
1. The unmet need in relapsed refractory head and neck carcinoma
Squamous cell carcinoma of head and neck (SCCHN) is the eighth most common cancer in the UK [Citation1], with a 4-year survival rate of only 56%. Currently, there are approximately 12,200 new cases and 4,000 deaths in the UK each year due to this cancer type [Citation1]. While metastatic disease is the main cause of death attributable to most cancers, loco-regional disease assumes this dubious distinction in patients with SCCHN. About 10% of the patients present with locally advanced disease, rendering them ineligible for any form of conventional therapy. Moreover, 60%-70% of patients who receive treatment with surgery and/or radiation experience disease relapse. Consequently, there is a clear and unmet need for improved treatment options for patients with locally advanced or recurrent SCCHN.
2. The rationale for ErbB targeting in head and neck carcinoma
In SCCHN, the ErbB family of receptor tyrosine kinases (RTK) represents a highly attractive target for novel therapies [Citation2]. The ErbB family comprises four members, namely epidermal growth factor receptor (EGFR or ErbB-1), ErbB-2 (HER2/neu), ErbB-3 and ErbB-4. These receptors provide a molecular network that plays a fundamental role in many biological systems. Individual ErbB molecules bind 2–8 distinct ligands with the exception of the orphan receptor, ErbB2. Signal complexity is diversified by the ability of ErbB RTK to undergo ligand-driven homo- or hetero-dimerization [Citation3]. Although all possible binary ErbB combinations have been detected, ErbB2 is the preferred dimerization partner for all other family members, owing to its constitutively ‘open’ ectodomain [Citation3]. In the adult, ErbB receptors are expressed at low levels in several non-hemopoietic tissues [Citation3]. However, increased synthesis of ErbB family members correlates strongly with the development of several solid tumors, particularly squamous cell carcinomas [Citation4,Citation6].
Overwhelming evidence implicates dysregulated ErbB signaling in the pathogenesis of SCCHN [Citation2]. This tumor represents a classical model of EGF-driven oncogenesis since it strongly over-expresses EGFR in >90% of the cases. Over-expression of EGFR is implicated in resistance to radiotherapy and is a strong prognostic marker for poor survival and metastasis [Citation3,Citation7,Citation9]. Furthermore, since the level of EGFR expression increases with tumor progression, this molecule represents an increasingly appropriate target with disease evolution. Disappointingly, however, clinical data indicate that only a minority of patients with SCCHN benefit from EGFR-targeted therapies [Citation10]. In part, this may result from the frequency with which other ErbB family members are co-expressed, conferring a worsened prognosis [Citation11]. Moreover, in many tumors, therapeutic resistance to ErbB-directed therapies is mediated by upregulated activity of non-targeted family members. In agreement with this, resistance of SCCHN cell lines to EGFR-targeted antibody or small molecule agents has been associated with increased ErbB2/ErbB3 signaling [Citation10,Citation12].
While small molecule inhibitors of ErbB signaling such as gefitinib have been disappointing, treatment with the EGFR-targeted antibody cetuximab has improved overall survival in both early and advanced SCCHN [Citation13]. This suggests that immunological effects such as antibody-dependent cell-mediated cytotoxicity have particular therapeutic impact in this disease. Moreover, prior cetuximab use (and resistance to cetuximab) seems to hamper the efficacy of PD1 immune checkpoint blockade [Citation13]. These considerations provide a justification for the evaluation of cellular approaches such as CAR T-cell immunotherapy, either alone or in combination with PD1 inhibition.
3. T4 Immunotherapy: a panErbB-targeted chimeric antigen receptor
Chimeric antigen receptors (CARs) are tailored fusion molecules, which couple an antigen recognition moiety to a bespoke signaling domain, allowing for HLA-independent retargeting of T-cell cytotoxicity. Immunotherapy using CAR T-cells has achieved unprecedented response rates in patients with refractory B-cell malignancies and myeloma. However, similar clinical success has not been replicated in the setting of solid tumors. Lack of cancer-specific antigens, poor trafficking and penetration of CAR-expressing T-cells into malignant deposits, and a hostile anti-inflammatory, hypoxic and metabolically compromised tumor microenvironment all contribute to this challenge [Citation14].
In an attempt to address these issues, pan-ErbB-targeted ‘T4 immunotherapy’ was developed. T4 immunotherapy describes the co-expression of a second-generation (CD28 + CD3ζ) CAR designated T1E28z with a chimeric cytokine receptor (CCR) termed 4αβ (). Stoichiometric expression of both transgenes is achieved in patient-derived T-cells using a single oncoretroviral vector (SFG) that contains an intervening Thosea asigna ribosomal skip peptide. The T1E28z CAR incorporates the promiscuous ErbB ligand, T1E, placed upstream of CD28 (hinge/transmembrane/endodomain) and CD3ζ endodomain motifs [Citation15]. T1E is a chimeric polypeptide in which the N-terminal seven amino acids from transforming growth factor (TGF)-α has been fused to the C-terminal 48 amino acids of epidermal growth factor (EGF). The T1E28z CAR simultaneously directs T-cell specificity against all EGFR homo- and heterodimers, the ErbB 2/3 heterodimer and all ErbB4 homo- and heterodimers [Citation16]. By this means, T-cells acquire potent cytotoxic activity against a broad range of tumor types, with decreased risk of immune evasion through antigen escape.
Figure 1. T-cell co-expressing 4ab and T1E28z interacting with ErbB dimers expressed on a tumor cell.
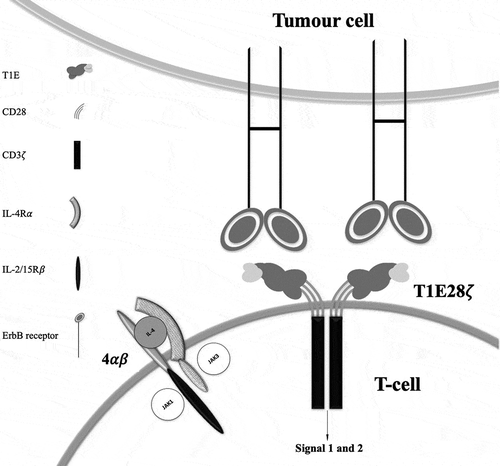
Patients who are candidates for CAR T-cell therapy are often lymphopenic and immunocompromised, which may hinder efficient T-cell transduction using integrating viral vectors. To circumvent this, the 4αβ CCR was engineered by fusing the interleukin (IL)-4 receptor α (IL-4Rα) ectodomain to the transmembrane and endodomain of the common beta chain (βc) subunit incorporated into the receptors for IL-2 and IL-15 (). Whereas IL-4 is normally a poor T-cell mitogen, a potent IL-2/IL-15-like growth signal is delivered by 4αβ, which associates in an IL-4 dependent manner with the common gamma chain (γc) [Citation17]. Use of the 4αβ system provides a robust method to ensure that cell products contain a predominance of genetically modified T-cells [Citation17]. T4-engineered T-cells expand exponentially when cultured with IL-4 over a period of 7–10 days, a process that results in selective CAR T-cell enrichment [Citation17].
4. Predicting and mitigating risk of on-target off-tumor toxicity of T4 immunotherapy
Due to low-level target expression in healthy tissues, risk of on-target off-tumor toxicity is a major theoretical concern associated with ErbB-targeted CAR T-cell immunotherapy. Illustrating this, intravenous (i.v.) administration of 10 billion HER2 re-targeted CAR T-cells to a lymphodepleted patient resulted in the rapid onset of pulmonary toxicity accompanied by cytokine release syndrome (CRS), leading to fatal multi-organ failure [Citation18]. At necropsy, it was concluded that death had resulted from low-level target engagement in the lungs, highlighting the need for careful toxicity testing of other experimental ErbB-targeted immunotherapies.
Preclinical risk assessment of T4 immunotherapy benefitted from the ability of the CAR to recognize mouse ErbB dimers with broadly similar efficiency to their human orthologues [Citation19]. When human T4+ CAR T-cells were administered to immunocompromised SCID Beige mice i.v. or using the intratumoral route, tumor regression was observed in the absence of histopathologic toxicity [Citation19]. However, T-cell dose- and tumor burden-dependent CRS was induced by intraperitoneal (i.p.) administration of T4+ T-cells [Citation19], establishing a mouse model that has since been replicated by other groups. Importantly, intratumoral administration of T4+ T-cells resulted in disease regression and persistence of the T-cells for several days at the site of injection, without CRS or other toxicity [Citation20]. These findings raised the possibility that T4 immunotherapy could be safely administered in man using the intratumoral route.
5. Phase I clinical evaluation of T4 immunotherapy
A Phase I clinical trial was designed to evaluate the safety of intratumoral T4 immunotherapy in patients with locally advanced or recurrent SCCHN (EudraCT number: 2012–001654-25; Clinicaltrials.gov number: NCT01818323) [Citation21]. This single center study has a 3 + 3 dose escalation design, in which fresh cell product is administered directly into the tumor at multiple points within one or more tumor target lesions [Citation21]. Dose escalation in five sequentially recruited cohorts of three patients each has proceeded from an initial dose level of 10 million to a maximum deliverable dose of 1 billion T4+ CAR T-cells. Patients enrolled in these five cohorts did not receive any form of lymphodepleting chemotherapy. No dose-limiting toxicities have been observed in any of 15 patients treated in the dose escalation phase of the study, nor has leakage of CAR T-cells into the circulation been detected. The commonest adverse reactions observed were fever and sustained swelling of the injected target lesion(s). Stable disease was achieved by RECIST (Response evaluation criteria in solid tumors) criteria at 6 weeks post CAR T-cell treatment in 10 of 16 patients. One individual achieved complete remission following subsequent treatment with pembrolizumab and T-VEC, and which remains durable to the time of writing (>3 years). A full manuscript providing further detail of the dose escalation phase of the trial is currently in preparation. Initially, it had been proposed to administer low dose oral cyclophosphamide to a sixth cohort in order to achieve the semi-selective depletion of regulatory T-cells [Citation21]. More recently, the protocol has been amended to substitute this with lymphodepleting chemotherapy using fludarabine (25 mg/m2) and cyclophosphamide (250 mg/m2), each administered i.v. for 3 days.
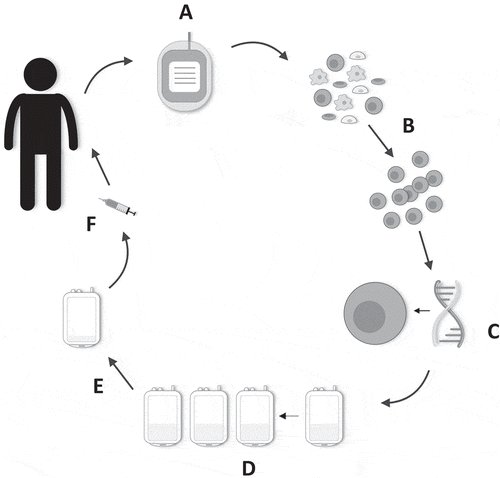
Manufacture of T4 immunotherapy is distinct to other approaches in common use in CAR T-cell clinical trials (). Eligible patients are often elderly with substantial disease and other co-morbidities. Therefore, a blood draw, rather than a more invasive leukapheresis procedure, is the chosen starting material from which to manufacture CAR T-cell batches. Using as little as 40–120 mL blood, between 2 and 7.5 billion CAR T-cells can be manufactured in 2 weeks, using IL-4 as the sole cytokine support for the cells post retroviral gene transfer. In contrast to other manufacturing processes, the addition of IL-4 provides a strong and selective growth signal via the 4αβ receptor, allowing preferential expansion and enrichment of T4+ T-cells ex vivo. Moreover, T-cells retain a type 1 polarized profile and comparable differentiation status to cells expanded using traditional IL-2-based methods [Citation17]. No batch failures have occurred in 16 sequentially treated patients, despite lymphopenia in most cases.
Figure 2. Manufacture of T4 immunotherapy. (a) 40–120 mL of whole blood is harvested from the patient. (b) Patient-derived peripheral blood mononuclear cells are isolated and activated. (c) Gene transfer is performed after 48 h. (d) Preferential expansion and enrichment of T4+ T-cells ex vivo is achieved with the addition of IL-4 to the cell culture bags. (e) Directly prior to administration, the cell culture is volume-reduced. (f) Cells are contained in a final 1–4 mL dosing volume and drawn up into a syringe. The dose is administered intratumorally using ultrasound guidance where required.
6. Expert Opinion
T4 immunotherapy has thus far proven to be safe when administered intratumorally in patients with SCCHN at a dose of up to 1 × 109 cells. Our experience also illustrates the fact that CAR T-cells can be reliably manufactured from a blood sample, obviating the need for leukapheresis. The absence of on-target off-tumor toxicity observed to date correlates with the fact that T-cells cannot be detected in the systemic circulation. Disease stabilization has been achieved in 10 of 16 patients. Nonetheless, it is acknowledged that even with direct delivery of T-cells to the site of disease, there is a need to consider combinatorial strategies that may help to mitigate some of the obstacles presented by the tumor microenvironment. Illustrating this, chemotherapy has been shown to further sensitize disease to T4 immunotherapy, even when agents are used against which tumors are already completely resistant [Citation22]. Alternatively, it can be envisioned that combination therapy with radiation and/or immune checkpoint blockade may prove useful in this setting. In this regard, it should be noted that neoantigens generated using radiation or chemotherapy have distinct half-lives that may sustain immunotherapeutic response [Citation13,Citation23,Citation25]. Moreover, conditioning of patients with lymphodepleting chemotherapy may prove useful in facilitating the persistence of CAR T-cells at the site of delivery. We are currently conditioning patients who receive T4 immunotherapy with fludarabine and cyclophosphamide and have plans to add PD1 inhibition in subsequent cohorts to be recruited.
Building on this clinical experience, additional studies may warrant consideration in which T4 immunotherapy is administered using the intracavity route in patients with selected solid tumor types. In preclinical models, we have demonstrated satisfactory efficacy and safety of this intervention in mouse models of epithelial ovarian cancer [Citation22] and malignant pleural mesothelioma [Citation26]. In each case, regional delivery has been used to maximize T-cell delivery to the site of disease and restrict systemic T-cell absorption.
Declaration of interest
J Maher is chief scientific officer, shareholder and scientific founder of Leucid Bio, which is a spinout company focused on development of cellular therapeutic agents. D Larcombe-Young is a consultant to Leucid Bio. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- UK CR Head and Neck Cancer Statistics. 2017; Available from: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/head-and-neck-cancers#heading-Zero.
- Byeon HK, Ku M, Yang J. Beyond EGFR inhibition: multilateral combat strategies to stop the progression of head and neck cancer. Exp Mol Med. 2019;51(1):1–14.
- Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7(7):505–516.
- Morgan S, Grandis JR. ErbB receptors in the biology and pathology of the aerodigestive tract. Exp Cell Res. 2009;315(4):572–582.
- Olayioye MA., R.M. Neve, H.A. Lane, et al. The ErbB signaling network: receptor heterodimerization in development and cancer. Embo J. 2000;19(13):3159–3167..
- Rogers SJ, Harrington KJ, Rhys-Evans P, et al. Biological significance of c-erbB family oncogenes in head and neck cancer. Cancer Metastasis Rev. 2005;24(1):47–69..
- Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21(2):177–184.
- Arteaga CL. ErbB-targeted therapeutic approaches in human cancer. Exp Cell Res. 2003;284(1):122–130.
- Wei Q, Sheng L, Shui Y, et al. EGFR, HER2, and HER3 expression in laryngeal primary tumors and corresponding metastases. Ann Surg Oncol. 2008;15(4):1193–1201..
- Erjala, K., M. Sundvall, T.T. Junttila, et al. Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin Cancer Res. 2006;12(13):4103–4111..
- Sacco AG, Worden FP. Molecularly targeted therapy for the treatment of head and neck cancer: a review of the ErbB family inhibitors. Onco Targets Ther. 2016;9:1927–1943.
- Wheeler DL, Huang S, Kruser TJ, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27(28):3944–3956..
- Ferris RL, Licitra L, Fayette J, et al. Nivolumab in patients with recurrent or metastatic squamous cell carcinoma of the head and neck: efficacy and safety in checkmate 141 by prior cetuximab use. Clin Cancer Res. 2019;25(17):5221–5230..
- Kosti P, Maher J, Arnold JN. Perspectives on chimeric antigen receptor T-cell immunotherapy for solid tumors. Front Immunol. 2018;9:1104.
- Maher J, Brentjens RJ, Gunset G, et al. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. 2002;20(1):70–75..
- Davies, D.M., J. Foster, S.J.C. Van Der Stegen, et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Molecular medicine (Cambridge. Mass.). 2012;18(1):565–576.
- Wilkie S, Burbridge SE, Chiapero-Stanke L, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. 2010;285(33):25538–25544.
- Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851..
- van der Stegen SJC, Davies DM, Wilkie S, et al. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: identifying a window of therapeutic opportunity? J Immunol. 2013;191(9):4589–4598..
- Parente-Pereira AC, Burnet J, Ellison D, et al. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. J Clin Immunol. 2011;31(4):710–718..
- van Schalkwyk, M.C., S.E. Papa, J.P. Jeannon, et al. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum Gene Ther Clin Dev. 2013;24(3):134–142.
- Parente-Pereira, A.C., L.M. Whilding, N. Brewig, et al. Synergistic chemoimmunotherapy of epithelial ovarian cancer using erBB-retargeted T cells combined with carboplatin. J Immunol. 2013;191(5):2437–2445.
- Burtness B, Harrington KJ, Greil R, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394(10212):1915–1928..
- Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21–28..
- Biau J, Bourhis J. Combining immunotherapy and radiotherapy in head and neck squamous cell cancers: which perspectives? Curr Opin Oncol. 2020;32(3):196–202.
- Klampatsa, A., D.Y. Achkova, D.M. Davies, et al. Intracavitary ‘T4 immunotherapy’ of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. 2017;393:52–59.