs.organomet.6b00633
- Hayakawa N , Sadamori K , Tsujimoto S , et al . Cleavage of a P=P double bond mediated by N-heterocyclic carbenes. Angew Chem Int Ed. 2017;56(21):5765–5769.10.1002/anie.201701201
- Otani T , Miyoshi M , Shibata T , et al . Thermally stable monosubstituted thiophene 1-oxide and 1-imides stabilized by a bulky rind group at their 3-position: synthesis, structure, and inversion barriers on the sulfur atom. Bull Chem Soc Jpn. 2017;90(6):697–705.10.1246/bcsj.20170042
- Ito M , Hashizume D , Fukunaga T , et al . Isolated monomeric and dimeric mixed diorganocuprates based on the size-controllable bulky ‘Rind’ ligands. J Am Chem Soc. 2009;131(50):18024–18025.10.1021/ja9071964
- Nishigaki J , Tsunoyama R , Tsunoyama H , et al . A new binding motif of sterically demanding thiolates on a gold cluster. J Am Chem Soc. 2012;134(35):14295–14297.10.1021/ja305477a
- Tanifuji K , Yamada N , Tajima T , et al . A convenient route to synthetic analogues of the oxidized form of high-potential iron–sulfur proteins. Inorg Chem. 2014;53(8):4000–4009.10.1021/ic402890 k
- Goda S , Nikai M , Ito M , et al . Synthesis and magnetic properties of linear two-coordinate monomeric diaryliron(II) complexes bearing fused-ring bulky ‘Rind’ groups. Chem Lett. 2016;45(6):634–636.10.1246/cl.160216
- Taguchi H , Sasaki D , Takeuchi K , et al . Unsymmetrical PNP-pincer type phosphaalkene ligands protected by a fused-ring bulky Eind group: synthesis and applications to Rh(I) and Ir(I) complexes. Organometallics. 2016;35(10):1526–1533.10.1021/acs.organomet.6b00113
- Kanazawa S , Ohira T , Goda S , et al . Synthesis and structural characterization of lithium and titanium complexes bearing a bulky aryloxide ligand based on a rigid fused-ring s-hydrindacene skeleton. Inorg Chem. 2016;55(13):6643–6652.10.1021/acs.inorgchem.6b00762
- Yoshimoto T , Hashimoto H , Hayakawa N , et al . Synthesis of a tungsten–silylyne complex via stepwise proton and hydride abstraction from a hydrido hydrosilylene complex. Organometallics. 2016;35(7):921–924.
- Takeuchi K , Taguchi H , Tanigawa I , et al . A square-planar complex of platinum(0). Angew Chem Int Ed. 2016;55(49):15347–15350.10.1002/anie.v55.49
- Matsuo T , Tamao K . Fused-ring bulky ‘Rind’ groups producing new possibilities in elemento-organic chemistry. Bull Chem Soc Jpn. 2015;88(9):1201–1220.
- Burroughes JH , Bradley DDC , Brown AR , et al . Light-emitting diodes based on conjugated polymers. Nature. 1990;347(6293):539–541.10.1038/347539a0
- Junkers T , Vandenbergh J , Adriaensens P , et al . Synthesis of poly(p-phenylene vinylene) materials via the precursor routes. Polym Chem. 2012;3(2):275–285.10.1039/C1PY00345C
- Martin RE , Diederich F . Linear monodisperse π-conjugated oligomers: model compounds for polymers and more. Angew Chem Int Ed. 1999;38(10):1350–1377.10.1002/(ISSN)1521-3773
- Gierschner J , Cornil J , Egelhaaf H-J . Optical bandgaps of π-conjugated organic materials at the polymer limit: experiment and theory. Adv Mater. 2007;19(2):173–191.10.1002/(ISSN)1521-4095
- Praveen VK , Ranjith C , Bandini E , et al . Oligo(phenylenevinylene) hybrids and self-assemblies: versatile materials for excitation energy transfer. Chem Soc Rev. 2014;43(12):4222–4242.10.1039/C3CS60406C
- Bejan I , Scheschkewitz D . Two Si=Si double bonds connected by a phenylene bridge. Angew Chem Int Ed. 2007;46(30):5783–5786.10.1002/(ISSN)1521-3773
- Fukazawa A , Li Y , Yamaguchi S , et al . Coplanar oligo(p-phenylenedisilenylene)s based on the octaethyl-substituted s-hydrindacenyl groups. J Am Chem Soc. 2007;129(46):14164–14165.10.1021/ja0764207
- Scheschkewitz D . A silicon analogue of vinyllithium: structural characterization of a disilenide. Angew Chem Int Ed. 2004;43(22):2965–2967.10.1002/(ISSN)1521-3773
- Jeck J , Bejan I , White AJP , et al . Transfer of a disilenyl moiety to aromatic substrates and lateral functional group transformation in aryl disilenes. J Am Chem Soc. 2010;132(48):17306–17315.10.1021/ja107547s
- Li L , Matsuo T , Hashizume D , et al . Coplanar oligo(p-phenylenedisilenylene)s as Si═Si analogues of oligo(p-phenylenevinylene)s: Evidence for extended π-conjugation through the carbon and silicon π-frameworks. J Am Chem Soc. 2015;137(47):15026–15035.10.1021/jacs.5b10113
- Frisch MJ , Trucks GW , Schlegel HB , et al . Gaussian 09, Revision B.01 and Revision D.01. Wallingford CT: Gaussian; 2010.
- Meier H . Conjugated oligomers with terminal donor–acceptor substitution. Angew Chem Int Ed. 2005;44(17):2482–2506.10.1002/(ISSN)1521-3773
- Meier H , Stalmach U , Kolshorn H . Effective conjugation length and UV/vis spectra of oligomers. Acta Polym. 1997;48(9):379–384.10.1002/actp.1997.010480905
- Zhu XZ , Tsuji H , Navarrete JTL , et al . Carbon-bridged oligo(phenylenevinylene)s: stable π-systems with high responsiveness to doping and excitation. J Am Chem Soc. 2012;134(46):19254–19259.10.1021/ja309318s
- Hayakawa N , Nishimura S , Kazusa N , et al . π-Conjugation between a Si═Si double bond and thiophene rings: synthesis, structural characteristics, and photophysical properties of 1,2-bis(thiophen-2-yl)disilene and 1,2-bis(2,2′-bithiophen-5-yl)disilene. Organometallics. 2017;36(17):3226–3233.10.1021/acs.organomet.7b00370
- Sasamori T , Yuasa A , Hosoi Y , et al . 1,2-Bis(ferrocenyl)disilene: a multistep redox system with an Si═Si double bond. Organometallics. 2008;27(14):3325–3327.10.1021/om8003543
- Yuasa A , Sasamori T , Hosoi Y , et al . Synthesis and properties of stable 1,2-bis(metallocenyl)disilenes: novel d–π conjugated systems with a Si=Si double bond. Bull Chem Soc Jpn. 2009;82(7):793–805.10.1246/bcsj.82.793
- Sato T , Mizuhata Y , Tokitoh N . 1,2-dialkynyldisilenes: silicon analogues of (E)-enediyne. Chem Commun. 2010;46(24):4402–4404.10.1039/c0cc00406e
- Kobayashi M , Matsuo T , Fukunaga T , et al . Air-stable, room-temperature emissive disilenes with π-extended aromatic groups. J Am Chem Soc. 2010;132(43):15162–15163.10.1021/ja108094 m
- Kobayashi M , Hayakawa N , Nakabayashi K , et al . Highly coplanar (E)-1,2-di(1-naphthyl)disilene involving a distinct CH–π interaction with the perpendicularly oriented protecting Eind group. Chem Lett. 2014;43(4):432–434.10.1246/cl.131043
- Watanabe H , Takeuchi K , Fukawa N , et al . Air-stable tetrakis(2,4,6-triisopropylphenyl)disilene. Direct synthesis of disilene from dihalomonosilane. Chem Lett. 1987;16(7):1341–1344.10.1246/cl.1987.1341
- Shizuka H , Tanaka H , Okazaki K , et al . Emission from the excited state of tetraneopentyldisilene. J Chem Soc, Chem Commun. 1986;10:748–750.10.1039/c39860000748
- Tamao K , Kobayashi M , Matsuo T , et al . The first observation of electroluminescence from di(2-naphthyl)disilene, an Si=Si double bond-containing π-conjugated compound. Chem Commun. 2012;48(7):1030–1032.10.1039/C1CC16067B
- Groenendaal L , Jonas F , Freitag D , et al . Poly(3,4-ethylenedioxythiophene) and its derivatives: past, present, and future. Adv Mater. 2000;12(7):481–494.10.1002/(ISSN)1521-4095
- Tokyo Chemical Industry Co. Ltd . website for customers in Japan. Available from http://www.tcichemicals.com/ja/jp/disilene.html. (EMind)Br (B4379), (Eind)Br (B4380), (E)-1,2-di(1-naphthyl)disilene (B4421) and (E)-1,2-di(2-naphthyl)disilene (B4422).
- Kobayashi M , Hayakawa N , Matsuo T , et al . (Z)-1,2-di(1-pyrenyl)disilene: synthesis, structure, and intramolecular charge-transfer emission. J Am Chem Soc. 2016;138(3):758–761.10.1021/jacs.5b11970
- Christian R , Thomas W . Solvent and solvent effects in organic chemistry. Weinhelm: Wiley-VCH; 2011. p. 549.
- Reed AE , Curtiss LAM , Weinhold F . Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev. 1988;88(6):899–926.10.1021/cr00088a005
- Iwamoto T , Kobayashi M , Uchiyama K , et al . Anthryl substituted trialkyldisilene showing distinct intramolecular charge transfer transition. J Am Chem Soc. 2009;131(9):3156–3157.10.1021/ja8093313
- Kosai T , Ishida S , Iwamoto T . Heteroaryldisilenes: heteroaryl groups serve as electron acceptors for Si=Si double bonds in intramolecular charge transfer transitions. Dalton Trans. 2017;46(34):11271–11281.10.1039/C7DT02357J
- Obeid NM , Klemmer L , Maus D , et al . (Oligo)aromatic species with one or two conjugated Si=Si bonds: near-IR emission of anthracenyl-bridged tetrasiladiene. Dalton Trans. 2017;46(27):8839–8848.10.1039/C7DT00397H
- Hayakawa N , Matsuo T . Unpublished results.
- Sekiguchi A , Yatabe T , Kabuto C , et al . The missing hexasilaprismane: synthesis, X-ray analysis and photochemical reactions. J Am Chem Soc. 1993;115(13):5853–5854.10.1021/ja00066a075
- Tsurusaki A , Iizuka C , Otsuka K , et al . Cyclopentasilane-fused hexasilabenzvalene. J Am Chem Soc. 2013;135(44):16340–16343.10.1021/ja409074 m
- Szilvási T , Veszprémi T . Molecular tailoring: reaction path control with bulky substituents. Organometallics. 2012;31(8):3207–3212.10.1021/om201246 g
- Weidenbruch M , Willms S , Saak W , et al . Hexaaryltetrasilabuta-1,3-diene: a molecule with conjugated Si=Si double bonds. Angew Chem Int Ed English. 1999;36(22):2503–2504.
- Ichinohe M , Sanuki K , Inoue S , et al . Disilenyllithium from tetrasila-1,3-butadiene: a silicon analogue of a vinyllithium. Organometallics. 2004;23(13):3088–3090.10.1021/om040056s
- Uchiyama K , Nagendran S , Ishida S , et al . Thermal and photochemical cleavage of Si=Si double bond in retrasila-1,3-diene. J Am Chem Soc. 2007;129(35):10638–10639.10.1021/ja0741473