
ABSTRACT
Introduction: Mesothelin (MSLN) is a tumor differentiation antigen normally restricted to the body’s mesothelial surfaces, but significantly overexpressed in a broad range of solid tumors. For this reason, MSLN has emerged as an important target for the development of novel immunotherapies. This review focuses on anti-MSLN chimeric antigen receptor (CAR) T cell immunotherapy approaches.
Areas covered: A brief overview of MSLN as a therapeutic target and existing anti-MSLN antibody-based drugs and vaccines is provided. A detailed account of anti-MSLN CAR-T cell approaches utilized in preclinical models is presented. Finally, a comprehensive summary of currently ongoing and completed anti-MSLN CAR-T cell clinical trials is discussed.
Expert opinion: Initial trials using anti-MSLN CAR-T cells have been safe, but efficacy has been limited. Employing regional routes of delivery, introducing novel modifications leading to enhanced tumor infiltration and persistence, and improved safety profiles and combining anti-MSLN CAR-T cells with standard therapies, could render them more efficacious in the treatment of solid malignancies.
1. Mesothelin
Mesothelin (MSLN) is a cell surface-bound, glycosylphosphatidylinositol (GPI) anchored protein, whose normal expression is restricted to the mesothelial cells of the pleura, pericardium, peritoneum, and tunica vaginalis in men, whereas very low MSLN expression (trace amounts) has also been observed in the epithelial lining of the ovaries, fallopian tubes, and rete testis [Citation1].
In contrast to its limited expression on mesothelial cells under normal physiological conditions, MSLN has been found to be overexpressed in a plethora of cancers [Citation1–4], including malignant mesothelioma [Citation4–9], ovarian cancer [Citation5,Citation9–13], breast cancer (and more specifically triple-negative breast cancer (TNBC)) [Citation14–16], pancreatic cancer [Citation9,Citation17–20], lung cancer [Citation21–23], gastric cancer [Citation24–28], endometrial cancer [Citation12], cervical cancer [Citation29], biliary cancer [Citation30–32], uterine serous carcinoma [Citation33], cholangiocarcinoma [Citation34,Citation35] and pediatric acute myeloid leukemia [Citation36]. Increased MSLN expression has been associated with a poorer prognosis for patients with TNBC [Citation14,Citation15], ovarian cancer [Citation10], lung adenocarcinoma [Citation22,Citation23], cholangiocarcinoma [Citation30,Citation35], and pancreatic adenocarcinoma [Citation17,Citation19].
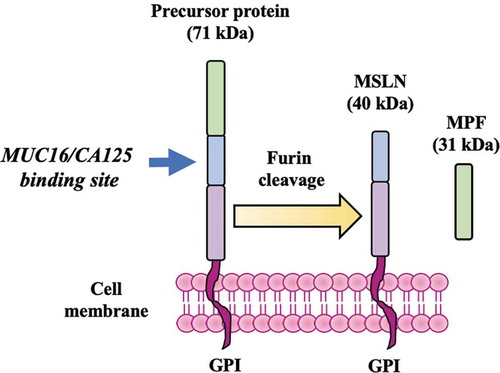
MSLN and its identity as a tumor-associated antigen (TAA) were discovered over 20 years ago when Chang et al. at the National Cancer Institute (NCI, Bethesda, MD) initially isolated (via mice immunization) and characterized a monoclonal antibody named K1, reactive with human ovarian carcinoma cells as well as with the normal mesothelium [Citation37]. A few years later, the same researchers used molecular cloning to identify and isolate the cDNA and protein recognized by the K1 antibody. They named the protein ‘mesothelin’ as it is normally produced by mesothelial cells and provided early evidence that this protein could be involved in cellular adhesion [Citation5]. The human MSLN gene is located at chromosome 16 and contains a 1884-base pair open reading frame and a total of 15 exons [Citation38]. The human MSLN gene was shown to encode a ~ 71-kDa precursor protein consisting of ~628 amino acids [Citation5]. The precursor is cleaved (via furin cleavage) at Arginine 295 (Arg295) into two products; a ~ 31-kDa N-terminal soluble protein known as mature megakaryocyte potentiating factor (MPF) and a ~ 40-kDa GPI-anchored mesothelin protein, bound on the cell surface [Citation5,Citation39,Citation40] (). In mesothelioma and ovarian cancer, the levels of membrane-bound MSLN that is released in the serum have been found to be elevated and serum MSLN is considered to be a tumor marker for mesothelioma and ovarian cancer patients41, 42.
Figure 1. MSLN and MPF. A precursor protein of ~71 kDa is divided via furan cleavage into a ~ 40 kDa GPI-anchored, membrane-bound protein which contains a MUC16/CA125 binding site known as mesothelin (MSLN), and a soluble protein of ~31 kDa known as megakaryocyte-potentiating factor (MPF). MSLN expression is normally restricted on the surface of mesothelial cells but it is also overexpressed in a broad range of cancers
The physiological/biological function of MSLN remains uncertain, since the lack of MSLN in an MSLN knockout mouse model did not affect the development, growth, or reproduction of those mice when they were compared to homozygous and heterozygous wild-type mice [Citation41]. Nevertheless, MSLN is considered to be involved in several mechanisms of cancer pathogenesis. In epithelial ovarian carcinoma, in patients who exhibited higher levels of MSLN mRNA expression in surgery-resected ovarian cancer tissues, showed resistance to chemotherapy with platinum and cyclophosphamide when compared to chemo-sensitive patients who expressed lower MSLN levels [Citation42]. MSLN has also been found to bind with high affinity to the surface mucin MUC16 (or CA125) [Citation43]. This binding could potentially mediate adhesion of ovarian cancer cells to the mesothelial cells residing in the peritoneal cavity and promote intraperitoneal ovarian cancer metastasis [Citation44].
MSLN is further involved in tumor progression and cell survival and proliferation in pancreatic cancer in vitro [Citation45] and in vivo [Citation46]. MSLN-overexpressing pancreatic cells were found to produce higher levels of interleukin-6 (IL-6) via NF-κB constitutive activation, further stimulating cancer cell proliferation in an autocrine manner [Citation47]. Furthermore, MSLN-overexpression in pancreatic cancer cell lines has been associated with resistance to tumor necrosis factor-alpha (TNF-α)-mediated apoptosis [Citation48], anoikis-induced apoptosis [Citation49] and resistance to paclitaxel treatment [Citation50].
Over the years, evidence has accumulated with regards to the importance of mesothelin as a TAA overexpressed in almost one-third of human cancers. The fact that MSLN is expressed primarily on dispensable tissues under normal conditions and is abnormally expressed in various tumors, makes it a highly appealing target when it comes to the development of novel anti-tumor therapies with a low risk of ‘on target-off tumor’ toxicity. For these reasons, various types of anti-mesothelin therapies have been developed, including; antibodies, antibody-drug conjugates (ADCs), immunotoxins, cancer vaccines, and chimeric antigen receptor (CAR)-T cell immunotherapies. We will briefly review these approaches, but will focus on an overview of experimental models and clinical trials for anti-MSLN CAR-T cell therapies.
1.1. Overview of anti-MSLN antibody-based and cancer vaccine therapies
Along with CAR-T cell immunotherapies, a variety of other types of novel anti-MSLN therapies have been developed over the last two decades such as antibody-based therapies (monoclonal antibodies, ADCs, immunotoxins) and vaccines.
The action of antibody-based drugs involves several mechanisms; antibody-mediated neutralization of the TAA leading to complement-mediated killing of the tumor cell, antibody-dependent cellular cytotoxicity (ADCC) via the NK cells leading to tumor cell death, antibody-dependent cellular phagocytosis (ADCP) of the tumor cells via monocyte-derived macrophages, and finally, hampering of the tumor cells’ proliferative capacity and induction of apoptosis via TAA targeting with antibodies conjugated to toxins or inhibitors.
1.2. Anti-MSLN antibodies, immunotoxins and antibody-drug conjugates
1.2.1. SS1 antibody and SS1P immunotoxin
SS1, is a murine-derived anti-MSLN antibody (stable single-chain Fv), able to recognize MSLN with higher affinity than the K1 monoclonal antibody [Citation51,Citation52]. SS1 fusion to Pseudomonas exotoxin A (PE38) led to the development of the recombinant immunotoxin (RIT) SS1P, able to be internalized by the targeted cells and induce apoptosis [Citation53]. SS1P has been tested in several clinical trials for advanced cancers such as mesothelioma, pancreatic and ovarian cancer (NCT01445392, NCT01362790, NCT01051934, NCT00066651, NCT00006981). These studies have either been completed or terminated, most of them at Phase I. SS1P did not lead to pericardial toxicity, but was found to be immunogenic in clinical trial NCT00006981; neutralizing antibodies to SS1P were also found to be produced [Citation54].
When SS1P was tested in combination therapies (pentostatin and cyclophosphamide for patients with chemotherapy-refractory mesothelioma; NCT01362790 and pemetrexed and cisplatin for chemotherapy-naïve MPM patients; NCT01445392), SS1P- and combination therapy-related toxicities were still observed, however partial responses and stable disease were observed and pentostatin and cyclophosphamide were able to delay the formation of SS1P neutralizing antibodies.
1.2.2. LMB-100/RG7787
LMB-100/RG7787 is a humanized antibody based on SS1 but is conjugated to a less immunogenic pseudomonas exotoxin, PE24, in an attempt to induce lower toxicity levels [Citation55].
1.2.3. Amatuximab (MORAb-009)
Since the development of SS1 antibody, several other SS1-based drugs have been developed, including humanized antibodies. Amatuximab (MORAb-009) is a chimeric anti-MSLN humanized monoclonal antibody. It consists of the SS1 single-chain fragment variable (scFv) domain fused to human IgG1/κ [Citation56]. A number of Phase I and II clinical trials for amatuximab, now completed or terminated, have taken place (NCT00738582, NCT02357147, NCT01413451, NCT01018784, NCT00325494, NCT00570713, NCT01521325). Two of these studies, NCT00738582 for patients with pleural mesothelioma and NCT00570713 for pancreatic cancer patients, reported no severe drug hypersensitivity events, and amatuximab was found to be well tolerated, no complete or partial responses were observed for the majority of patients and approximately half of the patients had stable disease [Citation57].
When pemetrexed and cisplatin were combined with amatuximab (NCT00738582), severe adverse effects occurred but also more partial responses were observed and more patients exhibited stable disease [Citation58]. Amatuximab was found to block interaction between MSLN and MUC16 (CA125), and this could prevent metastasis in ovarian cancer and mesotheliomas [Citation59]. In MPM patients, an amatuximab pharmacokinetics and exposure-response study (NCT02357147) showed that higher exposure to amatuximab combined with chemotherapy was associated with longer overall survival [Citation60].
1.2.4. Anetumab ravtasine (BAY94-9343)
Anetumab ravtasine or BAY94–9343, a novel drug-conjugated antibody, is a human anti-MSLN antibody conjugated to the maytansinoid tubulin inhibitor DM4 which disrupts the division, growth, and proliferation of tumor cells [Citation61,Citation62]. Anetumab ravtasine binds MSLN with strong affinity and high selectivity, leading to efficient antigen internalization. Several Phase I or II clinical trials for anetumab ravtasine have now been completed or terminated (NCT02696642, NCT02751918, NCT03455556, NCT02824042, NCT03023722, NCT02839681, NCT02639091, NCT02610140, NCT02485119), but four Phase I or II studies (NCT03126630, NCT03102320, NCT03587311, NCT03816358) are currently recruiting subjects whereas a Phase II study is currently enrolling by invitation (NCT03926143). Results from the first open-label multi-center Phase I dose-escalation and expansion study (NCT01439152) for anetumab ravtasine in patients with multiple solid tumor types (including MPM and ovarian cancer) have now been published, reporting that this antibody showed a manageable safety profile and favorable pharmacokinetics, it was well tolerated and had an encouraging preliminary clinical activity, thus paving the way for further Phase II studies [Citation63].
1.2.5. MMAE, BMS-9861 and BAY2287411
Other important ADCs include [Citation58];, a humanized anti-MSLN antibody conjugated to the anti-mitotic agent monomethyl auristatin E (MMAE) which has been administered to pancreatic and ovarian cancer patients in a Phase I clinical trial [Citation9,Citation64], BMS-986,148 conjugated to the alkylating agent duocarmycin (MED2460) which has been tested in a clinical trial for patients with mesothelioma, NSCLC, ovarian, pancreatic and gastric cancer [Citation56], and BAY2287411, a thorium-227 labeled antibody-chelator conjugate that is being tested in a Phase I clinical trial for patient with PDAC, serous ovarian cancer, and epithelioid mesothelioma (NCT03507452).
1.2.6. HPN536
HPN536 is the most recently developed anti-MSLN antibody currently being tested in a Phase I/II clinical trial (NCT03872206) for patients with advanced MSLN-expressing cancers. The novelty of this agent lies in the fact that it is a tri-specific antibody, which apart from the MSLN recognizing domain also contains an anti-albumin domain that confers HPN536 an extended half-life, as well as an anti-CD3ε scFv that mediates T cell recruitment and killing of the MSLN-expressing tumor cells.
1.3. Anti-MSLN cancer vaccines
Along with antibody-based drugs, anti-MSLN cancer vaccines have been developed, aiming to induce tumor-specific immune system activation leading to selective killing of the tumor cells. Two bacteria-based vaccines have been tested in clinical trials; CRS-207 that contained a live-attenuated Listeria monocytogenes strain and was engineered to express MSLN [Citation65] and JNJ-64,041,757 (ADU-214), containing a live-attenuated, double-deleted Listeria monocytogenes strain and expressing MSLN. CRS-207 was tested in patients with PDAC, mesothelioma, NSCLC and ovarian cancer and was found to be well tolerated and to induce immune activation [Citation65]. CRS-207 in combination with cyclophosphamide and another vaccine for pancreatic cancer (GVAX, engineered to secrete GM-CSF), was found to confer longer overall survival to patients with metastatic pancreatic adenocarcinoma compared to GVAX and cyclophosphamide alone [Citation65]. JNJ-64,041,757 was used to treat NSCLC patients alone or in combination with nivolumab in two clinical trials (NCT03371381 and NCT02592967) which were, however, terminated due to lack of a favorable clinical effect. Despite the generally disappointing clinical results and small numbers of clinical trials, neoantigen DNA vaccines are being tested and further preclinical efforts are being made with regards to the development of other types of vaccines, such as Meso-VAX in combination with adeno-associated virus (AAV)-IL-12 and CTGF/MSLN DNA vaccine [Citation66,Citation67].
A more detailed overview of anti-MSLN antibody-based drugs and vaccines can be found in a recent review by Lv J. & Li P [Citation68].
2. MSLN CARs: design and preclinical studies
2.1. Four Generations of MSLN CARs
A CAR consists of an extracellular antibody-derived single-chain fragment variable (scFv) domain (fusion protein containing a heavy and light Ig chain connected by a short linker) that recognizes the target antigen, followed by a hinge (or spacer) region (usually CD8α-derived) that improves the expansion of CAR-T cells [Citation69] and connects the scFv to a transmembrane domain (usually CD8α-derived) which, in turn, is connected to an intracellular domain (derived from CD3z and costimulatory molecules), responsible for T cell activation signaling [Citation70]. A number of MSLN-specific scFv’s have been studied, in CAR constructs of all CAR generations, depending on their intracellular costimulatory domain repertoire ().
Figure 2. The CAR generations. The first-generation CARs consisted of an extracellular scFv region (VL connected to VH via a linker) responsible for the recognition of the target tumor antigen, connected via a spacer or hinge to a transmembrane domain (both spacer/hinge and transmembrane domain usually CD8?-derived), which finally led to an intracellular CD3? domain responsible for CAR-T cell activation (signal 1). Along with CD3?, an intracellular co-stimulatory domain (CD28 or 4–1BB; signal 2) was added to generate the 2nd generation CAR-T cells and a second co-stimulatory domain (CD28, 4–1BB, OX40, DAP10 or TLR2) was included to generate the 3rd generation CAR-T cells. 4th generation CAR-T cells consist of CD3? along with co-stimulatory domain 1 but are also engineered to express and secrete an inflammatory cytokine such as IL-12 (but they can also secrete an inflammatory chemokine or an antibody targeting immune checkpoint inhibitors) which can modify the tumor microenvironment. Finally, 2nd generation CARs may contain, apart from the intracellular CD3? (signal 1) and co-stimulatory domain 1 (signal 2), an IL-1 R? domain which mediates JAK-STAT signaling (signal 3), providing the third signal for full T cell activation
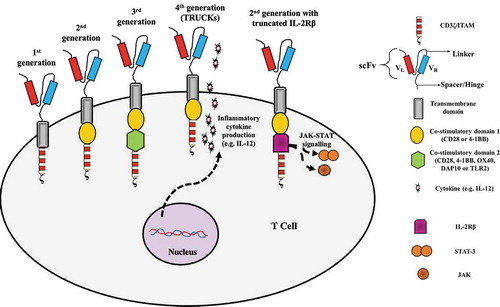
First-generation CARs consist only of a CD3ζ domain bearing the immunoreceptor tyrosine-based activation motif (ITAM) that is able to induce T cell activation and cytotoxicity, but do not maintain long-term proliferative capacity or sufficient cytotoxicity levels [Citation70–72].
Second-generation CARs that add co-stimulatory domains such as CD28, 4–1BB, or OX40 along with CD3z, lead to amplified activation, increased persistence, and enhanced cytotoxicity of CAR-T cells [Citation15,Citation71,Citation73–77] (). This was first demonstrated in an in vivo model using NSG (NOD/scid/IL2rγ(-/-)) mice bearing large pre-established mesothelioma tumor xenografts [Citation74]. Intravenously injected T cells lentivirally transduced with anti-MSLN (SS1 scFv) CD3ζ/CD28/4-1BB CARs, exhibited good persistence and multifactorial cytokine secretion leading to striking anti-tumor activity. Intratumoral injection of the same CAR-T cells led to a more rapid reduction in tumor mass [Citation76]. Second-generation anti-MSLN CAR-T cells have been widely used. In an ovarian cancer murine model using NSG mice, intraperitoneally injected anti-human MSLN mRNA electroporated CAR-T cells (CARMA-hMeso; CD3ζ/4-1BB) were able to induce dose-dependent suppression of tumor growth and conferred improved mice survival [Citation78]. A study using MSLN-expressing (SiHa) and non-expressing (Caski) human cervical squamous carcinoma cells, showed that lentivirally transduced anti-human MSLN CAR-T cells (‘MESO CAR-T’; CD3ζ/4-1BB) were able to specifically kill the SiHa cells and produced higher levels of pro-inflammatory cytokines in the presence of the SiHa cells [Citation29]. In addition, when SiHa cells were implanted into NSG mice, MESO CAR-T tumor-killing activity after two consecutive intratumoral injections was superior to that of control anti-MSLN CAR-T cells that did not include the 4–1BB co-stimulatory domain [Citation29].
Third-generation CARs consist of two co-stimulatory domains (such as CD28, 4–1BB, OX40, DNAX-activating protein 10 (DAP10), Toll-like receptor-2 (TLR2)) alongside CD3ζ [Citation76,Citation79] (). Lentivirally transduced anti-MSLN CD3ζ/CD28/TLR2 CAR-T cells were found to have superior killing capacity of MSLN-expressing human lung cancer cells compared to CD3ζ/CD28 CAR-T cells lacking TLR2 co-stimulation and to produce higher levels of IL-2, IFN-γ, and GM-CSF and similarly, the TLR2-containing CAR-T cells were able to more efficiently inhibit the growth of the MSLN-expressing lung cancer cells xenografted on NSI mice (NOD/scid/IL2rγ(-/-)) [Citation80]. Similarly, anti-MSLN CD3ζ/CD28 CARs in which DAP10 was also incorporated led to the generation of CAR-T cells with superior anti-tumor activity against MSLN-expressing lung cancer cell lines as well as in xenografted NSI mice [Citation81]. When the same anti-MSLN CAR mRNA constructs were electroporated into autologous T cells from tumor mesothelioma patients, multiple injections of these CAR-T cells were able to promote tumor regression in NSI mice bearing matching patient tumor xenografts [Citation82].
Fourth-generation CARs, or TRUCKs (T cells redirected for universal cytokine-mediated killing) are most often second-generation CARs (but can also be third-generation CARs) engineered to secrete immunomodulatory molecules such as interleukins (IL-12, IL-15, IL-7, IL-21), chemokines (CCL19) or immune checkpoint inhibitors such as antibodies against the programmed cell death protein 1 (PD-1) [Citation70,Citation83–86] (). Anti-MSLN TRUCKs have been found to exhibit a favorable anti-tumor effect. In a murine PDAC model expressing MSLN, intravenously injected anti-mouse CD3ζ/CD28/4-1BB MSLN CAR-T cells (mouse MSLN is 70% similar to the human MSLN) which were also expressing IL-7 and CCL19 were found to have an improved survival in the tumor site and exhibited superior anti-tumor activity compared to conventional CAR-T cells; they led to complete tumor regression, increased mouse survival and promoted increased infiltration of the hosts’ dendritic and T cells in the tumor sites [Citation83].
Another modification of CARs is being explored recently, based on the second-generation CARs (including a CD3ζ and CD28 domain) to which a truncated IL-2 receptor β chain which contains a binding site for the transcription factor STAT-3 is added [Citation87,Citation88] (). This addition to provide a cytokine (IL-2)-driven JAK-STAT signaling which, together with the TCR signaling and CD28 co-stimulation, provides all three signals for full T cell activation and improves survival and proliferation [Citation87,Citation88]. The CAR-T cells employ a variety of methods to exert their anti-tumor activity, including perforin and granzyme secretion, activation of the Fas/Fas Ligand and TRAIL pathway and secretion of pro-inflammatory cytokines such as IFN-γ, TNF-α, IL-12, and IL-18 [Citation88,Citation89].
2.2. Engineering approaches to MSLN CARs
The most widely and commonly used method for efficient T cell engineering for CAR-T cell generation is via lentiviral transduction, which allows for permanent and stable CAR expression although a potential danger for insertional mutagenesis exists [Citation70,Citation90–92]. For this reason, improved lentiviral vectors or non-viral methods are also being developed and tested. Bi-component vector systems such as the Sleeping Beauty and the piggyBac transposon systems have been efficiently used for non-viral for CAR-T cell engineering via electroporation, leading to stable, nonrandom integration [Citation91,Citation93,Citation94]. Second-generation CD3ζ/4-1BB ‘mesoCAR-T cells’ engineered with the piggyBac transposon system, were shown to exert significant cytotoxic activity against MSLN-expressing bile duct carcinoma (BDC) human cell lines and to significantly suppress tumor xenograft growth after intratumoral injection in NOD/SCID mice previously transplanted with MSLN-expressing human BDC cells, in contrast to control T cells [Citation95]. Similarly, piggyBac-modified CD3ζ/CD28 CAR-T cells exhibited increased IFN-γ production and a robust killing effect against both MSLN-expressing human PDAC cells in vitro and higher IFN-γ levels as well as suppressed tumor growth in PDAC xenograft NSG mouse models [Citation96]. A more recent study utilized the PiggyBac system to construct meso1 and meso3 CD3ζ/CD28 CAR-T cells; meso1 CAR recognizes the membrane-distal region (region I) of MSLN whereas meso3 CAR recognizes the membrane-proximal region (region III) of MSLN. The meso3 CAR was found to have superior performance compared to meso1 CAR in gastric and ovarian cancer in vivo models [Citation97]. There is also interest in viral-based targeted integration into the T cell receptor locus [Citation98]. Additionally, RNA electroporation methods have been used to generate CAR-T cells. This method leads to transient CAR expression due to eventual degradation of the CAR mRNA, which is a safety advantage, but requires multiple cycles of CAR-T cell injections [Citation82,Citation91]. Despite the lack of long-term persistence of the transiently expressed CARs, this approach can aid toward minimizing the risks of ‘on-target/off-tumor’ toxicities.
2.3. MSLN CAR Strategies to overcome solid tumor limitations
Unfortunately, CAR-T cell therapeutic approaches against solid tumors have had limited success. This is likely due to a number of factors.
For most solid tumors, the TAAs that have been used as potential therapeutic targets are not exclusively expressed in malignant tissues, but are also expressed in low levels in normal healthy tissues – this is the case for MSLN-, increasing the danger for CAR-T cell recognition and subsequent attack and damage of healthy tissues, and effect known as ‘on-target/off-tumor toxicity’ [Citation99]. ‘On-target/off-tumor toxicity’ events often occur in gastrointestinal, pulmonary, and hematological tissues and can highly vary in severity from manageable manifestations to severe toxicity and death [Citation100]. To prevent the detrimental effects of normal tissue antigen recognition, trans-signaling CAR-T cells have been experimentally tested. The rationale behind this approach is that dissociation of the activation and co-stimulation signals in two different CAR constructs, each of them targeting a different TAA, could lead to generation of CAR-T cells with higher tumor specificity, requiring tumor cell expression of both TAAs to achieve full activation and thus, sparing the healthy tissues which might lowly express only one of the TAAs. In a xenograft NSG mouse model of ovarian cancer, an anti-MSLN CD3ζ CAR providing signal 1 and an anti-folate receptor-alpha (FRα) CD28 CAR providing the co-stimulatory signal 2 were lentivirally transduced in primary human T cells; the trans-signaling CAR-T cells generated exhibited superior anti-tumor activity and increased persistence in vivo, similar to that of conventional second-generation CAR-T cells [Citation72]. Importantly though, the dual specificity trans-signaling CAR-T cells did not exhibit potent activity against MSLN-only expressing cells in vivo, in contrast to conventional second-generation anti-MSLN CD3ζ/CD28 CAR-T cells [Citation72]. In a more recent study, targeting MSLN and carcinoembryonic antigen (CEA) using dual CAR constructs (CEA/CD3ζ and MSLN/4-1BB) led to high in vivo anti-tumor activity whilst sparing the cognate healthy tissues [Citation101]. Thus, experimental evidence suggests that dual CARs could more precisely target and kill tumor cells, reducing the risk of ‘on-target/off-tumor’ effects.
Interpatient and intratumoral genetic heterogeneity, tumor antigen escape, inefficient trafficking, poor migration, and inadequate infiltration of the CAR-T cells to the tumor site, and a highly immunosuppressive and hostile tumor microenvironment (hypoxia, suppressive immune cells, immunosuppressive cytokines, immune checkpoint molecules, abnormal tumor vascularization), are all significant obstacles in the development of robust and efficient CAR-T cell therapies [Citation99]. Vigorous efforts are being made to overcome those obstacles and novel CAR constructs and different modes of CAR-T cell delivery (regional vs intravenous/systemic delivery) are currently being tested to determine optimal CAR-T cell therapy conditions.
One reason for inefficient CAR-T cell migration and homing to the tumor is the lack of chemokine receptor expression by the CAR-T cells that matches the chemokines expressed by the tumor cells. CCL2 is one of the most highly and uniformly expressed and secreted chemokines in MPM, but its corresponding receptor, CCR2, was found to be lowly expressed in CD3ζ/4-1BB mesoCAR-T cells [Citation74]. Moon et al. lentivirally transduced mesoCAR-T cells with CCR2b, the most dominant CCR2 isoform, and the CCR2b mesoCAR-T cells were intravenously injected in NSG mice bearing subcutaneous tumors of human MPM naturally expressing MSLN (M108) [Citation74]. The CCR2b CAR-T cells were able to infiltrate the tumor site and suppress tumor growth in a significantly more robust manner compared to mesoCAR-T cells without CCR2b expression [Citation74]. Superior chemotactic function, migratory properties and in vitro cytotoxic killing of M108 cells was also observed for the CCR2b mesoCAR-T cells [Citation74].
The presence of immunosuppressive mediators in the tumor microenvironment can also significantly hamper the activity of CAR-T cells. In pancreatic cancer patients, IL-10 has been shown to suppress MSLN-specific CD4+ and CD8 + T cell responses [Citation102]. In a recent in vitro study, lentivirally transduced anti-human MSLN CD3ζ/CD28/4-1BB CAR-T cells were co-cultured with human pancreatic cancer cells, in human pancreatic cancer cell conditioned media, with or without the presence IL-10 blocking antibody [Citation103]. In the absence of IL-10 blockade, IFN-γ and Granzyme B secretion by the CAR-T cells, as well as their anti-tumor cytotoxic activity, was significantly suppressed compared to conditioned media-free co-cultures of CAR-T cells and pancreatic cells. This suppressive effect, however, was significantly reversed when the CAR-T cells and pancreatic cells were co-cultured in conditioned media including IL-10 blocking antibody [Citation103]. Newick et al addressed CD3-ζ chain inactivation caused by protein kinase A (PKA), a protein activated by prostaglandin E2 (PGE2) and adenosine in the tumor microenvironment, by generating MSLN CAR T cells expressing a small peptide called the ‘regulatory subunit I anchoring disruptor’ (RIAD) that blunts the negative effects of PKA. The resulting MSLN CAR-RIAD T cells showed better efficacy than conventional MSLN CAR T cells both in vitro and in vivo and improved trafficking to the tumor site in vivo [Citation104].
Adenosine is an important immunosuppressive metabolite of the tumor microenvironment that through binding to its cognate receptor A2aR expressed by activated T cells leads to blockade of T cell anti-tumor activity [Citation105]. For this reason, a recent study tested the activity of a fully human CD3ζ/4-1ΒΒ anti-MSLN CAR-T cell in combination with an A2aR knockdown approach via addition of anti-A2aR shRNA sequences in the CAR vector or in combination with an A2aR pharmacological antagonist [Citation105]. The anti-MSLN CAR-T cells containing the anti-A2aR shRNA sequences were protected from the immunosuppressive and inhibitory effects of adenosine signaling and were able to exert their proliferative, cytokine-producing, and cytotoxic functions but the pharmacological inhibition of A2aR, although able to protect the proliferative and cytokine-producing functions of the CAR-T cells, failed to rescue their cytotoxic ability [Citation105].
Within the solid tumor microenvironment, upregulation of intrinsic T cell inhibitory enzymes such as diacylglycerol kinase (dgk), that negatively regulates TCR signaling and limits the activation of the Ras/ERK pathway, as well as increased expression levels of inhibitory checkpoint molecules such as PD-1, CTLA-4, TIM-3, LAG-3 and/or their corresponding receptors, can lead to rapid loss of CAR-T cell functional activity, significantly limiting their therapeutic efficacy [Citation75]. When murine CD8 + T cells derived from dgk-deficient mice (dgkα-deficient, dgkζ-deficient, or both) were transduced with anti-human MSLN CD3ζ/4-1BB CARs, they exhibited significantly improved cytokine production (IFN-γ) and enhanced cytotoxic activity, decreased sensitivity to TGF-β and upregulated expression of TRAIL and FasL in vitro [Citation73]. The murine CAR-T cells deficient in both dgk isoforms also demonstrated a superior performance in vivo [Citation73]. Moreover, pharmacologic inhibition of dgks, led to improved anti-tumor activity and function of human anti-MSLN CAR-T cells [Citation73].
Upon antigen encounter, activated T cells can upregulate inhibitory receptors, such as PD-1, and in response to activated T cell-derived pro-inflammatory cytokines, cancer cells upregulate expression of their respective inhibitory ligands, such as PD-L1; these events eventually lead to T cell anergy and hypofunction, suggesting that this could also be a limiting factor in CAR-T cell efficacy [Citation75,Citation106–110]. This inhibitory loop has been blocked in a number of ways. Liu et al. created MSLN-CAR- CD3ζ/41BB T cells that also expressed a PD1-CD28 chimeric ‘switch receptor’ [Citation111]. Upon interaction with its ligands, PD-L1, this receptor (which contained the extracellular domain of PD1 linked to the transmembrane domain of CD28), both bound and blocked PDL1 and delivered an additional activation signal (via the CD28 domain) instead of an inhibitory signal. These MSLN-CAR/PD1-CD28 CAR T cells showed increased efficacy in an animal model of mesothelioma [Citation111].
In another study using fully human anti-MSLN CD3ζ/CD28 and CD3ζ/4-1BB CARs, it was shown that intrapleural injection of CAR-T cells in an MPM mouse orthotopic model, led the CD28 CAR-T cells in a state of anergy/exhaustion much faster than the 4–1BB CAR-T cells rendering CD28 CAR-T cells more prone to relapse, although upon initial antigen stimulation in vitro both CAR-T cell types had demonstrated similar proliferation and effector cytokine secretion levels [Citation110]. In this study, tumor cells were found to overexpress both PD-1 ligands (PD-L1, PD-L2) and tumor-infiltrating CD28 CAR-T cells isolated from progressing tumors, were found to express PD-1, TIM-3, and LAG-3 [Citation110]. When, however, the CD28 CAR-T cells were co-administered with PD-1 blocking antibody or when CD28 CAR-T cells were co-transduced with a PD-1 dominant negative receptor (DNR) which conferred the CAR-T cells with an intrinsic PD-1 checkpoint blockade, the in vivo function of the anti-MSLN CD28 CAR-T cells was rescued [Citation110]. In another recent study, CRISPR/Cas9-mediated disruption of PD-1 in anti-MSLN CD3ζ/4-1BB CAR-T cells led to improved and enhanced capacity of the CAR-T cells to control large established tumors in TNBC NSG orthotopic xenograft mouse models, created via mammary gland injection of human MSLN-expressing and PD-L1-expressing TNBC cells (BT549) [Citation112].
Other experimental approaches to improve CAR-T cell activity within the hostile tumor microenvironment include: depleting the immunosuppressive myeloid-derived suppressor cells (MDSCs) via blocking antibodies or via the use of immunotoxins such as Gemtuzumab ozogamicin in in vivo murine models [Citation113]; combination of anti-MSLN CAR-T cells (CD3ζ/4-1BB) with an oncolytic adenovirus expressing cytokines such as IL-2 and TNF-α, which has been shown to improve CAR-T cell anti-tumor efficacy and survival in both xenograft and syngeneic PDAC mouse models [Citation114]; targeting of the immunosuppressive cytokine TGF-β via the use of an oncolytic adenovirus which was recently shown to improve the anti-tumor activity of human MSLN-targeting CD3ζ/CD28/4-1BB CAR-T cells in an TNBC xenograft NSG mouse model [Citation115]. Another recent study that targeted the metabolism of CD3ζ/CD28/4-1BB MSLN-targeting CAR-T cells via siRNA knockdown of cholesterol acyltransferase 1 (ACAT-1), led to increased cytotoxicity and pro-inflammatory cytokine production (IL-2, IFN-γ) both in vitro and in in vivo mouse xenograft pancreatic adenocarcinoma models [Citation116].
The mode of CAR-T cell administration could also play a role in the efficacy and success of this therapeutic intervention [Citation117]. In a mesothelioma xenograft mouse model, intratumoral injection of anti-MSLN CD3ζ/CD28/4-1BB CAR-T cells was shown to decrease tumor size faster than intravenous injection of the same CAR-T cells [Citation76]. In another study using an orthotopic NSG mouse model of mesothelioma, intrapleural administration of anti-MSLN CD3ζ/CD28 CAR-T cells led to robust in vivo CAR-T cell activation and expansion, as well as enhanced persistence and anti-tumor efficacy, outperforming the intravenously administered CAR-T cells [Citation71]. Therefore, tumor-specific, regional delivery instead of systemic delivery of CAR-T cells, could be key to overcoming some important solid tumor obstacles that are currently limiting their therapeutic efficacy. Another important advantage of regional or intratumoral CAR-T cell administration could be the prevention of ‘on-target/off-tumor’ toxicities.
It is finally important to mention that, often, the construction of anti-human MSLN CAR-T cells employs the use of murine-derived scFv (for example SS1) and this could potentially lead to adverse effects and immunogenicity. In a xenogeneic model of human ovarian cancer [Citation77], as well as in a patient-derived xenograft (PDX) model of pancreatic cancer [Citation118], the use of fully human anti-MSLN CAR-T cells led to efficient in vivo anti-tumor activity, suggesting that the use of fully human anti-MSLN could be a feasible option in clinic.
2.4. MSLN CAR-T cell clinical trials
A number of early-stage clinical trials for anti-MSLN CAR-T cells have taken place over the last decade, commencing in 2011. According to ClinicalTrials.gov, these trials are targeting several MSLN-positive solid tumor malignancies such as malignant pleural mesothelioma (MPM), lung cancer, ovarian cancer, pancreatic ductal adenocarcinoma (PDAC), triple-negative breast cancer (TNBC), endometrial cancer, gastric cancer, colorectal cancer, hepatoma (hepatocellular carcinoma), glioma, esophagus cancer, neuroendocrine tumors/Merkel cell carcinoma, and squamous cell cancer. Along with the anti-MSLN CAR-T cell clinical trials, an anti-MSLN CAR NK cell Early Phase I clinical trial in epithelial ovarian cancer (NCT03692637) is also taking place. A list of these trials is presented in . Despite this long list, there is relatively little published data yet about the results of these studies. The section below summarizes what has been published in either manuscript or abstract form.
Table 1. Phase I/II clinical trials of MSLN-targeted CAR T cells
The initial clinical trials tested a CD3ζ/4-1BB-based CAR that targeted MSLN using the SS1-based single chain anti-MSLN antibody (University of Pennsylvania: NCT01355965, NCT01897415). To maximize safety, these trials used mRNA electroporated CAR-T cells. The trials were based on encouraging preclinical data (discussed earlier in this review), which provided evidence for efficient anti-tumor activity of CAR-T cells generated via mRNA electroporation [Citation82]. The first-in-human NCT1355965 Phase I study involved multiple infusions of meso-RNA-CAR-T cells given to four patients, three MPM and one PDAC patient [Citation119]. In terms of safety, no ‘on-tumor/off-target’ toxicities such as pericarditis, pleuritic, and peritonitis nor cytokine release syndrome (CRS) symptoms were observed; however, one MPM patient experienced a severe anaphylaxis reaction within minutes of CAR T cell intravenous infusion that was delivered 6 weeks after the first series of injections. This was attributed to the development of anti-IgE antibodies induced by the immunogenic murine-derived anti-MSLN scFv in the CAR construct [Citation119].
The NCT01897415 study was conducted in chemotherapy-refractory metastatic PDAC patients (10 patients enrolled; 6 patients treated) [Citation120]. Patients were given intravenous CAR-Tmeso cells 3 times weekly for 3 weeks. None of the patients developed cytokine release syndrome or neurologic symptoms and there were no dose-limiting toxicities. Two patients had stable disease, with progression-free survival times of 3.8 and 5.4 months. 18F-2-fluoro-2-deoxy-D-glucose (FDG)-positron emission tomography/computed tomography imaging was used to monitor the metabolic active volume (MAV) of individual tumor lesions. The total MAV remained stable in three patients and decreased by 69.2% in 1 patient with biopsy-proven mesothelin expression. Transient CAR expression was detected in patients’ blood after infusion and led to expansion of new immunoglobulin G proteins. These results were stated to provide evidence for the potential antitumor activity of messenger RNA CARTmeso cells, as well as PDAC resistance to the immune response [Citation120].
Based on the good safety profiles of the studies (NCT01355965 and NCT01897415) using transiently expressed anti-MSLN CARs), a clinical trial using the same anti-MSLN (murine SS1 scFv) CD3ζ/4-1BB CAR construct was generated, but used lentiviral transduction (NCT02159716, University of Pennsylvania). 15 patients with malignant pleural mesothelioma (MPM), ovarian carcinoma and pancreatic ductal adenocarcinoma (PDAC) were given a single infusion of mesothelin CAR T cells, with or without cyclophosphamide [Citation121]. Lentiviral-transduced CART-meso cells were well tolerated; one dose-limiting toxicity (grade 4, sepsis) occurred at 1–3 × 10 7/m 2 CART-meso without cyclophosphamide [Citation121]. The best overall response was stable disease (11/15 patients). CART-meso cells expanded in the blood and reached peak levels by days 6–14 but persisted transiently. Cyclophosphamide pre-treatment-enhanced CART-meso expansion, but did not improve persistence beyond 28 days. CART-meso DNA was detected in 7/10 tumor biopsies. Human anti-chimeric antibodies (HACA) were detected in the blood of 8/14 patients. CART-meso cells were well tolerated and expanded in the blood of all patients but showed limited clinical activity [Citation121].
One reason for this lack of response was postulated to be the relatively short persistence of the CARTs, perhaps due to immunologic reactions (antibody and T cell-based) against the murine scFV antibody fragment. Accordingly, two follow-up clinical trials were initiated (NCT03054298, NCT03323944), using a fully humanized and more active anti-MSLN scFv CD3ζ/4-1BB CAR construct (called ‘M5’), hoping to enhance CAR-T cell persistence and anti-tumor activity [Citation121]. In the NCT03054298 study, both intravenous and intrapleural CAR-T administration will be tested in mesothelioma, lung cancer, and ovarian cancer patients. In the NCT03323944 trial, intravenous infusions will be given to patients with pancreatic cancer. Both trials are currently active and recruiting.
Investigators at Memorial Sloan Kettering Cancer Center (New York, US) are using retrovirally transduced human scFv CD3ζ/CD28 CAR-T cells that also contain an inducible Caspase 9 (iCasp9) suicide gene in an ongoing Phase I/II clinical trial (NCT02414269, currently recruiting) for patients with MPM, lung cancer, and breast cancer. The suicide gene was included as an effective ‘switch-on/switch-off’ mechanism to tightly regulate the activity of CAR-T cells if adverse ‘on-target/off-tumor’ events or CRS were noted [Citation122]. These meso-CAR-T cells are being administered intrapleurally, based on preclinical evidence suggesting that intrapleural administration conferred superior CAR-T cell anti-tumor activity and enhanced persistence compared to intravenous administration [Citation71,Citation117]. The investigators’ abstract for the 2019 American Association for Cancer Research (AACR) conference stated that 21 patients with MPD (19 MPM, 1 lung cancer, 1 breast cancer) were treated (40% received ≥3 lines of prior therapy) with meso-CAR T cells at doses from 3 × 105 to 1 × 107 CAR T cells/kg. Eighteen of the 21 patients received cyclophosphamide. No CAR T-cell-related toxicities higher than grade 2 were observed. CAR T cells were detected in the peripheral blood of 13 patients (day 1 to 38 weeks), as evidenced by vector copy number. T-cell persistence was associated with decreased serum SMRP levels (>50% compared to pretreatment) and evidence of tumor regression on imaging studies. Once lack of toxicity had been established (6–17 weeks after CAR T-cell infusion), 14 patients received anti-PD1 checkpoint blockade agents (1–21 cycles), off protocol, with no toxicity. The best responses among the 19 MPM patients were that 2 patients had complete metabolic response on PET scan (60 and 32 weeks ongoing), 5 had partial responses and 4 had stable disease. The trial is ongoing.
Another ongoing Phase I study (NCT03608618, currently recruiting) taking place in the US National Cancer Institute is also using CD3ζ/4-1BB (patent: US20190211109A1) mRNA electroporated anti-MSLN CAR-T cells with a human scFV (termed as the MCY-M11 CAR). Advanced ovarian cancer and peritoneal mesothelioma patients are being treated and the CAR-T-cells are administered intraperitoneally [Citation123]. No results have yet been reported.
Despite the lack of reported data on human mesothelin CAR T cell trials, a number of other trial designs have been reported and are accessible in Clinical.Trials.Gov. A trial is underway in China (NCT02959151), in which lentivirally transduced anti-MSLN CAR-T cells are intra-tumorarly administered in patients with hepatocellular carcinoma, metastatic pancreatic, and metastatic colorectal cancer. Transcatheter arterial infusion (TAI) of anti-MSLN CARs in pancreatic cancer patients has also been proposed as a mode of regional delivery (NCT02706782) that could enhance CAR-T cell performance and decrease the risk of ‘on-target/off-tumor toxicities.’
As was seen in the MSKCC trial (NCT02414269), the combination therapy of anti-MSLN CAR-T cells along with immune checkpoint inhibitors is under active study. This includes a study with the PD-1 blocking antibody pembrolizumab (NCT03393858, NCT02414269), engineering of anti-MSLN CAR-T cells to secrete anti-PD-1 and anti-CTLA-4 antibodies (NCT03182803) and development of CRISPR/Cas9 mediated PD-1 knocked-out CAR-T cells redirected against MSLN (NCT03916679).
Another strategy to enhance anti-MSLN CAR-T cell efficacy (and CAR-T cell efficacy in general) is CAR-T combination with lymphodepleting drugs such as cyclophosphamide and/or fludarabine (see or list of these trials), or by combining with chemotherapy drugs such as paclitaxel (NCT03747965) or with aldesleukin/IL-2 (NCT01583686) which promotes T-cell activation.
3. Conclusion
Clinical trial data, to date, have shown that anti-MSLN CAR T cells appear to be safe. However, despite good preclinical evidence of anti-tumor activity, they have not shown persistence or efficacy when intravenously infused into patients with a wide variety of mesothelin-expressing solid tumors.
Given these disappointing results, a range of approaches to improve the persistence and efficacy of this treatment are being proposed and tested in new clinical trials. This includes regional CAR-T cell delivery, utilization of fully human anti-MSLN scFV, and combination with checkpoint inhibitors and chemotherapies. Other strategies, still in preclinical testing, such as augmentation of bystander effects [Citation124], blockade of inhibitory factors (such as TGFβ, adenosine, or PGE2), CAR-T cells engineered to secrete inflammatory cytokines or tumor homing chemokine receptors, or CAR T cells with genetic manipulations that make them more resistant to chronic T cell activation and hypofunction may be needed to enhance their performance [Citation74,Citation83,Citation112,Citation115].
4. Expert Opinion
CAR-T cell immunotherapies are an important advancement in the treatment of blood malignancies and significant efforts are being made for their successful implementation in combatting solid tumors. To this end, the selection of TAAs to be targeted by CAR-T cells is crucial. Solid tumors, however, are often characterized by lack of unique TAA expression and although CAR-T cell action is not limited by MHC antigen recognition, it still requires expression of TAAs on the tumor cell surface. Thus, the selection of a TAA that is overexpressed by the tumor cells whilst its physiological expression is as confined as possible, is the best option for a therapeutic target. Mesothelin (MSLN), with its normal expression pattern being restricted primarily to the mesothelial surfaces of the pleura, peritoneum, pericardium, and the tunica vaginalis (in men), while significantly overexpressed in a broad range of solid tumors, is such a potential antigen.
Indeed, anti-MSLN CAR-T cells have been evaluated in several preclinical models and clinical trials, presented in this review. Along with anti-MSLN CAR-T cells, other therapeutic approaches with different tumor-killing mechanisms such as antibody-based drugs and anti-cancer vaccines targeting MSLN have been developed and tested both experimentally and in clinical trials. Compared to anti-MSLN antibody-based therapies and CAR-T cells, anti-MSLN vaccines have shown moderate and generally less favorable effects in clinic and fewer clinical trials for anti-MSLN vaccines are currently taking place.
One advantage of anti-MSLN CAR-T cells over other types of anti-MSLN immunotherapies, is the fact that CAR-T cells are highly sensitive ‘living drugs,’ combining the antigen specificity of a monoclonal antibody together with their inherent effector functions. CAR-T cells have an intrinsic ability to migrate and infiltrate toward the tumor tissues as well as to proliferate and prolong their survival upon target interception, while secreting pro-inflammatory factors that will attract and activate other immune cells in the tumor microenvironment. Excessive CAR-T cell activation, however, could lead to adverse effects such as CRS and therefore anti-MSLN CAR-T cell engineering with suicide genes such as iCasp9 could ameliorate their safety profile, if this is needed. Furthermore, given that CAR-T cells have in general a higher sensitivity for low antigen expression levels compared to monoclonal antibodies, ‘on-target/off-tumor effects’ against nonpathogenic tissues should be more often anticipated following CAR-T cell administration. The use of anti-MSLN CAR-T cells bearing a suicide gene or the use of inhibitory CARs, could confer protection against healthy tissue damage.
Anti-MSLN CAR-T cells can have a longer half-life over anti-MSLN antibody-based drugs, however CAR-T cells targeting solid tumors develop ‘exhaustion’ before being able to exert their anti-tumor effects at their full capacity. Thus, despite their longer presence at the tumor site, CAR-T cell hypofunction might be a significant obstacle toward effective tumor killing. For this reason, immune checkpoint inhibition approaches can conceivably improve anti-MSLN CAR-T cell function and could be a significant advancement toward the engineering of more potent CAR-T cells. The use of humanized or fully human anti-MSLN scFv is also a promising strategy that could enhance CAR-T cell survival and mitigate the risk of immunogenic responses.
Similarly, humanized or fully human anti-MSLN antibody-based drugs such as amatuximab and anetumab ravtansine have improved half-life, good anti-tumor efficacy, better safety profiles and reduced immunogenicity [Citation58,Citation61,Citation63,Citation125]. Thus, several Phase I/II clinical trials treating patients with MSLN-expressing solid tumors with amatuximab (and anetumab ravtansine) have been completed or are currently taking place. In contrast, antibody-based drugs with a murine anti-MSLN moiety such as SS1P have been shown to be highly immunogenic and patients developed neutralizing antibodies against SS1P [Citation125]. Therefore, an immunotoxin consisting of a humanized anti-MLSN antibody and a less immunogenic bacterial exotoxin, LMB-100/RG7787, has been more effective than SS1P [Citation55,Citation125].
Both anti-MSLN CAR-T cells and anti-MSLN antibody-based drugs have faced significant challenges in effectively reaching the solid tumor sites. Antibodies are usually administered intravenously and CAR-T cell intravenous administration is still quite common, leading often to ‘on-target/off-tumor’ toxicities as well as ineffective anti-tumor activity. Thus, for both therapies, a regional or intratumoral approach could potentially enhance their performance and reduce the risk of off-tumor recognition of targeted antigen. In cases where localized administration might not be feasible or effective, or in cases of a metastasis leading to multiple tumor niches, intravenous infusion of anti-MSLN CAR-T cells engineered to express tumor homing chemokines or chemokine receptors could have an advantage over intravenously administered anti-MSLN antibody-based drugs.
It is finally important to note that the use of chemotherapeutic and lymphodepleting agents in conjunction with anti-MSLN immunotherapies (CAR-T cells, antibody-based drugs, vaccines) could be necessary to achieve optimal results. For anti-MSLN CAR-T cells especially, a combinatorial approach using both established anti-cancer regimens and locally administered novel anti-MSLN CAR-T cells, the use of fully human scFv, blockade of inhibitory checkpoint molecules, expression of suicide genes, inflammatory cytokine secretion, expression of tumor homing cytokines and cytokine receptors, dual CARs-able to overcome the challenges of the tumor microenvironment while maintaining high safety profiles, will likely prove necessary for successfully treating solid tumors.
Article highlights
Mesothelin (MSLN) is a tumor differentiation antigen overexpressed in a broad range of solid tumors including mesothelioma, ovarian cancer, triple-negative breast cancer, lung cancer, pancreatic cancer, and gastric cancer.
Several novel anti-cancer immunotherapies targeting MSLN have been developed and are being tested in both preclinical settings and clinical trials.
Anti-MSLN immunotherapies include; antibody-based drugs (monoclonal antibodies, antibody-drug conjugates, immunotoxins), vaccines, and CAR-T cell therapies.
A significant number of clinical trials using anti-human MSLN CAR-T cells have been taking place since 2011; however, further work is required so as to develop anti-MSLN CAR-T cells with high anti-tumor efficacy.
Evidence from both preclinical models and clinical trials has demonstrated that further CAR-T cell modifications and novel approaches in CAR-T cell engineering are necessary to generate a successful anti-MSLN CAR-T cell therapy for solid tumors.
This box summarizes key points contained in the article.
Declaration of interest
A Klampatsa is supported by the CRIS Cancer Foundation. SM Albelda has received grant support from NCI GRANT PO1-CA217805 and prior research funding from Novartis. SM Albelda currently receives research funding from Tmunity. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Ordonez NG. Application of mesothelin immunostaining in tumor diagnosis. Am J Surg Pathol. 2003;27(11):1418–1428.
- Lamberts LE, de Groot DJ, Bense RD, et al. Functional genomic mRNA profiling of a large cancer data base demonstrates mesothelin overexpression in a broad range of tumor types. Oncotarget. 2015;6(29):28164–28172.
- Frierson HF Jr., Moskaluk CA, Powell SM, et al. Large-scale molecular and tissue microarray analysis of mesothelin expression in common human carcinomas. Hum Pathol. 2003;34(6):605–609.
- Ordonez NG. Value of mesothelin immunostaining in the diagnosis of mesothelioma. Mod Pathol. 2003;16(3):192–197.
- Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci U S A. 1996;93(1):136–140.
- Clarke J, Panwar B, Madrigal A, et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med. 2019;216(9):2128–2149.
- Galloway ML, Murray D, Moffat DF. The use of the monoclonal antibody mesothelin in the diagnosis of malignant mesothelioma in pleural biopsies. Histopathology. 2006;48(6):767–769.
- Kushitani K, Takeshima Y, Amatya VJ, et al. Immunohistochemical marker panels for distinguishing between epithelioid mesothelioma and lung adenocarcinoma. Pathol Int. 2007;57(4):190–199.
- Scales SJ, Gupta N, Pacheco G, et al. An antimesothelin-monomethyl auristatin e conjugate with potent antitumor activity in ovarian, pancreatic, and mesothelioma models. Mol Cancer Ther. 2014;13(11):2630–2640.
- Cheng WF, Huang CY, Chang MC, et al. High mesothelin correlates with chemoresistance and poor survival in epithelial ovarian carcinoma. Br J Cancer. 2009;100(7):1144–1153.
- Hassan R, Kreitman RJ, Pastan I, et al. Localization of mesothelin in epithelial ovarian cancer. Appl Immunohistochem Mol Morphol. 2005;13(3):243–247.
- Obulhasim G, Fujii H, Matsumoto T, et al. Mesothelin gene expression and promoter methylation/hypomethylation in gynecological tumors. Eur J Gynaecol Oncol. 2010;31(1):63–71.
- Yen MJ, Hsu CY, Mao TL, et al. Diffuse mesothelin expression correlates with prolonged patient survival in ovarian serous carcinoma. Clin Cancer Res. 2006;12(3 Pt 1):827–831.
- Li YR, Xian RR, Ziober A, et al. Mesothelin expression is associated with poor outcomes in breast cancer. Breast Cancer Res Treat. 2014;147(3):675–684.
- Tchou J, Wang LC, Selven B, et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat. 2012;133(2):799–804.
- Tozbikian G, Brogi E, Kadota K, et al. Mesothelin expression in triple negative breast carcinomas correlates significantly with basal-like phenotype, distant metastases and decreased survival. PLoS One. 2014;9(12):e114900.
- Shimizu A, Hirono S, Tani M, et al. Coexpression of MUC16 and mesothelin is related to the invasion process in pancreatic ductal adenocarcinoma. Cancer Sci. 2012;103(4):739–746.
- Swierczynski SL, Maitra A, Abraham SC, et al. Analysis of novel tumor markers in pancreatic and biliary carcinomas using tissue microarrays. Hum Pathol. 2004;35(3):357–366.
- Winter JM, Tang LH, Klimstra DS, et al. A novel survival-based tissue microarray of pancreatic cancer validates MUC1 and mesothelin as biomarkers. PLoS One. 2012;7(7):e40157.
- Argani P, Iacobuzio-Donahue C, Ryu B, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res. 2001;7(12):3862–3868.
- Ho M, Bera TK, Willingham MC, et al. Mesothelin expression in human lung cancer. Clin Cancer Res. 2007;13(5):1571–1575.
- Kachala SS, Bograd AJ, Villena-Vargas J, et al. Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clin Cancer Res. 2014;20(4):1020–1028.
- Thomas A, Chen Y, Steinberg SM, et al. High mesothelin expression in advanced lung adenocarcinoma is associated with KRAS mutations and a poor prognosis. Oncotarget. 2015;6(13):11694–11703.
- Baba K, Ishigami S, Arigami T, et al. Mesothelin expression correlates with prolonged patient survival in gastric cancer. J Surg Oncol. 2012;105(2):195–199.
- Einama T, Homma S, Kamachi H, et al. Luminal membrane expression of mesothelin is a prominent poor prognostic factor for gastric cancer. Br J Cancer. 2012;107(1):137–142.
- Ito T, Kajino K, Abe M, et al. ERC/mesothelin is expressed in human gastric cancer tissues and cell lines. Oncol Rep. 2014;31(1):27–33.
- Sotoudeh M, Shirvani SI, Merat S, et al. MSLN (Mesothelin), ANTXR1 (TEM8), and MUC3A are the potent antigenic targets for CAR T cell therapy of gastric adenocarcinoma. J Cell Biochem. 2019;120(4):5010–5017.
- Lv J, Zhao R, Wu D, et al. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J Hematol Oncol. 2019;12(1):18.
- He Y, Li XM, Yin CH, et al. Killing cervical cancer cells by specific chimeric antigen receptor-modified T cells. J Reprod Immunol. 2020;139:103115.
- Kawamata F, Kamachi H, Einama T, et al. Intracellular localization of mesothelin predicts patient prognosis of extrahepatic bile duct cancer. Int J Oncol. 2012;41(6):2109–2118.
- Zhao H, Davydova L, Mandich D, et al. S100A4 protein and mesothelin expression in dysplasia and carcinoma of the extrahepatic bile duct. Am J Clin Pathol. 2007;127(3):374–379.
- Alvarez H, Rojas PL, Yong KT, et al. Mesothelin is a specific biomarker of invasive cancer in the Barrett-associated adenocarcinoma progression model: translational implications for diagnosis and therapy. Nanomedicine. 2008;4(4):295–301.
- Dainty LA, Risinger JI, Morrison C, et al. Overexpression of folate binding protein and mesothelin are associated with uterine serous carcinoma. Gynecol Oncol. 2007;105(3):563–570.
- Yu L, Feng M, Kim H, et al. Mesothelin as a potential therapeutic target in human cholangiocarcinoma. J Cancer. 2010;1:141–149.
- Nomura R, Fujii H, Abe M, et al. Mesothelin expression is a prognostic factor in cholangiocellular carcinoma. Int Surg. 2013;98(2):164–169.
- Steinbach D, Onda M, Voigt A, et al. Mesothelin, a possible target for immunotherapy, is expressed in primary AML cells. Eur J Haematol. 2007;79(4):281–286.
- Chang K, Pastan I, Willingham MC. Isolation and characterization of a monoclonal antibody, K1, reactive with ovarian cancers and normal mesothelium. Int J Cancer. 1992;50(3):373–381.
- Urwin D, Lake RA. Structure of the Mesothelin/MPF gene and characterization of its promoter. Mol Cell Biol Res Commun. 2000;3(1):26–32.
- Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur J Cancer. 2008;44(1):46–53.
- Yamaguchi N, Hattori K, Oh-eda M, et al. A novel cytokine exhibiting megakaryocyte potentiating activity from a human pancreatic tumor cell line HPC-Y5. J Biol Chem. 1994;269(2):805–808.
- Bera TK, Pastan I. Mesothelin is not required for normal mouse development or reproduction. Mol Cell Biol. 2000;20(8):2902–2906.
- Tang Z, Qian M, Ho M. The role of mesothelin in tumor progression and targeted therapy. Anticancer Agents Med Chem. 2013;13(2):276–280.
- Rump A, Morikawa Y, Tanaka M, et al. Binding of ovarian cancer antigen CA125/MUC16 to mesothelin mediates cell adhesion. J Biol Chem. 2004;279(10):9190–9198.
- Gubbels JA, Belisle J, Onda M, et al. Mesothelin-MUC16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol Cancer. 2006;5(1):50.
- Bharadwaj U, Li M, Chen C, et al. Mesothelin-induced pancreatic cancer cell proliferation involves alteration of cyclin E via activation of signal transducer and activator of transcription protein 3. Mol Cancer Res. 2008;6(11):1755–1765.
- Li M, Bharadwaj U, Zhang R, et al. Mesothelin is a malignant factor and therapeutic vaccine target for pancreatic cancer. Mol Cancer Ther. 2008;7(2):286–296.
- Bharadwaj U, Marin-Muller C, Li M, et al. Mesothelin overexpression promotes autocrine IL-6/sIL-6R trans-signaling to stimulate pancreatic cancer cell proliferation. Carcinogenesis. 2011;32(7):1013–1024.
- Bharadwaj U, Marin-Muller C, Li M, et al. Mesothelin confers pancreatic cancer cell resistance to TNF-alpha-induced apoptosis through Akt/PI3K/NF-kappaB activation and IL-6/Mcl-1 overexpression. Mol Cancer. 2011;10:106.
- Uehara N, Matsuoka Y, Tsubura A. Mesothelin promotes anchorage-independent growth and prevents anoikis via extracellular signal-regulated kinase signaling pathway in human breast cancer cells. Mol Cancer Res. 2008;6(2):186–193.
- Chang MC, Chen CA, Hsieh CY, et al. Mesothelin inhibits paclitaxel-induced apoptosis through the PI3K pathway. Biochem J. 2009;424(3):449–458.
- Chowdhury PS, Viner JL, Beers R, et al. Isolation of a high-affinity stable single-chain Fv specific for Mesothelin from DNA-immunized mice by phage display and construction of a recombinant immunotoxin with anti-tumor activity. Proc Natl Acad Sci U S A. 1998;95(2):669–674.
- Chowdhury PS, Pastan I. Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat Biotechnol. 1999;17(6):568–572.
- Hilliard TS. The impact of Mesothelin in the ovarian cancer tumor microenvironment. Cancers (Basel). 2018;10(9):277.
- Kreitman RJ, Hassan R, Fitzgerald DJ, et al. Phase I trial of continuous infusion Anti-Mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15(16):5274–5279.
- Hollevoet K, Mason-Osann E, Liu XF, et al. In vitro and in vivo activity of the low-immunogenic Antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol Cancer Ther. 2014;13(8):2040–2049.
- Baldo P, Cecco S. Amatuximab and novel agents targeting Mesothelin for solid tumors. Onco Targets Ther. 2017;10:5337–5353.
- Hassan R, Cohen SJ, Phillips M, et al. Phase I clinical trial of the chimeric anti-Mesothelin monoclonal antibody MORAb-009 in patients with Mesothelin-expressing cancers. Clin Cancer Res. 2010;16(24):6132–6138.
- Hassan R, Kindler HL, Jahan T, et al. Phase II clinical trial of amatuximab, a chimeric Antimesothelin antibody with pemetrexed and cisplatin in advanced unresectable pleural mesothelioma. Clin Cancer Res. 2014;20(23):5927–5936.
- Hassan R, Schweizer C, Lu KF, et al. Inhibition of Mesothelin-CA-125 interaction in patients with mesothelioma by the Anti-Mesothelin monoclonal antibody MORAb-009: implications for cancer therapy. Lung Cancer. 2010;68(3):455–459.
- Gupta A, Hussein Z, Hassan R, et al. Population pharmacokinetics and exposure-response relationship of amatuximab, an Anti-Mesothelin monoclonal antibody, in patients with malignant pleural mesothelioma and its application in dose selection. Cancer Chemother Pharmacol. 2016;77(4):733–743.
- Golfier S, Kopitz C, Kahnert A, et al. Anetumab ravtansine: a novel Mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol Cancer Ther. 2014;13(6):1537–1548.
- Chen H, Lin Z, Arnst KE, et al. Tubulin inhibitor-based antibody-drug conjugates for cancer therapy. Molecules. 2017;22(8):1281.
- Hassan R, Blumenschein GR Jr, Moore KN, et al. First-in-human, multicenter, phase i dose-escalation and expansion study of Anti-Mesothelin antibody-drug conjugate Anetumab Ravtansine in advanced or metastatic solid tumors. J Clin Oncol. 2020;38(16):1824-1835.
- Weekes CD, Lamberts LE, Borad MJ, et al. Phase I study of DMOT4039A, an antibody-drug conjugate targeting Mesothelin, in patients with unresectable pancreatic or platinum-resistant ovarian cancer. Mol Cancer Ther. 2016;15(3):439–447.
- Le DT, Brockstedt DG, Nir-Paz R, et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing Mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin Cancer Res. 2012;18(3):858–868.
- Chang MC, Chen YL, Chiang YC, et al. Mesothelin-specific cell-based vaccine generates antigen-specific immunity and potent antitumor effects by combining with IL-12 immunomodulator. Gene Ther. 2016;23(1):38–49.
- Chen YL, Chang MC, Chiang YC, et al. Immuno-modulators enhance antigen-specific immunity and anti-tumor effects of Mesothelin-specific chimeric DNA vaccine through promoting DC maturation. Cancer Lett. 2018;425:152–163.
- Lv J, Li P. Mesothelin as a biomarker for targeted therapy. Biomark Res. 2019;7:18.
- Qin L, Lai Y, Zhao R, et al. Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J Hematol Oncol. 2017;10(1):68.
- Zhang C, Liu J, Zhong JF, et al. Engineering CAR-T cells. Biomark Res. 2017;5:22.
- Adusumilli PS, Cherkassky L, Villena-Vargas J, et al. Regional delivery of Mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6(261):261ra151.
- Lanitis E, Poussin M, Klattenhoff AW, et al. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. 2013;1(1):43–53.
- Riese MJ, Wang LC, Moon EK, et al. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. 2013;73(12):3566–3577.
- Moon EK, Carpenito C, Sun J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a Mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17(14):4719–4730.
- Moon EK, Wang LC, Dolfi DV, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20(16):4262–4273.
- Carpenito C, Milone MC, Hassan R, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106(9):3360–3365.
- Lanitis E, Poussin M, Hagemann IS, et al. Redirected antitumor activity of primary human lymphocytes transduced with a fully human Anti-Mesothelin chimeric receptor. Mol Ther. 2012;20(3):633–643.
- Hung CF, Xu X, Li L, et al. Development of anti-human Mesothelin-targeted chimeric antigen receptor messenger RNA-Transfected peripheral blood lymphocytes for ovarian cancer therapy. Hum Gene Ther. 2018;29(5):614–625.
- Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int J Cancer. 2011;129(12):2935–2944.
- Lai Y, Weng J, Wei X, et al. Toll-like receptor 2 costimulation potentiates the antitumor efficacy of CAR T Cells. Leukemia. 2018;32(3):801–808.
- Zhao R, Cheng L, Jiang Z, et al. DNAX-activating protein 10 co-stimulation enhances the anti-tumor efficacy of chimeric antigen receptor T cells. Oncoimmunology. 2019;8(1):e1509173.
- Zhao Y, Moon E, Carpenito C, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70(22):9053–9061.
- Adachi K, Adachi K, Kano Y, et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36(4):346–351.
- Li S, Siriwon N, Zhang X, et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res. 2017;23(22):6982–6992.
- Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115(17):3508–3519.
- Pegram HJ, Lee JC, Hayman EG, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119(18):4133–4141.
- Kagoya Y, Tanaka S, Guo T, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. 2018;24(3):352–359.
- Tokarew N, Ogonek J, Endres S, et al. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. 2019;120(1):26–37.
- Benmebarek MR, Karches CH, Cadilha BL, et al. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. 2019;20(6).
- Milone MC, O’Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32(7):1529–1541.
- Miliotou AN, Papadopoulou LC. CAR T-cell therapy: a new era in cancer immunotherapy. Curr Pharm Biotechnol. 2018;19(1):5–18.
- Feins S, Kong W, Williams EF, et al. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol. 2019;94(S1):S3–S9.
- Woodard LE, Wilson MH. piggyBac-ing models and new therapeutic strategies. Trends Biotechnol. 2015;33(9):525–533.
- Jin Z, Maiti S, Huls H, et al. The hyperactive sleeping beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor. Gene Ther. 2011;18(9):849–856.
- Xu JY, Ye ZL, Jiang DQ, et al. Mesothelin-targeting chimeric antigen receptor-modified T cells by piggyBac transposon system suppress the growth of bile duct carcinoma. Tumour Biol. 2017;39(4):1010428317695949.
- He J, Zhang Z, Lv S, et al. Engineered CAR T cells targeting Mesothelin by piggyBac transposon system for the treatment of pancreatic cancer. Cell Immunol. 2018;329:31–40.
- Zhang Z, Jiang D, Yang H, et al. Modified CAR T cells targeting membrane-proximal epitope of Mesothelin enhances the antitumor function against large solid tumor. Cell Death Dis. 2019;10(7):476.
- Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–117.
- Heyman B, Yang Y. Chimeric antigen receptor T cell therapy for solid tumors: current status, obstacles and future strategies. Cancers (Basel). 2019;11(2):191.
- Bonifant CL, Jackson HJ, Brentjens RJ, et al. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011.
- Zhang E, Yang P, Gu J, et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J Hematol Oncol. 2018;11(1):102.
- Chen Y, Ayaru L, Mathew S, et al. Expansion of Anti-Mesothelin specific CD4+ and CD8+ T cell responses in patients with pancreatic carcinoma. PLoS One. 2014;9(2):e88133.
- Batchu RB, Gruzdyn OV, Mahmud EM, et al. Inhibition of Interleukin-10 in the tumor microenvironment can restore Mesothelin chimeric antigen receptor T cell activity in pancreatic cancer in vitro. Surgery. 2018;163(3):627–632.
- Newick K, O'Brien S, Sun J, et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein Kinase A localization. Cancer Immunol Res. 2016;4(6):541–551.
- Masoumi E, Jafarzadeh L, Mirzaei HR, et al. Genetic and pharmacological targeting of A2a receptor improves function of Anti-Mesothelin CAR T cells. J Exp Clin Cancer Res. 2020;39(1):49.
- McGray AJ, Hallett R, Bernard D, et al. Immunotherapy-induced CD8+ T cells instigate immune suppression in the tumor. Mol Ther. 2014;22(1):206–218.
- Spranger S, Spaapen RM, Zha Y, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5(200):200ra116.
- Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128.
- Tan KW, Chacko AM, Chew V. PD-1 expression and its significance in tumour microenvironment of hepatocellular carcinoma. Transl Gastroenterol Hepatol. 2019;4:51.
- Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130–3144.
- Liu X, Ranganathan R, Jiang S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016;76(6):1578–1590.
- Hu W, Zi Z, Jin Y, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human Mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. 2019;68(3):365–377.
- Fultang L, Panetti S, NG M, et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine. 2019;47:235–246.
- Watanabe K, Luo Y, Da T, et al. Pancreatic cancer therapy with combined Mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight. 2018;3(7):e99573.
- Li Y, Xiao F, Zhang A, et al. Oncolytic adenovirus targeting TGF-beta enhances anti-tumor responses of Mesothelin-targeted chimeric antigen receptor T cell therapy against breast cancer. Cell Immunol. 2020;348:104041.
- Zhao L, Liu Y, Zhao F, et al. Inhibition of cholesterol esterification enzyme enhances the potency of human chimeric antigen receptor T cells against pancreatic carcinoma. Mol Ther Oncolytics. 2020;16:262–271.
- Mayor M, Zeltsman M, McGee E, et al. A regional approach for CAR T-cell therapy for mesothelioma: from mouse models to clinical trial. Immunotherapy. 2016;8(5):491–494.
- Jiang H, Song B, Wang P, et al. Efficient growth suppression in pancreatic cancer PDX model by fully human Anti-Mesothelin CAR-T cells. Protein Cell. 2017;8(12):926–931.
- Maus MV, Haas AR, Beatty GL, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 2013;1(1):26–31.
- Beatty GL, O'Hara MH, Lacey SF, et al. Activity of Mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. 2018;155(1):29–32.
- Haas AR, Tanyi JL, O'Hara MH, et al. Phase I study of Lentiviral-transduced chimeric antigen receptor-modified T Cells recognizing Mesothelin in advanced solid cancers. Mol Ther. 2019;27(11):1919–1929.
- Adusumilli PS, Z. M, Rusch VW, et al. A phase I clinical trial of malignant pleural disease treated with regionally delivered autologous Mesothelin-targeted CAR T cells: safety and efficacy [abstract CT036]. In Proceedings of the American Association for Cancer Research Annual Meeting 2019;Atlanta, GA.
- Ghobadi A, T hakerP, Weng D, et al., A phase 1 study of intraperitoneal MCY-M11 therapy for women with platinum resistant high grade serous adenocarcinoma of the ovary, primary peritoneum, or fallopian tube, or subjects with peritoneal mesothelioma with recurrence after prior chemotherapy [abstract CT159]. In Proceedingsof the American Association for Cancer Research Annual Meeting 2019; Atlanta, GA.
- Klampatsa A, L eibowitzMS, Sun J, et al. Analysis and augmentation of the immunologic bystander effects of CAR T cell therapy in a syngeneic Mouse cancer model. Mol Ther Oncolytics. 2020;18:360-371.
- Zhao XY, Subramanyam B, Sarapa N, et al. Novel antibody therapeutics targeting Mesothelin in solid tumors. Clin Cancer Drugs. 2016;3(2):76–86.