
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Nitrogen-doped graphene oxide monoliths (GOMs) were readily constructed by crosslinking graphene oxide (GO) using triethylenetetramine (TETA) and hydroxyethyl cellulose (HEC). The addition of HEC was beneficial to the formation of a network structure compared to that in the absence of HEC. The generated monoliths have shown various morphologies with different d spacing, layer thickness, and micropore size. Fourier transform infrared (FTIR) and X-ray photoelectron spectroscopy (XPS) analyses provided evidences for the formation of covalent C–N bonds and some nitrogen-containing heterocyclic composition inside the graphene oxide sheet, indicating that the interaction of GO with the amine crosslinker involved the crosslinking reaction between GO epoxides and amine groups. HEC was also involved in the N-doping reaction via the partial reduction of oxygen in HEC molecules. Analysis of X-ray diffraction (XRD) results indicated that the lattice distance between GO sheets increased after TETA/HEC crosslinking. Thermogravimetric analysis (TGA) confirmed the successful incorporation of crosslinker moieties on the surface of GO sheets. The fabricated GOMs could be used to efficiently adsorb metal ions and arsenate by the introduced polar functional groups on GO sheets and porous structures based on hydrogen bonds, whose morphologies and compositions were confirmed via XRD, scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS).

Classification:
1. Introduction
Since their discovery in 2004, graphene materials continue to attract great attention due to their extraordinary properties such as ultrahigh specific surface area (theoretical value: 2630 m2g−1 [Citation1]), combined with superior chemical and thermal stability, thermal conductivity, mechanical, and electrical properties [Citation2]. These properties are attributed to the unique single-layer structure with two-dimensional graphene as a universal building block. Graphene derivatives modified by diverse molecules, such as oxides, metal atoms and ions, and polymers, provide more opportunities to develop new functionalities [Citation3–12] to be used as electrode materials, supercapacitors, sensors, anticorrosive coatings, catalysts, and a variety of absorbent and porous materials [Citation13–21]. For example, at the liquid/graphite interface, Tobe et al. [Citation13] found a twelve-membered phenylene-ethynylene macrocycle called dehydrobenzo annulene (DBA), substituted by six flexible alkoxy chains self-assembled to form hexagonal porous 2D molecular networks via van der Waals interactions between interdigitated alkyl chains as the directional intermolecular linkages. Graphene oxide (GO), an important derivative of graphene, has an additional functionality because of the presence of various oxygen-containing groups on its surface, such as –OH, –C=O, –COOH, and epoxides [Citation5]. These functional groups provide further opportunities for GO to react with other substances and potentially make GO much more adaptable for the above-mentioned applications. Salunkhe et al. [Citation14] reported an effective route for the preparation of layered reduced graphene oxide (rGO) with uniformly coated polyaniline (PANI) layers. These nanocomposites were synthesized by chemical oxidative polymerization of aniline monomer in the presence of layered rGO, which shown excellent capacitive performance with a high specific capacitance of 286 Fg−1 and high cycle reversibility of 94 % after 2000 cycles. Veeramani et al. [Citation15] synthesized graphene sheet-like porous activated carbon (GPAC) with a high specific surface area using Bougainvillea spectabilis as a precursor with the assistance of a facile and reliable chemical activation method. The as-synthesized GPAC materials were used to investigate their structural and porous properties as an excellent electrode material for supercapacitor and catechin sensing.
Nitrogen is one of the most common heteroatoms that could bond covalently with graphene-based materials. Thermal treatment in the presence of nitrogen precursors [Citation20–25] and in situ growth by chemical vapor deposition have been used to facilitate N-doped carbon nanomaterials, with improved catalytic and electrochemical performances [Citation22,26]. Two-step reaction involving carboxylic acid group activation and amidation is the conventional strategy to introduce primary amine radicals to GO surface [Citation27]; this method is tedious and not suitable for large-scale preparation of amine reduced GO. Lai et al. [Citation22] first reported a one-step solvothermal process to achieve both the reduction of GO and the introduction of primary amine to graphene in ethylene glycol medium. The resulting amine modified graphene is an excellent electrochemical material for supercapacitor applications and also shows high cycle stability, with negligible decrease of specific capacitance value after a thousand cycles of charging and discharging [Citation14–16,22]. However, it was reported that ethylene glycol may result in the stacking of graphene sheets because of the strong electrostatic interaction between hydroxyl groups of ethylene glycol and amine groups or other ionic groups [Citation28–30].
Three-dimensional (3D) GO-based monoliths (GOMs) have been developed with silica [Citation18,21], polymers [Citation7,11], oxides [Citation3,9,10], organic molecules [Citation4,5,12,17], and metal particles [Citation28,31,32] supporters, etc. The driving forces of self-assembly or formation of GOMs include covalent bonding, hydrogen bonding, π–π stacking, or electrostatic interaction [Citation32]. Zhao et al. prepared nitrogen-doped graphene aerogel materials as an efficient sorbent for gasoline through the hydrothermal treatment of GO with pyrrole [Citation33]. Andjelkovic et al. [Citation34] synthesized graphene aerogels decorated with a-FeOOH nanoparticles for the removal of arsenic from wastewater. A nanocomposite graphene hydrogel with three-dimensional nano-mesopores was reported by Gao et al. [Citation35] by employing vitamin C. New reduced graphene oxide (rGO)-based silver nanoparticle-containing composite hydrogels were successfully prepared in situ through the simultaneous reduction of GO and noble metal precursors within the GO gel matrix [Citation32]. Based on the above examples, the size and distribution of pores and the GOMs mechanical strength are the key points in order to facilitate the formation of 3D gels (hydrogel/aerogel) [Citation36–42]. Nonetheless, the density, polarity, and homogeneity of the GOMs are still a challenging issue. For example, in the field of pollutant removal from the contaminated water [Citation17], the structural properties of GOMs directly affect material performance. 3D gels are attracting sorbents because they can theoretically provide large solid/liquid or gas phase interfaces, where organic dyes [Citation36–41] or metal ions [Citation42] can be easily adsorbed due to the interconnected porous structures. However, there have been few reports to study the formation mechanism of N-doped graphene oxide monoliths, especially, when nitrogen heteroatom is in the form of primary amine or secondary amine on the fabrication of 3D GOMs. Although it is known that hydrogen bonding plays an important role in the formation of 3D graphene oxide monoliths, there have been no systematic studies on how hydrogen bonding affects the structure of GOMs.
In this work, in order to reduce the usage of nitrogen containing crosslinkers due to their toxicity and irritation properties, we synthesize N-doped graphene oxide hydrogel in the presence of eco-friendly hydroxyethyl cellulose (HEC), which contains a great number of hydroxyl groups. To the best of our knowledge, the usage of HEC in the fabrication of GOM is currently not reported in the literature. Triethylenetetramine has very high activity as a crosslinker, and it is possible to prepare N-doped graphene oxide monoliths through covalent bonding between GO and triethylenetetramine under mild reaction conditions. However, HEC tends to form a gel matrix due to a large amount of hydroxyl groups from glucose units, while the insertion of long glucose chains results in an increase in d-spacing of GO sheets. Therefore, our goal is to prepare GOM by a rapid and simple method, that is, the GO water solution is directly mixed with HEC solution, followed by the addition of triethylenetetramine crosslinker. This one-step reduction of GO and introduction of nitrogen are obtained simultaneously. The effect of the ratio of GO to crosslinker, and the effect of the concentration of HEC on the structural property are investigated in this work. Finally, the as-prepared hydrogel composite can be used as a highly efficient adsorbing material for toxic arsenic compound and copper ion.
2. Experimental details
2.1. Materials and preparation of graphene oxide
2.1.1. Materials
Deionized water was employed in the preparation of GO. Hydroxyethyl cellulose (HEC) (M w = 250,000, degree of substitution: 1.00, viscosity: 80~125 Pa.s), triethylenetetramine, sodium nitrate (NaNO3), sulfuric acid (H2SO4), potassium permanganate (KMnO4), and other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used as received.
2.1.2. Preparation of graphene oxide
GO was prepared by Hummers method [Citation12]. Graphite (5 g) and NaNO3 (3 g) were placed in a flask under stirring at room temperature, and concentrated H2SO4 (150 mL) was added under vigorous stirring in an ice-bath environment. KMnO4 (15 g) was slowly added into the mixture with vigorous stirring within 1.5 h, and the temperature was maintained below 10 °C. The mixture was then transferred to a water bath set at 35–40 °C and the reaction was allowed for 0.5 h until a thick paste was obtained. Next, 5 wt. % H2SO4 (200 mL) was added at 98 °C within 2 h, subsequently, the temperature was reduced to 50 °C and 30 wt. % H2O2 aqueous solution (30 mL) was slowly added until the color of solution turned from brown to yellow. The resulting mixture was purified by repeating the following procedures: firstly, washing the mixture with 3 wt. % H2SO4/deionized H2O to remove metal ions; secondly, dispersing GO by ultrasonication (Branson, Smith Kline Co., Danbury, CT, USA.) for 30 min; thirdly, centrifugation (4000 rpm, 30 min), then washing three times. When the supernatant fluid became colorless, the dark precipitate was collected and dried in a freeze dryer (BenchTop Pro, VirTis, Warminster, PA, USA) for 1 day.
2.2. Preparation of N-doped graphene monoliths supported by HEC
GO water solution (0.5 wt. %) was dispersed by ultrasonication for 30 min at room temperature. N-doped graphene monoliths were prepared according to the weight ratios in Table . For example, to prepare GOM-T1, GO solution (1.5 mL, containing GO 7.5 mg) was mixed with TETA (11.25 mg), and quickly dispersed by ultrasonication, then, transferred into an oil bath at 35 °C for 20 s. For GOM-TH1, HEC (3 mg) was added into the corresponding GO solution (1.5 mL). The mixture was fully dispersed for 1 h by ultrasonication. Finally, a relatively small amount of TETA (9 mg) was added into the above mixture. After 10 min of ultrasonication, GOM-TH1 was fabricated by heating the reaction mixture of GO and HEC containing TETA under the same condition as that of GOM-T1. Other GOMs (GOM-T2 and GOM-TH2) with component ratios listed in Table were also prepared using the same method. The formation of GOM could occur in water dispersion, and dehydrated GOM was obtained by freeze drying. In Table , the effect of TETA amounts on the crosslinking reaction was investigated by comparing GOM-T1 and GOM-T2; and GOM-TH1 and GOM-TH2 samples.
Table 1. GOMs prepared at different weight ratios of GO/TETA/HEC.
2.3. Removal of arsenate and metal ions
The stock solutions of As(V) and Cu(II) were prepared by dissolving the solids of CuCl2 (50 mg) (including a small amount of CuBr2 and other trace impurities), AlCl3 (0.2 mg), NaHSO4 (0.2 mg), and Na2HAsO4·12H2O (100 mg) in deionized water (3.5 mL). The prepared aerogel of GOM-TH1 (7 mg) was added into a glass vial containing a low concentration of As(V) and Cu(II) ions. The pH was adjusted to 10 by adding a small volume of 0.01 or 0.1 mol L−1 NaOH or HCl. The mixture was shaken slightly at 30 °C for a few minutes, and then settled for a few minutes. This shaking–settling recycle experiment was repeated about five times. Next, the mixture was kept undisturbed overnight to reach equilibrium. Finally, the element adsorbed GOM-TH1 was filtered, and washed with deionized water, then, freeze dried for analysis. The adsorbed elements were confirmed by SEM in the backscattered electron mode (BSE), energy-dispersive X-ray spectroscopy (EDX), and XRD analysis.
2.4. Characterization
The micro-morphology and cross-sectional structures of the monoliths were characterized by field emission scanning electron microscopy (Zeiss, Merlin, Jena, Germany). The Fourier transform-infrared (FTIR) spectrum was recorded with a Nicolet 6700 spectrometer (Thermo Scientific, Waltham, MA, USA). XRD patterns were collected using a Rigaku (Osaka, Japan) Smartlab powder diffractometer with Cu Kα radiation (λ = 1.5418 Å) at 45 kV, 200 mA, with a step size of 0.01° and a count speed of 1 degree per minute.
X-ray photoelectron spectroscopy (XPS) analysis was performed using an AXIS Nova spectrometer (Kratos Analytical Inc., Manchester, UK) with a monochromated Al Kα source at a power of 180 W (15 kV × 12 mA) and a hemispherical analyzer operating in the fixed analyzer transmission mode. The total pressure in the main vacuum chamber during analysis was typically between 10−9 and 10−8 mbar. Survey spectra were acquired at a pass energy of 160 eV. All elements presented were identified from the survey spectra. To obtain more detailed information about chemical structure, high resolution spectra were recorded for individual peak at 40 eV pass energy (yielding a typical peak width for polymers of 1.0 eV). Data processing was performed using CasaXPS processing software version 2.3.15 (Casa Software Ltd., Teignmouth, UK). The atomic concentrations of the detected elements were calculated using integral peak intensities and the sensitivity factors supplied by the manufacturer. Binding energies were referenced to the main C1s peak at 284.7 eV for aromatic hydrocarbon.
A Bruker (MA, USA) FastScan atomic force microscope (AFM) with Icon scanning head and NanoScope 9.3 and ScanAsyst-Air probes was employed in PeakForce QNM mode to analyze the particle size of GO. The grafted contents were determined using thermogravimetric analysis (TGA; TGA2 stare system, Mettler Toledo, Zurich, Switzerland) by heating the N-GOM from 25 to 600 °C at a rate of 10 °C/min in a nitrogen atmosphere.
3. Results and discussion
3.1. Fabrication of crosslinked GOMs
GO has a large number of highly reactive oxygen-containing functional groups including carboxylic and hydroxyl groups at the edge and epoxy groups on the basal planes [Citation43,44], which make GO a promising functional material for a wide range of applications [Citation45–48]. In particular, these functional groups can react with various crosslinkers such as diamines to form GO monoliths [Citation49]. In this work, we are the first to investigate the use of HEC in crosslinking process and to clarify the coordinative role of primary or secondary amine in fabricating GOMs in water. TETA is a short chain aliphatic amine, containing two primary and two secondary amine groups. HEC is a polysaccharide composed of a large number of D-glucose units linked by β-1, 4-glycosidic bond, containing many hydroxyl groups and ether bonds with good biocompatibility. To the best of our knowledge, HEC and TETA have not been used together for the fabrication of GOM before.
Figure (a) shows the digital photographs of GO solution (Figure (a), left) and GO/HEC mixed solution (Figure (a), right) resting for one week at room temperature. No gel was observed for GO/HEC solution, indicating that there was no formation of GOM in the presence of HEC, and without any amine reagent. It suggested that amine reagent was indispensable for the crosslinking reaction. With the addition of TETA, gelation occurred after heating the mixture of GO/TETA/HEC at 35 °C for 20 s. The crosslinked monolith series, as shown in Figure (b), all showed the integral crosslinked hydrogel and/or aerogel structures. For example, the corresponding aerogel of GOM-TH1 had a certain elastic strength, as shown in Figure (b) (right image), which can support the weight of an empty tube (6.6 g). This means that the TETA amine crosslinker and HEC molecules can promote the formation of crosslinked structure in a minimal period of time and at a lower temperature, compared to the conditions of 3D self-assembly of GO and DNA (5 min and 90 °C) [Citation12]. HEC plays a role as an assistant crosslinker in the fabrication of GOMs. Obviously, the driving force for the formation of GOMs is the curing reaction between epoxy and primary amine groups or secondary amine groups [Citation3,11,28]. There are likely other reactions, such as amidation reaction between amine groups and the carboxyl groups. Furthermore, the crosslinking reaction also likely occurs between the amine groups and HEC molecules, because a large number of active hydroxyl groups in HEC chains can promote the formation of stereoscopic structure in the form of hydrogen bonds.
Figure 1 . (a) Photographs of GO solution (left) and GO/HEC mixture solution (right); (b) Photographs of the reacted GOMs (GOM-T1, GOM-T2, GOM-TH1, and GOM-TH2, respectively, from left to right); SEM images of (c) GOM-T1, (d) GOM-T2, (e) GOM-TH1, (f) GOM-TH2; (g) AFM image of pristine GO sheets.


Figure (c)–(f) shows SEM images of four GOMs, at various magnifications. To compare, an AFM image of a few GO sheets prior to crosslinking is shown in Figure (g). The size of a single GO sheet is in the range of hundreds of nanometers in width and a few nanometers in thickness. When amine crosslinker (TETA) was introduced, a rough and laminar morphology composed of channels and pores was formed. As observed in Figure (c) and (d), GO sheets stack together with the size of structure blocks in the range of tens of micrometers. Although skeleton structures are dominant in GOMs, there are some large voids between GO sheets. The transformation from 2D GO sheets to a 3D GOM is dependent on the concentration of TETA. The thickness (~0.3 μm) of stacked GO sheets in GOM-T1 (Figure (c)) is approximately the same as that of GOM-T2 (Figure (d)), but the latter has more and larger interstices between GO sheets. Moreover, better laminated arrangement of GO sheets and more penetrable pores with continuous channels were also observed in Figure (d). These structural differences are likely related to the lower TETA concentration used in GOM-T2, resulting in a lower crosslink density.
In the presence of HEC, however, the large channels present in GOM-T1 and GOM-T2 (Figure (c) and (d)) are not observed, as shown in Figure (e) and (f). Although Figure (e) and (f) all show ample crosslinking, there are some subtle differences between them. For example, the stacking GO sheets in Figure (f) appear thicker than that in Figure (e), possibly due to the higher ratio of HEC/TETA (0.4/0.7 (f) vs. 0.4/1.2 (e) in Table ). The pores observed in Figure (f) all show intact morphology unlike those in Figure (e), which contains some broken and incomplete cavities. Moreover, there are some smaller pores with thinner walls (~0.1 μm) in Figure (e). The surface of the crosslinking structure indicates more compact and ordered morphology in Figure (f) than in Figure (e). This may result from a more favorable spatial coordinative interaction between HEC and TETA molecules. The ether bond (C–O–C) in HEC chain is σ bond, which can rotate around the symmetric axis of this bond in order to keep the alternate arrangement of D-glucose units. However, when the relative content of HEC is higher (Figure (f)), HEC molecules cannot remain in a free and extensible state. These HEC molecules overlap and intertwine with each other and possibly produce more hydrogen bonds between HEC and GO sheets, or among HEC molecules, or between HEC and TETA molecules. As a result, the surface morphology is smoother and more compact in spite of thicker stacked GO sheets, compared with the original thickness of GO (~2 nm, see AFM image in Figure (g)). Moreover, for GOM-TH2, the stacking of GO sheets is most likely attributed to hydrogen bonding and the formation of more compounds between sheets. As such, TETA can crosslink GO sheets alone and develop a rough and laminar structure composed of channels and pores. More compact and ordered pore structures can be attributed to the participation of a large number of hydroxyl groups in HEC in facilitating hydrogen bonds.
In summary, GOM-T1 and GOM-T2 can quickly form stereoscopic structure in the case of TETA. For GOM-TH1 and GOM-TH2 samples, HEC can promote the fabrication of GOMs as an assistant crosslinker due to a large number of active hydroxyl groups in HEC chains. More compact and ordered porous network structures are formed, accompanied with stacking in different degrees.
3.2. Structure analysis of crosslinked GOMs
As shown in Figure (a), the d spacing and corresponding 2θ angle are 1.411 nm (2θ = 6.26°), 1.255 nm (2θ = 7.04°), 1.868 nm (2θ = 4.73°), 1.624 nm (2θ = 5.44°), for GOM-T1, GOM-T2, GOM-TH1, GOM-TH2, respectively, as compared to 0.796 nm of pristine GO (2θ = 11.08°). There are two broad peaks from 6 to 8° and from 18 to 22° for HEC sample. The characteristic peak of pristine GO changed dramatically as an outcome of crosslinking reaction, resulting in the structure change of pristine GO [Citation7]. XRD patterns provide further evidence that different morphologies and structures are influenced by the different ratios of HEC and TETA. For example, the left shift of d spacing of GOM-TH1 sample is the largest, and the d spacing shift of GOM-T2 sample is the smallest, compared with that of pristine GO. It is obvious that the structural properties of the GOMs not only are related to the type of crosslinkers, but also to the ratio of TETA/GO in the presence or absence of HEC. This is because both TETA and HEC are involved in spatial coordinative interaction in the fabricating process of GOMs. TETA molecule has 4 nitrogen atoms and 6 active hydrogen atoms associated with primary amine and secondary amine groups. These active hydrogens are readily protonated in curing reactions or N-doping. Moreover, these protonated hydrogens prominently increase the electrophilic ability of the primary or secondary amine groups, resulting in the stronger crosslinking. The analysis details are described below.
Figure 2. (a) XRD patterns and (b) FTIR spectra of pristine GO, HEC, TETA, and GOMs, respectively.
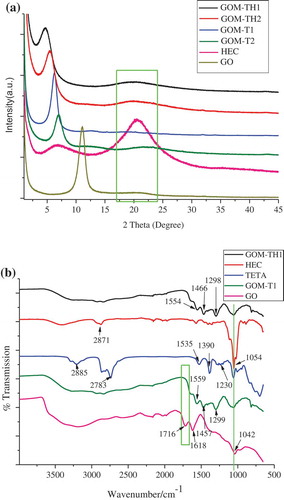
The XRD peak intensity of GOM-T1 is stronger than that of GOM-T2 and the d spacing is also larger with the increase of TETA/GO ratio, which agrees with reports by Lei et al. [Citation5] and Lai et al. [Citation22]. The higher ratio of crosslinker/GO can promote the fabrication of GOMs because of the increased doping ability of primary amine and the activity of hydrogen attached to primary and secondary amine groups.
An amorphous phase is observed in GOM-TH1 and GOM-TH2, as shown by a broad peak from 18 to 22° with reduced intensity compared with that of HEC. This provides evidence that HEC molecule took part in the crosslinking and changed the GOM structure. The 2θ values for GOM-TH1 or GOM-TH2 are smaller than those of GOM-T1 and GOM-T2, but the d spacing is larger, indicating different levels of stacking in the presence of HEC. The more distinct shift of XRD peak in the presence of HEC could be due to the fact that HEC provides a large number of hydroxyl groups in D-glucose structural units. In the presence of HEC, the crosslinking reaction not only occurs between TETA and GO sheets, but also likely between TETA and HEC molecules, even between HEC and GO sheets. As a result, more network structure was observed in GOM-TH1 and GOM-TH2, as shown in Figure (e) and (f).
With the increased ratio of HEC/TETA (0.4/0.7 (Figure (f)) vs. 0.4/1.2 (Figure (e)) in Table ), the d spacing of GOM-TH2 decreases from 1.868 to 1.624 nm, indicating the stacking of GO sheets is denser in GOM-TH2 than in GOM-TH1. The thickness of the stacking GO layers in GOM-TH2 is ~0.2 μm, which is thicker than that in GOM-TH1 (~0.1 μm). This may be due to the presence of a large number of hydroxyl groups in HEC that took part in the p–π conjugative integration with carbon p orbital attached to hexatomic ring of GO [Citation23]. In the meantime, π–π* stacking also plays an important role due to the participation of D-glucose units. In fact, the above-mentioned interactions are most likely the source of hydrogen bonds among GO sheets, TETA, and HEC because of a great number of negative charged functional groups such as –CN, –(C=O)–O, CO, and HO and the hydrogen atoms attached to HEC in the form of –CH or TETA in the form of –NH. Therefore, the hydrogen bonding is possibly formed among these positive and negatively groups. In addition, the network structures observed in SEM images (Figure (e), (f)) also confirm the different crosslinking performances due to the participation of HEC.
Thus, in the absence of HEC, the crosslinking reaction with TETA molecule results in N-doping or addition reaction supported by the protonated amine groups. If HEC is added, a network structure is preferred due to stronger hydrogen bonding and electrostatic interactions between GO sheets and sufficient number of pyranose rings in the form of π–π* stacking or van der Waals forces.
Figure (b) shows the FTIR spectra of GO, TETA, HEC, GOM-T1, and GOM-TH1. The stretching vibration of hydroxyl group in GO shows a broad band from 3000 to 3700 cm−1; the C=O stretching vibration is assigned at 1716 cm−1; the C–O–C vibration of epoxy group and the C=C vibration of aromatic ring are assigned at 1042 and 1618 cm−1, respectively. In the spectrum of TETA, the peaks above 3000 cm−1 are attributed to the stretching vibration of N–H, while the stretching vibrations of C–H are found at 2885 and 2783 cm−1. The peaks at 1535, 1390, and 1230 cm−1 are attributed to the in-plane bending vibration of N–H and C–H, and the stretching vibration of C–N, respectively. The spectrum of GOM-T1 suggests crosslinking reactions between TETA molecule and GO sheets. Firstly, the stretching vibrations of N–H, C–H, and C–N bonds (2885, 2783, and 1230 cm−1, respectively) become weaker, likely resulting from the formation of hydrogen bonds in network matrix. At the same time, due to the charge transfer, their bond length may also become longer, which also relates to the weaker vibration. Moreover, the absorption peaks of C=C double bonds in GO (1618 cm−1) disappear after crosslinking, indicating the possible involvement of a double bond in the reaction. These changes are most likely attributed to the participation of primary or secondary nitrogen in TETA. The lone pair electrons in nitrogen are involved in a conjugation effect with C=C double bonds, resulting in the average of electron cloud density of C=C bond [Citation50], which facilitates the addition reaction of the protonated nitrogen. The hydrogen bonds within the GO sheet or between GO sheets most likely result in the formation of N–H–N; N–H–O, and O–H–O [Citation51] because of the stronger electronegativity of nitrogen and oxygen. It is also possible that the formation of hydrogen bonds can cause the reduction of the electron cloud density of double bonds because of the charge transfer in the GO sheet. Therefore, the C=C double bond is most likely substituted by C–C single bond due to the additional reaction.
In addition, FTIR analysis shows the appearance of new peaks at 1559, 1457, and 1299 cm−1 for GOM-T1, which are consistent with the new C=N bonds, in-plane bending vibration of C–H bonds, and the new C–N bonds, respectively [Citation4,8]. These new peaks are the result of the crosslinking reaction of TETA with GO sheets. It is interesting to note that the in-plane bending vibration of C–H shifts from 1390 to 1457 cm−1, which may result from the aromatic contributions of hexatomic carbon rings. The characteristic absorption peak of C–O–C in epoxy group (1042 cm−1) in the pristine GO sheet is replaced by a wider peak in GOM-T1, which is probably attributed to the ring-opening of epoxy groups, generating more C–OH groups. The vibration frequencies of these groups are resulting in the vibration coupling effect, widening the peak.
Similar crosslinking structures were also observed in the spectrum of GOM-TH1. For example, the characteristic absorption peaks of N–H and the stretching vibration absorption peaks of C=C all become weaker. In addition, there are new peaks at 1554, 1466, and 1298 cm−1 in GOM-TH1, which are assigned as new C=N bonds, in-plane bending vibration of C–H bonds, and new C–N bonds, respectively [Citation12]. More importantly, the peak of C–O–C at 1054 cm−1 assigned as pyranose ring skeletal vibration becomes weaker after the addition of HEC to the system. The skeletal vibration of C–C and C–O (1320~1350 cm−1) and the stretching vibration of C–H (2871 cm−1) in HEC chain all become weaker in GOM-TH1. These observations indicate the involvement of HEC in the crosslinking process and that the physical structure and properties of the HEC molecule have changed after crosslinking.
The formation of new covalent bonds and the structure characteristics of the GOMs were further examined by XPS as shown in Figure .
Figure 3. O1s spectra of (a) HEC, (b) GOM-T1, (c) GOM-TH1, and (d) pristine GO.
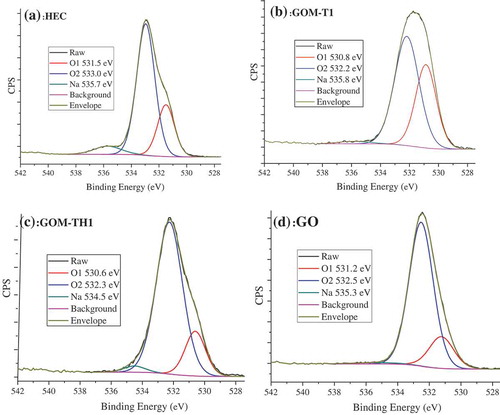
Figure shows the O1s spectra of HEC, GOM-T1, GOM-TH1, and pristine GO, and Tables and show the elemental composition in atomic percentage and the relative fraction of the O, C, and N contributions from XPS spectra, respectively. O1 and O2 are assigned as the contributions of different functional groups, respectively [Citation47], as shown in Table . Compared to that of GO sample, it can be seen that the values of O1 in GOM-T1 or GOM-TH1 samples shift to lower binding energies after crosslinking using TETA/HEC. For example, in GOM-T1 sample, the binding energy of O1 shifts to 530.8 eV resulting from the successful grafting of nitrogen to GO surface through the introduction of amide groups. In the case of using HEC (i.e. GOM-TH1), the binding energy of O1 further shifts to 530.6 eV. These shifts indicate the role of TETA and HEC as the peak position for O in N–C=O tends to sit at a lower binding energy than O in aliphatic C=O; while O in aromatic C=O will tend to sit at an even lower binding energy position [Citation47]. This point can be further explained by change in the ratio of O1 to O2. For example, the ratio of O1 to O2 for GOM-T1 and GOM-T2 increases compared to that of GO, indicating an increase in the concentration of N–C=O and C=O groups relative to the C–O groups. This phenomenon provides the evidence of the crosslinking reaction such as the amidation or ring-opening reaction of epoxy groups. In the case of using HEC, for GOM-TH1 and GOM-TH2, the ratio of O1 to O2 is lower compared to those in GOM-T1 and GOM-T2 and closer to the ratio for GO. This indicates a greater inclusion of C–O groups relative to N–C=O and C=O in GOM-TH1 and GOM-TH2 compared with GOM-T1 and GOM-T2, which would be associated with the glucose units in HEC.
Table 2. Elemental composition in atomic percentage (%) derived from XPS survey spectra.
Table 3. The relative fraction of the O, C, and N contributions derived from peak fitting of high resolution O1s, C1s, and N1s XPS spectra.
The XPS spectra further confirmed the role of amine groups after crosslinking, where the survey spectra for all GOMs showed the presence of O, N, and C, and the elemental composition was presented in Table . It is found that the ratio of O/C in the GOM samples is 0.312, 0.314, 0.349, and 0.408 for GOM-T1, GOM-T2, GOM-TH1, and GOM-TH2 samples, respectively. However, the ratio of O/C in pristine GO is 0.504, indicating that the added TETA can partially reduce oxygen in the fabrication of GOMs in the presence of a small quantity of HEC. A ratio increase of HEC/TETA results in the largest O/C ratio for GOM-TH2, which is attributed to the higher oxygen content of HEC (0.623). By comparing the values of O/C for these samples, the removal of oxygen after crosslinking is mainly attributed to a high activity of TETA because of the participation of primary and secondary amines. This crosslinking mechanism results from the participation of nitrogen in the form of addition reaction, the heterocyclic nitrogen compounds (such as pyrrolic N, imine, imide groups, and graphitic N, etc., Table ), the hydrogen bonds between GO sheets [Citation11], etc. The participation of secondary amine groups may also promote the construction of GOMs at different levels, because these N-H groups in TETA molecule can strongly attract the GO sheet, which contain the π bonding of heteroatomic carbon ring and some negatively charged groups, such as (C=O)–O, H–O, etc. The addition of HEC promotes the formation of GOMs as HEC is made up of a large number of D-glucose units, containing many hydroxyl groups and ether bonds. A great number of hydroxyl groups take part in the p–π conjugative effect with carbon p orbital attached to hexatomic ring of GO [Citation23]. Moreover, π–π* stacking effect will also play an important role in the formation of GOMs due to the participation of D-glucose units.
Fitting of the high resolution N1s and C1s spectra was undertaken to provide additional information of chemical bonding. All samples displayed a significant amount of component N1, which can be associated with pyrrolic N, amine, amide, or imines. Regarding the relative fraction of N2 assigned to imide and graphitic N, all samples displayed a similar amount. The samples with only TETA (GOM-T1 and GOM-T2) exhibited larger values for N3. The addition of HEC resulted in the reduction in intensity associated with N3, thus indicating that HEC influences the fraction of N associated with the remaining N+ after synthesis of GOM. For example, the value of N3 (13.88) for GOM-TH2 is the smallest, because there are plenty of negatively charged functional groups in HEC chain, such as hydroxyl and glycosidic bonds [Citation52]. Therefore, charge transfer most likely occurs between HEC and TETA crosslinker, which means that the N doping also occurs within HEC molecules.
As shown in Table , C1s spectra were assigned to the contributions including carbon bond (C–C) and carbon hydrogen (C–H) (C1), epoxy and hydroxyl (C2), carbonyl (C3), and carboxyl (C4), etc., respectively. The newly formed nitrogen-containing groups are resulted from the introduced TETA, which have the potential to contribute to components C2 to C5 due to a range of functional groups that can be generated in the fabrication of GOMs. The relative contribution (39.5%) of C1 component (C–C, C–H) in GOM-TH1 is the largest, while the value of 22.36% for GOM-TH2 is the smallest. The reason may be due to the introduction of a greater fraction of glucose groups, which reduces the C–H component. This point can be further testified based on the ratio of C2 to C1, for example, the ratio of C2 to C1 for GOM-TH2 is 2.954, compared to 1.20 of GOM-TH1 indicating that the contribution of C2 (C–O) increases dramatically due to the increase of the ratio of HEC relative to TETA (Table ). In the cases of GOM-T1 and GOM-T2, the ratio of C2 to C1 increases after crosslinking compared with that of GO, as shown in Table . This is attributed in part to the inclusion of the contribution from C–N or C=N as a result of TETA. The atomic ratios for N (N/C) were 0.226, 0.224, 0.207, and 0.179 for GOM-T1, GOM-T2, GOM-TH1, and GOM-TH2, respectively, which indicates a decrease of the relative concentration of TETA, in good agreement with the weight ratios in Table . The higher ratio of HEC/TETA is also beneficial to the formation of GOMs because the primary and secondary amine groups can take part in the crosslinking reaction. In the case of a higher ratio of HEC/TETA, the ability of forming hydrogen bonds in HEC can promote the formation of GOMs even if the N-doping is affected to a certain degree.
In summary, the varied components of C1s are attributed to the different structure of curing reagents, resulting in different ratio of C1, C2, C3, C4, and C5. These have provided evidence for the successful crosslinking construction between GO and amine molecule through the formation of covalent bonding and/or heterocyclic nitrogen and hydrogen bonding because of the introduction of HEC. The improved crosslinking structure in GOMs is expected as the results of synergic effect between HEC and TETA. The crosslinking reaction includes the formation of C–N bond and some heterocyclic nitrogen compounds in GO sheets due to the N-doping of TETA, and the formation of the network structure is due to the participation of hydroxyl groups of HEC in crosslinking reaction. In the presence of both TETA and HEC, the crosslinked network structure is formed between GO sheets, GO sheet and HEC, and TETA and HEC molecules. The successful combination of TETA/HEC with GO was further confirmed by TGA.
TGA and differential thermogravimetry (DTG) results of GO, HEC, and GOMs are shown in Figure . There are three stages of decomposition and the total mass loss from 25 to 600 °C is 45.5, 48.8, 53.5, and 82.5 wt. % for pristine GO, GOM-T1, GOM-TH1, and HEC, respectively (Figure (a)). Firstly, the mass loss around 100 °C is 7.5, 5.5, 5.1, and 4.5 wt. % for GO, HEC, GOM-T1, and GOM-TH1, respectively, which is related to the evaporation of adsorbed water molecules [Citation45]. The lowest water absorption is found in GOM-TH1 due to the synergic effect of TETA and HEC. TETA reacts with labile groups that lead to a higher thermal stability. HEC provides more hydrogen bonds within GO sheet or between GO sheets, further restricting the immobile water. Secondly, above 100 °C, GO shows two main degradation stages, around 30.0 wt. % weight loss between 110 and 300 °C related to CO, CO2, and H2O release from the most labile functional groups and 8.0 wt. % weight loss between 300 and 600 °C related to the degradation of more stable oxygen functionalities [Citation45]. Two endothermic peaks are observed at the temperatures of 163.6 and 246.7 °C, respectively, as shown in Figure (b). After crosslinking GO with TETA (GOM-T1), the degradation temperature changed from 163.6 to 178.3 °C with a mass loss of 27.0 wt. %. Next, an endothermic peak is observed at 290.3 °C for the GOM-T1 instead of 246.7 °C for GO alone, possibly due to degradation of amine groups [Citation45]. Subsequently, a slower degradation continues until 600 °C. This is attributed to the degradation of triethylene moieties covalently attached to the surface of GO sheets [Citation53].
Figure 4. (a) TGA and (b) DTG curves of pristine GO, HEC, GOM-T1, and GOM-TH1.
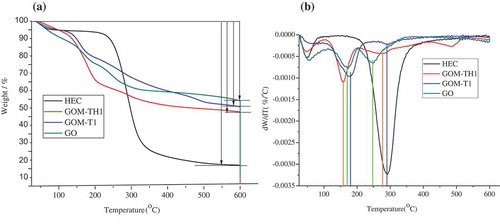
For the sample GOM-TH1, faster and more weight loss is observed below 300 °C than those of GO and GOM-T1 (Figure (a)). Two endothermic peaks at 158.5 and 276.3 °C are also observed, respectively (Figure (b)). The mass loss of 38.0 wt. % is significantly higher than those of GOM-T1 and GO. Especially, the endothermic peak at 158.5 °C is much lower than that of GO (at 163.6 °C). It is also different from the DTG curve of HEC, which has only one large endothermic peak at 292 °C above 100 °C due to the presence of D-glucoses through β-1, 4-glycosidic bond in HEC [Citation52]. This result provides evidence of the successful attachment of HEC to GO sheet surface, because the unstable compositions most likely show the cleavage between glucose units via the ether bond. It is also noteworthy that after attaching HEC onto GO sheets, there is a small endothermic peak at 483.3 °C (Td, max), indicating thermal stability improvement for GOM-TH1. This is attributed to the formation of more stable structures between the primary or secondary amine moieties from TETA and HEC. Most possibly TETA crosslinked with GO layers and also bound to D-glucose of the HEC chain, resulting in some new structures. In summary, the improved thermal stabilities of GOM-T1 and GOM-TH1 indicate the successful crosslinking between TETA, HEC, and GO sheets.
3.3. Adsorption of arsenate and metal ions
A piece of GOM-TH1 (7 mg) was soaked in the solution of copper ions and arsenate overnight, as mentioned in Section 2.3. The color change was observed as shown in Figure S1(a), indicating effective removal of metal ions. Figure shows SEM images, XRD pattern, and EDX spectra of GOM-TH1 after adsorption of As(V) and Cu(II). As shown in Figure (a) and (b), the surface morphology reveals a large number of ridges or veins on the rough surface of GOM resulting from the wrinkling of GO sheets. There are also a large number of gray or black spherical dots on the leaf-like GOM sheets, which were not observed in GOM-TH1 before metal adsorption.
Figure 5. (a), (b) SEM images at different magnification, (c) XRD pattern, (d) backscattered electron (BSE) image, and (e) EDX spectra (the data in Table) obtained at positions 7 and 11 of freeze dried GOM-TH1 after adsorption of As(V) and Cu(II).
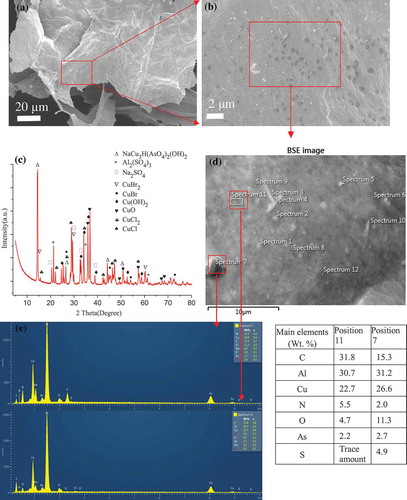
Figure (c) indicates the characteristic XRD peaks of the absorbed substances, with some new substances such as NaCu3H(AsO4)2(OH)2, Cu(OH)2, CuCl found on the surface of GO sheets. This possibly indicates some chemical reactions in the adsorption process including the sedimentation of Cu(OH)2 and the reduction reaction of Cu(II) to Cu(I) [Citation54]. This may be because some negative functional groups on the surface of GO sheets provide charge transfer, such as the hydroxyl, oxygen in glucose units or glycosidic bonds. A possible reaction mechanism is shown in Figure S1. Copper ions possibly take part in the coordination or chemical bond with negative charged groups such as oxygen-containing and nitrogen-containing groups due to the vacant 3d orbital [Citation49–51]. Moreover, the reduction and precipitation reaction of copper ions also likely occur under alkaline condition (pH = 10), as described below.(1)
(1)
(2)
(2)
Therefore, there are main characteristic peaks for Cu(II) in the form of CuBr2 (2θ = 28°, 60°), CuO (2θ = 36°), Cu(OH)2 (2θ = 24°, 32°, 36°), CuCl2 (2θ = 37.5°), and the generated Cu(I) is in the form of CuBr (2θ = 25.5°, 45°) and CuCl (2θ = 27.5°) [Citation54–58].
Arsenate ions can precipitate in the form of a double salt with other ions including copper and hydroxyl because of the electrostatic interaction from these charged groups [Citation59–61]. Moreover, for NaCu3H(AsO4)2(OH)2 the intensity of XRD peak (2θ = 14°) is stronger. The coordination ability of aluminum ions is weaker than that of copper ions due to their smaller radius, but aluminum ions take the form of [Al(OH)4]− in the alkaline environment. Therefore, after freeze drying, aluminum ions are precipitated in the form of salt, such as Al2(SO4)3. The other possibly extracted salt is Na2SO4.
The adsorption capacity (A
c
) was obtained according to the following equation [Citation62]:(3)
(3)
where C
0 (mol/L) represents the initial concentration of arsenate or copper metal ions, C
t
(mol/L) is the centration at a given time; W (g) is the weight of porous GOM-TH1 monoliths; V (mL) denotes the volume of solution; and A
c
(mg/g) is the apparent adsorption capacity of the porous monoliths at a given time. If given a long enough adsorption period, the saturation adsorption capacity A
cs (mg/g) can be obtained [Citation62]. Therefore, the adsorption can reach saturation overnight. Solaar 969 atomic absorption spectrophotometer (Thermo Fisher Scientific, USA) can be used to determine the concentration of the metal ions in the supernatant by a standard calibration curve. After repeating tests three times, the average A
cs of As(V) (35.5 mg/g) is larger than 23.78 mg/g reported by Zhang [Citation63]; the A
cs of Cu(II) is 52.9 mg/g, which is also more than 46.6 mg/g reported by Yang [Citation64]. It is noteworthy that the A
cs of As(V) and A
cs of Cu(II) are not the maximum as only one concentration of each metal ions is used in the experiment, moreover, the value A
cs of As(v) is not as high as 68 mg/g in Ref. [Citation65]. The possible reason is that the adsorbent surface could be negatively charged above pH 4.8 [Citation66]. Irrespective of the charge on the surface of GOMs, the As(V) adsorption can increase in acidic pH, as has been well reported in earlier studies. The anionic arsenic species ( or
) can be adsorbed onto the positively charged adsorbent (
,
) by electrostatic attraction [Citation66,67]. Adsorption capacity As(V) begins to decrease in alkaline (pH = 10) due to the high density of hydroxyl (OH−) ion. Therefore, this anion could compete with negatively charged arsenic species (
or
) with specific sites of adsorption. The other reason is that in our research system (arsenate and copper ions), both can react to produce the compound (NaCu3H(AsO4)2(OH)2) on the surface of GOMs, moreover, when the adsorbed GOM-TH1 was filtered, and washed with deionized water, it is most possible that some products (NaCu3H(AsO4)2(OH)2) could be washed off from the surface of GOMs. Therefore, this also makes the adsorption of Cu(II) decrease. However, the prepared GOMs show the effective adsorption of arsenate and copper ions. As known to all, adsorption kinetics depends on the interaction of adsorbate adsorbent in aqueous system. The effect factors are also complex, such as, pH, anions and cations, temperature, etc., and the adsorption kinetics will be exhaustively investigated in another work. Figure (e) shows EDX spectra of the analysis positions 7 and 11, with the results of other positions shown in Figure S2. The adsorption of As(V) and Cu(II) is confirmed in the EDX spectra. It is noteworthy that the characteristic peaks of Cu at about 1.0, 8.0, 8.9 keV [Citation48] are observed. The contents of elements vary among different positions (Table in Figure (e)).
4. Conclusions
In this study, we developed an improved method for macro-assembled graphene oxide monoliths using TETA as a main crosslinker and HEC as an assistant crosslinker. The reported material was characterized in detail and revealed an improved network structure attributed to the combined action among TETA/HEC/GO sheets. TETA played a role of N-doping reagent in the construction of GOMs resulting in the formation of heterocyclic nitrogen compound or new C–N bond. HEC molecule provided a large number of hydroxyl groups for the formation of hydrogen bonds, and also took part in the crosslinking reaction involving the oxygen reduction.
With the addition of different amounts of HEC, the interlayer space of GO sheets in GOM-TH1 increased to 1.868 nm compared to 1.225 nm in GOM-T2. The HEC and TETA could improve the thermal stability of GO by the formation of more stable structures among TETA/HEC/GO sheets. The analysis of TGA indicated that amine molecules were successfully attached into the structure of GO. The prepared GOMs showed successful adsorption of arsenate and copper ions, which was attributed to not only the porous structure of GOMs, but also a great number of polar functional groups attached to GO sheets that could form coordination compounds or reduce the metal ions. The simple process and mild conditions (low temperature and short reaction time) make this method especially important to environmentally friendly materials.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This research was supported by visiting scholar program supported by Shandong University of Science and Technology [20160323] and CSIRO Manufacturing.
Supplemental data
Supplemental data for this article is available online at https://doi.org/10.1080/14686996.2018.1457398.
The_revised_support_information.doc
Download MS Word (10.6 MB)support_information.doc
Download MS Word (11 MB)Acknowledgments
The authors are grateful to Prof. Dan Li (Melbourne University, Melbourne) for valuable advice and guidance. The authors acknowledge Mark Greaves (CSIRO, Clayton) for SEM and optical measurements. The authors are thankful to Aaron Seeber and Mark Styles (CSIRO, Clayton) for assistance with the XRD characterization and Zhaojun Han (CSIRO, Sydney) for valuable advice.
Related Research Data
References
- Novoselov KS , Geim AK , Morozov SV , et al . Electric field effect in atomically thin carbon films. Science. 2004;306:666–669.10.1126/science.1102896
- Li X , Tao Y , Li F , et al . Efficient preparation and characterization of functional graphene with versatile applicability. J Harbin Inst Technol (New Series). 2016;23:1–29.
- Li B , Cao H , Shao J , et al . Enhanced anode performances of the Fe3O4-Carbon-rGO three dimensional composite in lithium ion batteries. Chem Commun. 2011;47:10374–10376.10.1039/c1cc13462k
- Shin HJ , Kim KK , Benayad A , et al . Efficient reduction of graphite oxide by sodium borohydride and its effect on electrical conductance. Adv Funct Mater. 2009;19:1987–1992.10.1002/adfm.v19:12
- Lei S , Guo Q , Shi J , et al . Preparation of phenolic-based carbon foam with controllable pore structure and high compressive strength. Carbon. 2010;48:2644–2673.10.1016/j.carbon.2010.03.017
- Fan Z , Wang K , Wei T , et al . An environmentally friendly and efficient route for the reduction of graphene oxide by aluminum powder. Carbon. 2010;48:1686–1689.10.1016/j.carbon.2009.12.063
- Cote LJ , Cruz-Silva R , Huang J . Flash reduction and patterning of graphene oxide and its polymer composite. J Am Chem Soc. 2009;131:11027–11032.10.1021/ja902348k
- Zhang Z , Xu F , Yang W , et al . A facile one-pot method to high-quality Ag-graphene composite nanosheets for efficient surface-enhanced Raman scattering. Chem Commun. 2011;47:6440–6442.10.1039/c1cc11125f
- Zhou W , Ding C , Jia X , et al . Self-assembly of Fe2O3/reduced graphene oxide hydrogel for high Li-storage. Mater Res Bull. 2015;62:19–23.10.1016/j.materresbull.2014.11.010
- Zhang X , Jiang B , Guo JX , et al . Large and stable reversible lithium-ion storages from mesoporous SnO2 nanosheets with ultralong lifespan over 1000 cycles. J Power Sources. 2014;268:365–371.10.1016/j.jpowsour.2014.06.077
- Tang Y , Zhang Y , Deng J , et al . Mechanical force-driven growth of elongated bending TiO2-based nanotubular materials for ultrafast rechargeable lithium ion batteries. Adv Mater. 2014;26:6111–6118.10.1002/adma.201402000
- Xu Y , Wu Q , Sun Y , et al . Three-dimensional self-assembly of graphene oxide and DNA into multifunctional hydrogels. ACS Nano. 2010;4:7358–7362.10.1021/nn1027104
- Tobe Y , Tahara K , De Feyter S . Adaptive building blocks consisting of rigid triangular core and flexible alkoxy chains for self-assembly at liquid/solid interfaces. Bull Chem Soc Jpn. 2016;89:1277–1306.10.1246/bcsj.20160214
- Salunkhe RR , Hsu SH , Wu KCW , et al . Large-scale synthesis of reduced graphene oxides with uniformly coated polyaniline for supercapacitor applications. ChemSusChem. 2014;7:1551–1556.10.1002/cssc.201400147
- Veeramani V , Sivakumar M , Chen SM , et al . Lignocellulosic biomass-derived, graphene sheet-like porous activated carbon for electrochemical supercapacitor and catechin sensing. RSC Adv. 2017;7:45668–45675.10.1039/C7RA07810B
- Dutta S , Kim J , Ide Y , et al . 3D network of cellulose-based energy storage devices and related emerging applications. Mater Horiz. 2017;4:522–545.10.1039/C6MH00500D
- Samiey B , Cheng CH , Wu J . Organic-inorganic hybrid polymers as adsorbents for removal of heavy metal ions from solutions: a review. Materials. 2014;7:673–726.10.3390/ma7020673
- Liu Q , Shi J , Sun J , et al . Graphene and graphene oxide sheets supported on silica as versatile and high-performance adsorbents for solid-phase extraction. Angew Chem. 2011;123:6035–6039.10.1002/ange.v123.26
- Han Q , Liang Q , Zhang X , et al . Graphene aerogel based monolith for effective solid-phase extraction of trace environmental pollutants from water samples. J Chromatogr A. 2016;1447:39–46.10.1016/j.chroma.2016.04.032
- Eda G , Fanchini G , Chhowalla M . Large-area ultrathin films of reduced graphene oxide as a transparent and flexible electronic material. Nat Nanotechnol. 2008;3:270–274.10.1038/nnano.2008.83
- Zhang X , Chen S , Han Q , et al . Preparation and retention mechanism study of graphene and graphene oxide bonded silica microspheres as stationary phases for high performance liquid chromatography. J Chromatogr A. 2013;1307:135–143.10.1016/j.chroma.2013.07.106
- Lai L , Chen L , Zhan D , et al . One-step synthesis of NH2-graphene from in situ graphene-oxide reduction and its improved electrochemical properties. Carbon. 2011;49:3250–3257.10.1016/j.carbon.2011.03.051
- Kim YJ , Abe Y , Yanagiura T , et al . Easy preparation of nitrogen-enriched carbon materials from peptides of silk fibroins and their use to produce a high volumetric energy density in supercapacitors. Carbon. 2007;45:2116–2125.10.1016/j.carbon.2007.05.026
- Hulicova-Jurcakova D , Kodama M , Shiraishi S , et al . Nitrogen-enriched nonporous carbon electrodes with extraordinary supercapacitance. Adv Funct Mater. 2009;19:1800–1809.10.1002/adfm.v19:11
- Yang X , Wu D , Chen X , et al . Nitrogen-enriched nanocarbons with a 3D continuous mesopore structure from polyacrylonitrile for supercapacitor application. J Phys Chem C. 2010;114:8581–8586.10.1021/jp101255d
- Jurewicz K , Babel K , Ziolkowski A , et al . Ammoxidation of active carbons for improvement of supercapacitor characteristics. Electrochim Acta. 2003;48:1491–1498.10.1016/S0013-4686(03)00035-5
- Stevens JL , Huang A , Peng H , et al . Sidewall amino-functionalization of single-walled carbon nanotubes through fluorination and subsequent reactions with terminal diamines. Nano Lett. 2003;3:331–336.10.1021/nl025944w
- Petit C , Bandosz TJ . Enhanced adsorption of ammonia on metal-organic framework/graphite oxide composites: analysis of surface interactions. Adv Funct Mater. 2010;20:111–118.10.1002/adfm.v20:1
- Seredych M , Bandosz TJ . Mechanism of ammonia retention on graphite oxides: role of surface chemistry and structure. J Phys Chem C. 2007;111:15596–15604.10.1021/jp0735785
- Slabaugh WH , Seiler BC . Interactions of ammonia with graphite oxide. J Phys Chem. 1962;66:396–401.10.1021/j100809a004
- Huang Z , Lee HK . Micro-solid-phase extraction of organochlorine pesticides using porous metal-organic framework MIL-101 as sorbent. J Chromatogr A. 2015;1401:9–16.10.1016/j.chroma.2015.04.052
- Jiao T , Guo H , Zhang Q , et al . Reduced graphene oxide-based silver nanoparticle-containing composite hydrogel as highly efficient dye catalysts for wastewater treatment. Sci Rep. 2015;5:11873. doi: 10.1038/srep11873.
- Zhao Y , Hu C , Hu Y , et al . Versatile, ultralight nitrogen-doped graphene framework. Angew Chem. 2012;124:11533–11537.10.1002/ange.201206554
- Andjelkovic L , Tran DN , Kabiri S , et al . Graphene aerogels decorated with a-FeOOH nanoparticles for efficient adsorption of arsenic from contaminated waters. ACS Appl Mater Interfaces. 2015;7:9758–9766.10.1021/acsami.5b01624
- Gao M , Peh CKN , Ong WL , et al . Green chemistry synthesis of a nanocomposite graphene hydrogel with three-dimensional nano-mesopores for photocatalytic H-2 production. RSC Adv. 2013;3:13169–13177.10.1039/c3ra22950e
- Gao H , Xiao F , Ching C , et al . Flexible all-solid-state asymmetric supercapacitors based on free-standing carbon nanotube/graphene and Mn3O4 nanoparticle/graphene paper electrodes. ACS Appl Mater Interfaces. 2012;4:7019–7025.
- Sun J , Liang Q , Han Q , et al . One-step synthesis of magnetic graphene oxide nanocomposite and its application in magnetic solid phase extraction of heavy metal ions from biological samples. Talanta. 2015;132:557–563.10.1016/j.talanta.2014.09.043
- Han Q , Wang Z , Xia J , et al . Application of graphene for the SPE clean-up of organophosphorus pesticides residues from apple juices. J Sep Sci. 2014;37:99–105.10.1002/jssc.201301005
- Wang Z , Han Q , Xia J , et al . Graphene-based solid-phase extraction disk for fast separation and preconcentration of trace polycyclic aromatic hydrocarbons from environmental water samples. J Sep Sci. 2013;36:1834–1842.10.1002/jssc.v36.11
- Liu J , Zhang Q , Chen X , et al . Surface assembly of graphene oxide nanosheets on SiO2 particles for the selective isolation of hemoglobin. Chem Eur J. 2011;17:4864–4870.10.1002/chem.201003361
- Jiao T , Liu Y , Wu Y , et al . Facile and scalable preparation of graphene oxide-based magnetic hybrids for fast and highly efficient removal of organic dyes. Sci Rep. 2015;5:12451. doi:10.1038/srep12451.
- Chen J , Yao B , Li C , et al . An improved Hummers method for eco-friendly synthesis of graphene oxide. Carbon. 2013;64:225–229.10.1016/j.carbon.2013.07.055
- Jin T , Kong F , Bai R , et al . Anti-corrosion mechanism of epoxy-resin and differently content Fe2O3 coatings on magnesium alloy. Front Mater Sci. 2016;10:367–375.10.1007/s11706-016-0357-5
- Samadaei F , Salami-Kalajahi M , Roghani-Mamaqani H . Grafting of poly(acrylic acid) onto poly(amidoamine)-functionalized graphene oxide via surface-mediated reversible addition-fragmentation chain transfer polymerization. Int J Polym Mater Polym Biomater. 2016;65(6):302–309.10.1080/00914037.2015.1119686
- Khezri K , Najafi M , Roghani-Mamaqani H . Reversible addition fragmentation chain transfer polymerization of styrene from the edge of graphene oxide nanolayers. J Polym Res. 2017;34:24–34.
- Hellgren N , Haasch R , Schmidt S , et al . Interpretation of X-ray photoelectron spectra of carbon-nitride thin films: new insights from in situ XPS. Carbon. 2016;108:242–252.10.1016/j.carbon.2016.07.017
- Beamson G , Briggs D . High resolution XPS of organic polymers – the scienta ESCA 300 database. Chichester: Wiley Interscience; 1992. Appendices 3.1 and 3.2.
- Wan B , Yuan J , Feng Q , et al . Hydrothermal synthesis of K, Na doped Cu-S nanocrystalline and effect of doping on crystal structure and performance. Acta Phys Sin. 2013;62:178102–178109.
- Wan W , Li L , Zhao Z , et al . Ultrafast fabrication of covalently cross-linked multifunctional grapheme oxide monoliths. Adv Funct Mater. 2014;24:4915–4921.10.1002/adfm.201303815
- Jin T , Li X , Sun H . Interaction mechanisms between poly (amido-amine) and nano-silicon dioxide. Int J Quantum Chem. 2013;113:1213–1224.10.1002/qua.v113.8
- Jin T , Lü H . Ab initio study of complexation process between poly(amido-amine) and nano-silicon dioxide. Chin J Chem Phys. 2013;26:277–287.10.1063/1674-0068/26/03/277-286
- Kumar ASK , Jiang S . Chitosan-functionalized graphene oxide: a novel adsorbent an efficient adsorption of arsenic from aqueous solution. J Environ Chem Eng. 2016;4:1698–1713.10.1016/j.jece.2016.02.035
- Huynh VT , Nguyen D , Such CH , et al . Polymer coating of graphene oxide via reversible addition-fragmentation chain transfer mediated emulsion polymerization. J Polym Sci Part A Polym Chem. 2015;53:1413–1421.10.1002/pola.27596
- Singh K , Yadav BC , Singh VK . Electrical conductivity of cuprous bromide in the temperature range of 30–490 °C. Indian J Chem. 2012;51:1090–1094.
- Gu S , Sun H , Fan Y , et al . Synthesis of size tunable nanocopper oxide and its surface sulphidization. Chin J Inorg Chem. 2013;29:1185–1191.
- Wen X , Zhang W , Yang S . Synthesis of Cu(OH)2 and CuO nanoribbon arrays on o copper surface. Langmuir. 2003;19:5898–5903.10.1021/la0342870
- Wen X , Zhang W , Yang S . Solution phase synthesis of Cu(OH)2 nanoribbons by coordination self-assembly using Cu2S nanowires as precursors. Nano Lett. 2002;2:1397–1401.10.1021/nl025848v
- Du G , Van Tendeloo G . Cu(OH)2 nanowires, CuO nanowires and CuO nanobelts. Chem Phys Lett. 2004;393:64–69.10.1016/j.cplett.2004.06.017
- Jin T , Zhang F . Interaction mechanism of ultrafine silica and poly(amido-amine) and dispersibility of the complexes in coatings. Prog Org Coat. 2013;72:447–451.10.1016/j.porgcoat.2012.10.011
- Jin T , Kong F . Effect of differently terminal groups of poly(amido-amine) dendrimers on dispersion stability of nano-silica and ab initio calculations. Surf Interface Anal. 2015;47:474–481.10.1002/sia.5735
- Jin T , Kong F , Bai R . Structure and property investigations of the lowest energy poly(amidoamine)-CH2CH3 conformers. Chem Lett. 2015;44:943–945.10.1246/cl.150254
- Zhang N , Qiu H , Si Y , et al . Fabrication of highly porous biodegradable monoliths strengthened by graphene oxide and their adsorption of metal ions. Carbon. 2011;49:827–837.10.1016/j.carbon.2010.10.024
- Zhang K , Dwivedi V , Chi C , et al . Graphene oxide/ferric hydroxide composites for efficient arsenate removal from drinking water. J Hazard Mater. 2010;182:162–168.10.1016/j.jhazmat.2010.06.010
- Yang ST , Chang Y , Wang H , et al . Folding/aggregation of graphene oxide and its application in Cu2+ removal. J Colloid Interface Sci. 2010;351:122–127.10.1016/j.jcis.2010.07.042
- Sherlala AIA , Raman AAA , Bello MM , et al . A review of the applications of organo-functionalized magnetic graphene oxide anocomposites for heavy metal adsorption. Chemosphere. 2018;193:1004–1017.10.1016/j.chemosphere.2017.11.093
- Kumar ASK , Jiang SJ . Synthesis of magnetically separable and recyclable magnetic nanoparticles decorated with β-cyclodextrin functionalized graphene oxide an excellent adsorption of As(V)/(III). J Mol Liq. 2017;237:387–401.10.1016/j.molliq.2017.04.093
- Zare-Dorabei R , Ferdowsi SM , Barzin A , et al . Highly efficient simultaneous ultrasonic-assisted adsorption of Pb(II), Cd(II), Ni(II) and Cu(II) ions from aqueous solutions by graphene oxide modified with 2,20-dipyridylamine:central composite design optimization. Ultrason Sonochem. 2016;32:265–276.10.1016/j.ultsonch.2016.03.020