
ABSTRACT
In this study, we designed and synthesized an implantable anti-CD25 antibody-immobilized polyethylene (CD25-PE) mesh to suppress tumor growth by removing regulatory T cells (Tregs). The PE mesh was graft-polymerized with poly(acrylic acid), and the anti-mouse CD25 antibody was then immobilized using the 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide reaction. Immobilization of the antibody on the PE mesh was confirmed by immunostaining. The CD25-PE mesh could effectively and selectively capture CD25-positive cells through antigen-antibody interactions when the CD25-PE mesh was incubated with a suspension of mouse spleen cells, including CD25-positive cells. In addition, implantation of the CD25-PE mesh into mice subcutaneously demonstrated the Treg-capturing ability of the CD25-PE mesh with only a weak inflammatory reaction. In tumor-bearing mice, tumor growth was suppressed by subcutaneous implantation of the CD25-PE mesh near the tumor for 1 week. These results suggested that the anti-CD25 antibody-immobilized material could capture Tregs in vivo and inhibit tumor proliferation in a limited tumor-bearing mouse model. Further research is needed to facilitate cancer immunotherapy using implantable anti-CD25 antibody-immobilized material as a Treg-capturing device.
Graphical abstract
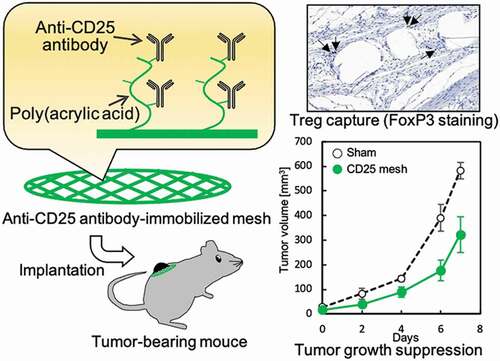
1. Introduction
Cancer treatment can be classified into surgical treatment, chemotherapy, radiotherapy, and immunotherapy. Cancer immunotherapy is a method for treating cancer using the immune system. To date, various cancer immunotherapies have been proposed, including vaccine therapy using autologous cancer vaccines [Citation1], dendritic cell vaccines [Citation2], and adoptive immunotherapy using natural killer (NK) cells and cytotoxic T cells [Citation3]. Among these approaches, cancer immunotherapy related to regulatory T cells (Tregs) has recently become a major research focus. Tregs, i.e., CD4-, CD25-, and FoxP3-positive T cells, are key players in immune suppression [Citation4] and function by controlling the activation of antigen-presenting cells via cytotoxic T lymphocyte antigen (CTLA)-4 and immunosuppressive cytokines (e.g., interleukin-10). In addition, Tregs play roles in suppressing the attack of T cells and other immune cells by modulating the production of transforming growth factor-β [Citation5]. Furthermore, in the tumor microenvironment, which is formed by various components, including cancer cells, immune cells, and the extracellular matrix, Treg accumulation is induced by secretion of the chemokine C-C motif chemokine ligand 22 (CCL22) from cancer cells and tumor-infiltrating macrophages, resulting in an antitumor immune response [Citation6,Citation7]. Several treatments that inhibit immunosuppressive signal transduction by immune checkpoint inhibitors (e.g., anti-CTLA-4 and anti-programmed death-1 antibodies) and depletion of Tregs by administration of anti-C-C motif chemokine receptor 4 antibodies have been proposed as Treg-related cancer immunotherapies [Citation8,Citation9]. The development of selective Treg removal methods is also proposed [Citation10,Citation11]. Although the efficacies of these treatments have been demonstrated, treatment with immune checkpoint inhibitors can induce serious side effects owing to activation of T cells [Citation12]. In addition, because Tregs are strongly related to autoimmunity, Treg-removing treatments may cause systemic autoimmune diseases. Therefore, the development of a method for local Treg removal at the tumor is essential.
In our previous reports, we developed an antibody-immobilized material for the selective capture of immune cells, including Tregs [Citation13–15]. The antibody-immobilized material consisted of a grafted polymer and an antibody, in which the selective capture of target cells was achieved based on the nonadhesive properties of polymer grafting and the antigen/antibody interaction.
In this study, for the development of novel cancer immunotherapies related to Tregs, we designed and synthesized an implantable anti-CD25 antibody-immobilized polyethylene mesh and investigated its properties, including selective Treg capture in vitro and in vivo and ability to suppress tumor growth.
2. Materials and methods
2.1. Materials
PE mesh (fiber diameter: 86 µm, pitch: 125 µm/163 µm) was purchased from Semitec Corp. (Osaka, Japan). Antibodies (anti-mouse CD25, anti-human CD25, anti-mouse CD4, fluorescein isothiocyanate [FITC]-labeled anti-mouse CD25, and allophycocyanin [APC]-labeled anti-mouse CD4) were purchased from BioLegend, Inc. (San Diego, California, USA). Monoclonal anti-FoxP3 antibodies were purchased from Thermo Fisher Scientific (Waltham, Massachusetts, USA). Immunostaining kit (POD conjugate anti-rat, for mouse tissue) was purchased from Takara Bio Inc. (Shiga, Japan). Simple Stain MAX-PO (Rat) and diaminobenzidine (DAB) Substrate Kit were purchased from Nichiren Bioscience Inc. (Tokyo, Japan). B16 melanoma cells were purchased from the JCRB cell bank (Osaka, Japan). C57BL/6 mice were purchased from Sankyo Lab Service Corp. (Tokyo, Japan).
2.2. Preparation of anti-CD25 antibody-immobilized mesh
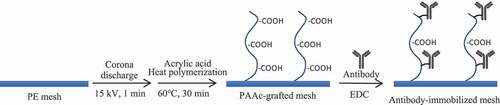
shows the preparation of an anti-CD25 antibody-immobilized mesh. After Soxhlet treatment with ethanol at 60°C for 8 h, corona discharge treatment was performed on both surfaces of the PE mesh (15 kV, 1 min). The mesh was immersed in 2% and 5% acrylic acid solutions. After degassing, the mesh was subjected to heat polymerization in a water bath at 60°C for 30 min. After washing the meshes with hot water, the poly(acrylic acid) (PAAc)-grafted PE meshes were immersed in a solution of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC, 10 mg/mL) and anti-mouse CD25 antibody (0.5, 5, 50 µg/mL) for 2 h at room temperature.
2.3. Evaluation of the antibody-immobilized PE mesh
The PAAc-grafted PE mesh was evaluated by methylene blue staining (0.02%) and weight measurements before and after polymerization. The anti-mouse CD25 antibody-immobilized PE mesh (CD25-PE) was stained using an immunostaining kit (POD Conjugate Anti Rat, for mouse tissue; TaKaRa) following the manufacturer’s instructions. The amount of antibody immobilization was measured using ImageJ software. The obtained color image of the antibody-immobilized PE mesh was converted to a monochrome image and inverted black and white. The gray values of the mesh were then measured.
2.4. Treg capture using the anti-CD25 antibody-immobilized PE mesh in vitro
All experiments involving mice were conducted using protocols approved by the Institutional Animal Care and Use Committee of Tokyo Medical and Dental University (approval number: A2019-313C). A spleen was harvested from a mouse (5 weeks of age) and placed into cell strainer (pore size: 70 µm), which was set on a dish of 6 cm diameter with 15 mL of red blood cell lysis buffer (BD Biosciences, San Jose, California, USA). The spleen was made some incisions and mashed through the cell strainer into the dish using a plunger end of a syringe. The spleen cells were transferred to a conical tube of 50 mL and centrifuged at 300xg, 4°C for 10 min. After removal of the supernatant, the cells were suspended with phosphate-buffered saline (PBS) including 2% bovine serum albumin (BSA) and were filtered with cell strainer (30 µm). A suspension of spleen cells (1.0 × 106 cells/mL; total volume: 0.5 mL) was prepared. The meshes were blocked with 2% BSA in PBS at room temperature for 30 min, and five meshes (diameter: 8 mm, area: 0.96 cm2) were placed in a tube of 2 mL Spleen cells were suspended and incubated with gentle shaking at 4°C for 1 h. The number of cells that had not adhered to the mesh was measured, and the cell capture rate was calculated by dividing the number of adhered cells by the number of seeded cells.
2.5. Flow cytometric analysis of spleen cells
Mouse spleen cells were mixed with FITC-labeled anti-mouse CD25 antibodies and APC-labeled anti-mouse CD4 antibodies and incubated for 30 min in an ice bath. The cells were washed twice with PBS containing 0.2% fetal bovine serum and subjected to flow cytometry analysis (Novocyte, Agilent Technologies Japan Ltd., Hachioji, Japan).
2.6. Implantation of the anti-CD25 antibody-immobilized PE mesh into healthy mice
Mice (5 weeks old) were anesthetized, and an anti-CD25 antibody-immobilized PE mesh was subcutaneously implanted into each mouse. After 7 days, the mice were euthanized, and the mesh and skin were extracted. The experimental protocol was approved by the animal ethics committee of our institution (Animal experiment approval number: A2019-313 C).
2.7. Hematoxylin-eosin (H-E) staining and FoxP3 immunostaining of implanted anti-CD25 antibody-immobilized PE meshes
The extracted mesh and skin were fixed with 10% neutral formalin buffer, embedded in paraffin, and sliced to a thickness of 4 µm. The slices of the samples were stained with H-E. The number of cells within 30 µm of the mesh fiber was then counted, and the slices were subjected to antigen activation treatment with ethylenediaminetetraacetic acid. Samples were immersed in 3% hydrogen peroxide solution at room temperature for 10 min, blocked with PBS (3% BSA) at room temperature for 1 h, and then mixed with anti-FoxP3 antibodies at 4°C overnight. DAB staining was performed using a Simple Stain MAX-PO (Rat) and DAB substrate kit. The numbers of cells and Tregs within 30 µm of the fiber were measured, and the Treg ratio was calculated.
2.8. Implantation of the anti-CD25 antibody-immobilized PE mesh into tumor-bearing mice
B16 melanoma cells (5.0 × 105 cells) were subcutaneously injected into mice (5 weeks of age). Tumor volume was calculated as (tumor width [mm]2 × tumor length [mm])/2 [Citation16]. When the tumor volume was approximately 25 mm3, the CD25-PE mesh was implanted subcutaneously under the tumor (Fig. S2).
2.9. Statistical analysis
Results were expressed as the mean ± standard error of the mean, with each experiment performed at least three times. Tukey’s multiple comparison test was used to test for statistical significance. A p-value < 0.05 was considered to be statistically significant.
3. Results
3.1. Preparation of an anti-CD25 antibody-immobilized mesh
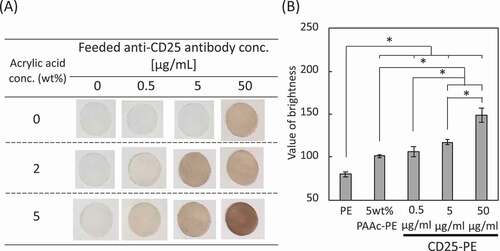
The PE mesh was subjected to corona discharge treatment and graft polymerization with acrylic acid. The PAAc-grafted mesh was evaluated by methylene blue staining and weight measurements before and after polymerization ()). The blue-stained mesh was observed at an acrylic acid concentration of 2 wt% and became dark blue at an acrylic acid concentration of 5 wt%. In addition, the weights of the mesh before and after graft polymerization increased with increasing acrylic acid concentration. Subsequently, anti-CD25 antibody was immobilized on 2 and 5 wt% PAAc-PE using the EDC reaction. DAB-stained meshes are shown in ). For PE meshes without PAAc graft-polymerization, white meshes were observed at anti-CD25 antibody concentrations of 0, 0.5, and 5 µg/mL, whereas brown-stained meshes were observed at an antibody concentration of 50 µg/mL, indicating the nonspecific absorption of the antibody on the PE mesh. For the 2 and 5 wt% PAAc-PE meshes, meshes became dark brown as the concentration of anti-CD25 antibody increased. Also, the anti-CD25 antibody-immobilized PE mesh grafted with 5 wt% PAAc was darker than that grafted with 2 wt% PAAc. So, we used the anti-CD25 antibody-immobilized PE mesh grafted with 5 wt% PAAc for below experiments. The DAB-stained meshes containing 5 wt% acrylic acid were analyzed using Image J ()). An increase in the immobilization amount of antibody was observed with increasing antibody concentrations.
Figure 1. (a) Methylene blue staining. (b) The amount of grafted poly(acrylic acid) on PE mesh (n = 5). *p < 0.01
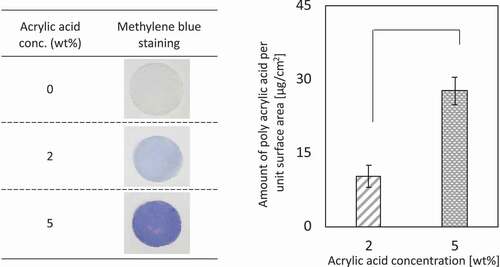
Figure 2. (a) DAB staining of CD25-PE. (b) Amount of the antibody immobilized on the 5 wt% PAAc-PE (n = 5). *p < 0.01
3.2. In vitro cell capture of antibody-immobilized mesh
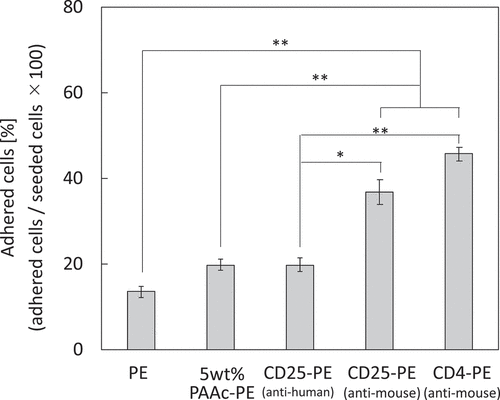
Mouse spleen cells were used to investigate the cell capture of anti-CD25 antibody-immobilized mesh. Mouse spleen cells were assessed by fluorescence-assisted cell sorting (FACS; Fig. S1). The percentages of CD25-positive and CD4-positive cells were approximately 8% and 18.5%, respectively. The anti-CD25 antibody-immobilized meshes (PAAc: 5 wt%, feed conc. of anti-CD25 antibody: 50 µg/mL) were set in suspensions of mouse spleen cells (1 × 106 cells/mL) and incubated with gentle shaking at 4°C for 1 h. Cells that had not adhered on the meshes were counted, and the cell adhesion rate was calculated (). For PE, 5% PAAc-PE, and anti-human CD25-PE meshes, there were no significant differences between cell capture rates, indicating that the cells adhered nonspecifically. In contrast, the number of adhered cells was increased for anti-mouse CD25-PE and anti-mouse CD4-PE compared with that of other meshes, indicating that cell capture on antibody-immobilized meshes involved an antigen-antibody interaction.
Figure 3. Capturing of mouse spleen cell by CD25-PE meshes (n = 5). *p < 0.05, **p < 0.01
3.3. Implantation of the anti-CD25 antibody-immobilized mesh into healthy mice
To investigate the in vivo reaction of the anti-CD25 antibody-immobilized mesh, the mesh was subcutaneously implanted into healthy mice ()). When PE and 5 wt% PAAc-PE meshes were implanted, red color, as a sign of the inflammatory reaction, was observed. In contrast, there were no significant differences between the sham operation and CD25-PE meshes when various concentrations of antibody were used.
Figure 4. (a) Implantation of PE meshes with and without anti-CD25 antibody subcutaneously into healthy mice. (b) H-E staining of PE meshes with and without anti-CD25 antibody implanted subcutaneously into healthy mice. (c) The number of cells aroud the fibers of the implanted meshes (n = 5). (d) FoxP3 immunostaining of PE meshes with and without anti-CD25 antibody implanted subcutaneously in healthy mice. (e) The ratio of Tregs to cells within 30 µm from mesh fibers (n = 9). *p < 0.05, **p < 0.01. (Scale bar: 100 µm, N.D.: Not detected)
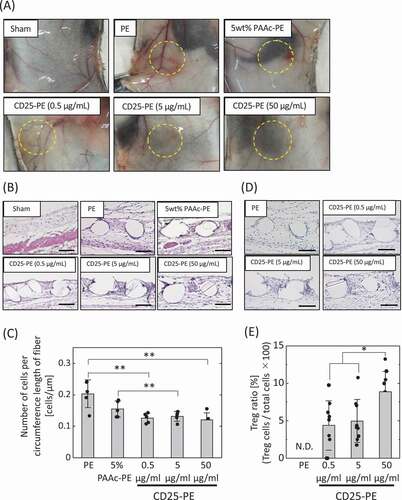
The implanted meshes were subjected to H-E staining ()). Cells within 30 µm of the mesh fiber were observed and counted ()). The highest cell accumulation was observed for the PE mesh. Compared with PE, the cell number decreased for the PAAc-PE and CD25-PE meshes. There were no differences between the CD25-PE meshes at various concentrations of antibody.
Subsequently, the implanted meshes were subjected to immunostaining with an antibody targeting FoxP3, a specific marker of Tregs ()). The number of Tregs within 30 µm of the mesh fiber was counted ()). Notably, Tregs were not observed around the PE fibers, but were observed for all CD25-PE meshes. The percentages of Tregs increased as the antibody concentration increased.
3.4. Suppression of tumor growth by implantation of the CD25 antibody-immobilized mesh into tumor-bearing mice
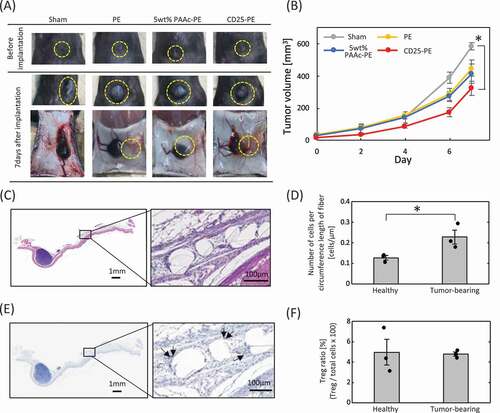
To examine the tumor-suppressive effects of the anti-CD25 antibody-immobilized mesh, CD25-PE mesh was implanted near the tumor in tumor-bearing mice ()). The implanted meshes were observed near the tumor after 7 days of implantation, although a slight shift in position was observed for the PE meshes. Changes in tumor volume over time are shown in ). For the sham operation, the tumor volume was increased to approximately 600 mm3 after 7 days. For PE and PAAc-PE mesh implantation, similar tumor growth was observed until 4 days after implantation, and tumor growth was inhibited at 6 and 7 days after implantation compared with that of the sham operation. For the CD25-PE mesh, the greatest suppression of tumor growth was found after 7 days, and the tumor size rapidly increased between days 6 and 7. The implanted CD25-PE mesh was subjected to H-E staining and FoxP3 immunostaining ()). Although the CD25-PE mesh was slightly farther away from the tumor, Tregs were observed around the fibers of the mesh. The number of cells around the fiber of the mesh was counted, and the Treg ratio was calculated (). Compared with the CD25-PE mesh implanted into healthy mice, the number of cells was increased for CD25-PE meshes implanted into tumor-bearing mice ()). There were no differences in Treg ratios between healthy and tumor-bearing mice ()).
Figure 5. (a) Implantation of PE meshes with and without anti-CD25 antibody into tumor-bearing mice. (b) Tumor growth following implantation of anti-CD25-PE meshes (n = 3). (c) H-E staining of the CD25-PE meshes at 7 days after subcutaneous implantation into tumor-bearing mice. (d) The number of cells around the fiber of the CD25-PE mesh implanted subcutaneously into healthy and tumor-bearing mice (n = 3). (e) FoxP3 immunostaining of the CD25-PE mesh at 7 days after subcutaneous implantation into tumor-bearing mice. (f) The ratio of Tregs to cells within 30 µm from mesh fibers (n = 3). *p < 0.01
4. Discussion
Cancer immunotherapy can involve enhancement of immune attack or release of immune tolerance [Citation17]. Importantly, FoxP3+ CD25+ CD4+ Tregs have been shown to strongly affect tumor immune tolerance [Citation4]. In the tumor microenvironment, Tregs infiltrate and accumulate through the secretion of chemokines, such as CCL22, from cancer cells and inhibit the attack of immune cells, such as NK cells and effector T cells, to cancer cells [Citation4,Citation5]. In this study, in order to remove Tregs from the tumor and release immune tolerance, we designed and prepared an antibody-immobilized mesh and subsequently investigated the effects of the mesh on tumor growth suppression following implantation in mice. The transcription factor FoxP3 is a specific marker of Tregs and is FoxP3 expressed inside the cells; thus, this specific marker could not be used in our system. CD25 is a surface marker of Tregs; therefore, we chose anti-CD25 antibodies in this study. A recent study showed that three types of Tregs, including naïve Tregs, effector Tregs, and non-Tregs, were present and exhibited variations in immunosuppression. Among these types of Tregs, effector Tregs, which show high immunosuppressive ability, are present as the CD25++FoxP3++ cell population. Moreover, effector Tregs have been identified in proliferative tumors [Citation18]. Therefore, in this study, we designed an anti-CD25 antibody-immobilized material to remove the effector Tregs from tumors.
The anti-CD25 antibody was immobilized on the surface of PAAc-PE mesh with acrylic acid concentrations of 2 and 5 wt% through the EDC reaction. The amount of immobilized antibody increased as the concentration of the antibody increased for both acrylic acid concentrations. However, the non-graft-polymerized PE was stained by immunostaining, suggesting nonspecific absorption of the antibody. Therefore, we used the anti-CD25 antibody-immobilized PE mesh at an antibody concentration of 5 µg/mL for subsequent experiments.
Analysis of the cell capture ability of the obtained CD25-PE mesh in mouse spleen cells (CD4−CD25−, CD4+CD25−, CD4−CD25+, and CD4+CD25+ cells) showed cell capture ratios of approximately 14% and 20% for PE and 5 wt% PAAc-PE meshes, respectively. We assumed that the cells adhered to the meshes nonspecifically because the cells were observed at the crossover region of the mesh fibers (data not shown). Although the cell capture ratio of the anti-human CD25-PE antibody was still approximately 20%, an increase in the cell capture ratio was found for anti-mouse CD25-PE (37%) and anti-mouse CD4-PE (46%) meshes. These results indicated that cell capture occurred through antigen/antibody interactions on the antibody-immobilized PE meshes in addition to nonspecific cell capture. For FACS analysis, the rates of CD25-positive and CD4-positive cells were approximately 8% and 18%, respectively. Unexpected increases in the cell capture ratios for the anti-mouse CD25-PE and anti-mouse CD4-PE meshes were observed. Thus, further studies are required to clarify the mechanisms involved in these effects.
Analysis of the Treg capture ability of the CD25-PE mesh in vivo demonstrated inflammation and cell accumulation around the mesh fibers of the PE mesh owing to the foreign body reaction. In contrast, for the CD25-PE mesh, the inflammation reaction was not observed, and sham operation suppressed cell accumulation. In addition, although Tregs were not observed for PE meshes immunochemically stained with FoxP3, Treg accumulation around the CD25-PE mesh was also achieved. Surface modification of implant materials with biomolecules, such as peptides, proteins, and antibodies, can control cell behavior and function [Citation19–22]. Thus, Tregs accumulated around the CD25-PE mesh may inhibit inflammation through immunosuppression.
Finally, we investigated the tumor-suppressive ability of the CD25-PE mesh in tumor-bearing mice. Our results showed that the PE and PAAc-PE meshes caused decreased tumor volumes only on days 6 and 7. It is assumed that the implantation of them induced changes of the tumor surroundings, such as micro-circulation state, capsulation, inflammation, and it is required to clear the mechanism in future. In contrast, CD25-PE resulted in high suppression of tumor growth until 7 days after implantation compared with the other meshes; the tumor volume decreased to 323 mm3, indicating that this mesh showed high tumor suppression ability. We assumed that the immune cells suppressed by Tregs in or around the tumor were activated by capturing Tregs on the CD25-PE mesh. This assumption was supported by the observation that the number of cells around the CD25-PE mesh of tumor-bearing mice was higher than that of healthy mice. In addition, spherical immune cells were observed for the CD25-PE mesh near the tumor, whereas the cells around CD25-PE meshes implanted into healthy mice were spindle-shaped cells, similar to fibroblasts. However, for FoxP3 immunostaining, the Treg ratio among the cells around the CD25-PE mesh was approximately 5%, similar to the CD25-PE ratio in healthy mice. Furthermore, although the tumor growth curves in mice in the CD25-PE mesh group gradually increased over 6 days after implantation, a remarkable increase in tumor volume was observed from days 6 to 7. These results suggest that it may be necessary to consider long-term application of CD25-PE mesh and that additional improvements to the CD25-PE mesh may be required.
5. Conclusion
In this study, we designed and synthesized an anti-CD25 antibody-immobilized PE mesh as an implantable Treg capture device to suppress tumor growth by releasing immune tolerance. The CD25-PE mesh effectively captured Tregs via antigen/antibody interactions in vitro and in vivo. Additionally, transplantation of the antibody-immobilized mesh into tumor-bearing mice suppressed tumor growth. These results suggest that such Treg control may have applications as a novel type of cancer treatment. Further research is needed to facilitate cancer immunotherapy using implantable anti-CD25 antibody-immobilized material as a Treg-capturing device.
Scheme 1. Preparation of the anti-CD25 antibody-immobilized PE mesh
Acknowledgments
We are grateful to Prof. Nishiyama and Dr. Takemoto, Tokyo Institute of Technology for suggestion and discussion for the preparation of the tumor-bearing mice. We would like to thank Editage (www.editage.com) for English language editing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. Nat Partner J Vaciines. 2019;4(7). DOI:https://doi.org/10.1038/s41541-019-0103-y
- Mougel A, Terme M, Tancho C. Therapeutic cancer vaccine and combinations with antiangiogenic therapies and iImmune checkpoint blockade. Front Immunol. 2019;10:467.
- Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281.
- Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–118.
- Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of Treg-mediated T cell suppression. Front Immunol. 2012;3:51.
- Siegler EL, Kim YJ, Wang P. Nanomedicine targeting the tumor microenvironment: therapeutic strategies to inhibit angiogenesis, remodel matrix, and modulate immune responses. J Cell Immunother. 2016;2:69–78.
- Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target?. Cancer Sci. 2019;110:2080–2089.
- Nair VS, Elkord E. Immne checkpoint inhibitors in cancer therapy: a focus on T-recularory cells. Immunol Cell Biol. 2019;96:21–33.
- Sugiyama D, Nishikawa H, Maeda Y, et al.. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci USA. 2013;110:17945–17950.
- Okuno Y, Ymazaki Y, Fukutomi H, et al.. A novel surface modification and immobilization method of anti-CD25 antibody on nonwoven fabric filter removing regulatory T cell selectively. ACS Omega. 2020;5:772–780.
- Ono S, Kimura A, Hiraki S, et al.. Removal of increased circulating CD4+CD25+Foxp3+ regulatory T cells in patients with septic shock using hemoperfusion with polymyxin B-immobilized fibers. Surgery. 2013;153:262–271.
- Martins F, Sofiya L, Sykiotis GP, et al.. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16:563–580.
- Kimura T, Nakamura N, Sasaki N, et al.. Capture and release of target cells using a surface that immobilizes an antibody via desthiobiotin–avidin interaction. Sens Mater. 2016;28:1255–1263.
- Kimura T, Nakamura N, Umeda K, et al.. Capture and release of cells using a temperature-responsive surface that immobilizes an antibody through DNA duplex formation. J Biomater Sci Polym Ed. 2017;28:1172–1182.
- Kimura T, Nakamura N, Hashimoto Y, et al.. Selective cell capture and release using antibody-immobilized polymer-grafted surface. Kobunshi Ronbunshu. 2018;75:155–163.
- Faustino-Rocha A, Oliveira PA, Pinho-Oliveira J, et al.. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim. 2013;42:217–224.
- Salemme V, Centoze G, Cavallo F, et al.. The crosstalk between tumor cells and the immune microenvironment in breast cancer: implications for immunotherapy. Front Oncol. 2021;11:610303.
- Tanaka A, Sakaguchi. Targeting Treg cells in cancer immunotherapy. European J Immunol. 2019;49:1140–1146.
- Morra M. Biomolecular modification of implant surfaces. Expert Rev Med Devices. 2007;4:361–372.
- Kakinoki S, Sakai Y, Fujisato T, et al.. Accelerated tissue integration into porous materials by immobilizing basic fibroblast growth factor using a biologically safe three-step reaction. J Biomed Mater Res Part A. 2015;103:3790–3797.
- Benvenuto P, Neves MAD, Blaszykowski C, et al.. Adlayer-mediated antibody immobilization to stainless steel for potential application to endothelial progenitor cell capture. Langmuir. 2015;31:5423–5431.
- Wu X, Yin T, Tian J, et al.. Distinctive effects of CD34- and CD133-specific antibody-coated stents on re-endothelialization and in-stent restenosis at the early phase of vascular injury. Regener Biomater. 2015;2:87–96.