
ABSTRACT
Introduction
The use of tumor-selectively replicating viruses is a rapidly expanding field that is showing considerable promise for cancer treatment. Retroviral replicating vectors (RRV) are unique among the various replication-competent viruses currently being investigated for potential clinical utility, because they permanently integrate into the cancer cell genome and are capable of long-term persistence within tumors. RRV can mediate efficient tumor-specific delivery of prodrug activator genes, and subsequent prodrug treatment leads to synchronized cell killing of infected cancer cells, as well as activation of antitumor immune responses.
Areas Covered
Here we review preclinical studies supporting bench-to-bedside translation of Toca 511, an optimized RRV for prodrug activator gene therapy, the results from Phase I through III clinical trials to date, and potential future directions for this therapy as well as other clinical candidate RRV.
Expert Opinion
Toca 511 has shown highly promising results in early-stage clinical trials. This vector progressed to a registrational Phase III trial, but the results announced in late 2019 appeared negative overall. However, the median prodrug dosing schedule was not optimal, and promising possible efficacy was observed in some prespecified subgroups. Further clinical investigation, as well as development of RRV with other transgene payloads, is merited.
1. Introduction
Cancer remains one of the leading causes of death worldwide, with the number of cancer-related deaths expected to increase by approximately 70% over the next 20 years [Citation1]. Standard cancer treatments include chemotherapy, radiation, and/or surgery, while more recent and promising advances utilize biological therapies such as interleukins, interferons, monoclonal antibodies (e.g. Herceptin, Rituaxan, Avastin), gene therapies, cell therapies, and oncolytic virotherapy.
Initially, gene therapy focused on the treatment of monogenetic hereditary disorders, but gene transfer approaches for more common acquired disorders such as cancer and cardiovascular disease were rapidly suggested and investigated in both preclinical studies and clinical trials. Cancer gene therapy has now become one of the most studied applications in the gene therapy field, accounting for over 65% of the clinical trials at various stages worldwide [Citation2]. The most fundamental issue for gene therapy strategies is efficient delivery of a therapeutic payload to the intended target cells; this was first enabled by the ability to manipulate and employ viruses as gene delivery vehicles, or ‘vectors’, as many viruses can naturally and effectively enter human cells and express their genes within the infected cells. Early efforts sought to improve the safety of viruses used as gene transfer vectors, by retaining the ability to enter cells and deliver a therapeutic gene, while rendering the viral vector incapable of further replication. However, despite extensive efforts and over a decade of clinical trials, it became clear that gene delivery to cancer cells by replication-defective vectors was often not efficient enough to be effective in vivo [Citation3,Citation4] and lacked clinical benefit in many indications.
The inefficiency of gene delivery led to renewed interest in methods that either amplified the vector effect through the immune system (such as CAR-T cells), improved delivery (such as some AAV chimeras), or both (such as replication-competent viruses). The use of replication-competent viruses represents the emergence of the field of oncolytic virotherapy [Citation5–12], or in fact, its re-emergence: the earliest report of the use of viruses for cancer treatment dates back to the early 1900s, where a patient displayed a dramatic remission of leukemia following influenza infection [Citation13]. During the mid-twentieth century, many different viruses were tested in patients with advanced cancers [Citation14,Citation15]. However, results were frequently discouraging, as initial tumor necrosis was typically followed by immune clearance of the virus and tumor recurrence. Advances in the understanding and manipulation of molecular mechanisms involved in viral infection have driven the renewed interest for the use of various replication-competent viruses to treat cancer.
The majority of viruses currently being clinically evaluated for cancer treatment, including retroviruses, are capable of selective replication in tumors due to cancer cell-intrinsic defects in innate immunity [Citation16,Citation17] and local suppression of adaptive immunity in the tumor microenvironment [Citation18,Citation19], and cause host cell lysis as a natural part of their infection cycle. Retroviral replicating vectors (RRV) are frequently classified with oncolytic viruses, and do take advantage of the same mechanisms of tumor-selectivity. Nonetheless, there are interesting and potentially useful differences between oncolytic virotherapy and RRV-based therapies.
2. Retroviral replicating vectors (RRV) for cancer gene therapy
RRV, currently being evaluated clinically for the treatment of recurrent glioblastoma multiforme and colorectal cancer, are derived from amphotropic murine leukemia virus (MLV), a gammaretrovirus [Citation20,Citation21]. RRV have an intact MLV viral genome (including long terminal repeats [LTR], packaging signal (ψ), and genes gag, pol, and env), enabling vector replication. In addition, RRV carry a transgene cassette between the env gene and the 3ʹuntranslated region (3ʹ UTR). In most RRV described so far, transgene expression has been driven with an internal ribosome entry site (IRES, see )but other expression systems have also been used successfully [Citation22,Citation23]. These vectors are unique compared to other oncolytic agents currently in development, as viral replication does not result in cell lysis. Rather, these vectors maintain viral persistence in tumors through the combined characteristics of non-lytic replication, stable integration into the cancer cell genome, and interferon-suppressive activity [Citation24–28].
Figure 1. Toca 511 design and mechanism of action: (a) Toca 511 is a retroviral replicating vector (RRV) based on an amphotropic mouse gamma-retrovirus which also encodes an optimized yeast cytosine deaminase (CD) and preferentially infects tumors (LTR: long terminal repeat sequences, gag/pol/env: retroviral structural genes, IRES-CD: transgene expression cassette consisting of internal ribosome entry site linked to CD gene). (b) Upon 5-FC administration, optimized CD metabolizes the antifungal prodrug Toca FC (a proprietary formulation of 5-FC) to the active anticancer drug 5-FU directly within infected cancer cells. (c) Illustration of mechanism of action of Toca 511 and Toca FC causing antitumor immune activation via the tumor microenvironment: (1) Gamma-retrovirus Toca 511 selectively infects tumor, persists, and further spreads through tumor while delivering the CD gene; (2) Toca FC prodrug locally converts to 5-FU chemotherapeutic drug within the tumor, killing cancer cells and resulting in activation of antigen-presenting cells and T cell priming; (3) ‘Bystander effect’ of locally generated intratumoral 5-FU also eliminates adjacent immunosuppressive myeloid cells (myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs)), thereby activating antitumor immunity
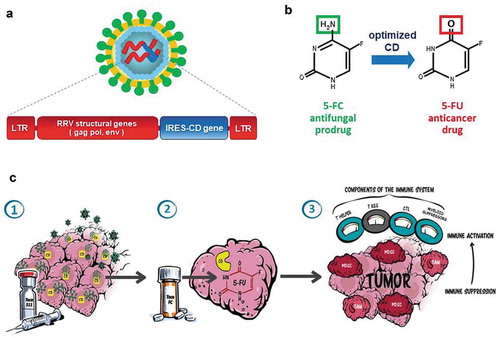
Design and construction of the RRV has been facilitated by the relatively small size of the genome, the availability of in-depth knowledge of mouse gammaretrovirus biology, and the relative ease with which high-titer vector preparations for both laboratory-based and clinical studies can be produced. For example, initial virus production can be achieved by transient transfection of 293T cells with a cloned full-length proviral vector genome encoded on a single plasmid. In addition, the technology of permanent producer cell lines [Citation29] allows relatively simple scale-up and purification from preclinical to clinical and commercial scale, as the virus can be harvested from culture supernatant, analogous to methods of harvesting monoclonal antibodies. This in turn has allowed development of serum-free suspension cultures for vector production, with continuous perfusion technology rather than batch production, and with single-use production materials [Citation30].
Upon infection, incoming RRV undergoes reverse transcription and integrates into genomic DNA of host cells undergoing mitosis, without causing cytolysis. However, as the MLV capsid lacks a nuclear localization signal for active transport across an intact nuclear membrane, the incoming viral capsid cannot enter nuclei in quiescent cells. Thus, integration into the host cell genome occurs only when the nuclear membrane dissolves during mitosis, and so RRV can only infect actively dividing cells [Citation31]. Although not normally cytolytic, RRV can, nevertheless, be engineered to directly kill cells by expression of a prodrug-activating transgene encoding an enzyme that catalyzes intracellular production of a toxic metabolite from a nontoxic substrate [Citation20,Citation21,Citation32–38]. Viral replication results in permanent integration of the transgene without significant inflammation. Subsequently, killing of infected tumor cells can then be triggered upon prodrug administration () at any appropriate time (e.g. upon recurrence of malignant cells that escaped initial treatment). Released tumor-associated antigens can contribute to triggering antitumor immune responses. Since prodrug conversion occurs only in infected cells (i.e. within the local tumor microenvironment (TME)), the adverse side effects caused by systemic chemotherapy can be minimized [Citation39,Citation40].
Earlier versions of RRV contained transgene sequences within the long terminal repeat (LTR) regions [Citation36,Citation41,Citation42]. However, during replication, these earlier RRV had a strong tendency to rapidly delete the inserted transgene sequences. By placing the transgene immediately downstream of the env stop codon, Logg et al. [Citation20] developed an RRV configuration with much greater genomic stability and vector replication kinetics comparable to that of wild-type retroviruses. A small inoculum of this vector mediated efficient delivery and spread of the encoded Green Fluorescent Protein (GFP) reporter gene throughout an entire population of cancer cells in vitro and in subcutaneous tumors in vivo in animal models [Citation20]. Subsequently, it was found that RRV with this configuration can efficiently transduce and stably propagate in a wide variety of cancer cell lines in vitro and tumor models in vivo [Citation20,Citation32–35,Citation37,Citation38,Citation43–56]. When RRV infect target tumor cells, each transduced cell itself becomes a virus-producing cell that promotes further transduction events; therefore, a small inoculum results in remarkable in situ amplification [Citation37,Citation38,Citation44,Citation49,Citation50,Citation54], achieving better therapeutic results in animal models than 1000-fold higher levels of replication-defective adenoviral vectors [Citation50], or 10,000-fold higher levels of highly concentrated non-replicating retroviral vectors [Citation57]. As noted, RRV are non-cytolytic, and can also inhibit interferon production [Citation28]. This renders them less likely to prematurely trigger local inflammation and innate immune responses leading to adaptive antiviral immunity during the process of viral replication and intratumoral spread, as compared to direct oncolytic viruses such as adenovirus or vaccinia.
We first developed and tested a therapeutic RRV by replacing the GFP transgene with yeast cytosine deaminase (CD) [Citation38,Citation44]. Yeast CD converts the well-tolerated prodrug 5-fluorocytosine (5-FC) into the active chemotherapeutic drug 5-fluorouracil (5-FU), achieving high local concentrations of the chemotoxic conversion product selectively within the infected cancer cells. We confirmed that enhanced tumor transduction by RRV encoding this CD prodrug activator enzyme (RRV-CD), in conjunction with 5-FC treatment, translates into significant therapeutic benefit in both human xenograft intracranial tumor models [Citation38,Citation44] and syngeneic rodent intracranial tumor models [Citation46]. For clinical use, two potential weaknesses with the vector had to be addressed, in order to maximize therapeutic efficacy in the larger tumors that would need to be treated in the clinical setting: (1) the limited genomic stability of the vector and (2) suboptimal activity of the yeast CD protein [Citation21]. This was accomplished through various modifications of the original vector backbone to remove repeat sequences prone to recombination events, and human codon optimization as well as introduction of thermostabilizing point mutations into the yeast CD gene. These modifications resulted in increased genomic stability of the vector over multiple passages combined with an increase in CD-specific activity in infected cells [Citation21]. These optimization steps led to the development of the enhanced vector, Toca 511 (also referred to as AC3-yCD2, or vocimagene amiretrorepvec), which is under ongoing clinical evaluation.
Vector spread and anticancer efficacy of RRV-CD have been demonstrated in various rodent brain cancer models. As noted above, in addition to its absolute requirement for active cell division, it is now recognized that innate and adaptive immune mechanisms inhibit and clear RRV infection in normal cells and tissues. As a result, systemic biodistribution of RRV is undetectable in immunocompetent hosts [Citation35,Citation44,Citation58]. However, these mechanisms are defective or suppressed in cancer, enabling RRV to replicate selectively in the permissive tumor microenvironment and efficiently transduce cancer cells in vivo. After intratumoral injection of RRV at multiplicities of infection (MOI, i.e. virus dose : glioma cell ratios) as low as 0.01 into pre-established intracranial brain tumor models, >98% transduction can be achieved throughout the entire tumor mass after a few weeks [Citation35,Citation38,Citation44,Citation50,Citation54]. In contrast, tumor transduction levels were ≤1% after injecting the same dose of conventional non-replicating retroviral vectors [Citation44,Citation50]. Furthermore, we found that the ability of RRV to stably integrate and persist in a reservoir of residual infected cancer cells proved highly advantageous, enabling re-infection of recurrent tumors. Thus, a single stereotactic intratumoral injection of RRV-CD followed by multiple cycles of prodrug treatment achieved 90% survival for >135-day follow-up period, compared to 0% survival of controls in <40 days (p < 0. 0001) [Citation38].
As noted, yeast cytosine deaminase expressed by RRV-CD catalyzes intracellular conversion of the antifungal prodrug 5-FC into the anticancer drug 5-FU. The primary mechanism of action by which 5-FU exerts its cytotoxicity is inhibition of thymidylate synthase, the key enzyme in the de novo synthesis of thymidine [Citation59]. Lack of thymidine, the only nucleotide unique to DNA, has a profound effect on actively dividing cells, ultimately resulting in ‘thymineless death’ [Citation60]. However, quiescent cells do not require high levels of thymidine to support DNA replication, and are able to maintain DNA repair by upregulation of the salvage pathway enzyme thymidine kinase [Citation61]. Thus, cancer cells that were previously infected during mitosis, but which then enter quiescence, would survive prodrug treatment. In fact, 5-FU inhibition of thymidylate synthase has even been reported to drive cancer cells into quiescence to escape cell death [Citation62,Citation63]. These infected quiescent cancer cells represent a reservoir of virus-producing cells which continue to constitutively bud off virus progeny from the integrated provirus even in quiescence, enabling viral persistence and continued viral transmission to as yet uninfected cancer cells as they resume mitosis during tumor recurrence. Notably, long-term control of tumor growth was achieved in human xenograft tumor models in immunodeficient mice, but recurrence was still observed upon cessation of prodrug treatment [Citation38].
In contrast, complete tumor eradication was observed in syngeneic intracranial tumor models in immunocompetent mice after a few prodrug cycles [Citation35,Citation58], and these mice survived tumor-free without any further treatment for over 360 days until study termination [Citation58]. These results indicated that antitumor immune responses contributed to elimination of residual disease in immunocompetent hosts.
Immune responses after Toca 511/Toca FC treatment have been evaluated in both subcutaneous and orthotopic intracerebral glioma models [Citation58,Citation64], and in a liver metastasis model of colorectal cancer [Citation65]. These studies have demonstrated that Toca 511/5-FC treatment also exerts an extended immunotherapeutic effect (). Specifically, in preclinical glioma models, notable findings include: (1) intratumoral immune cell population changes, analyzed sequentially at serial time points, showed a ‘bystander effect’ of tumor-localized myelotoxicity caused by 5-FU generated in infected glioma cells. This alters the tumor microenvironment by eliminating myeloid-derived suppressor cells (MDSC) and tumor-associated macrophages (TAM) [Citation64]; (2) after initial reduction in intratumoral CD8+ and CD4+ T cells and B cells, there is a new influx of these lymphocyte populations immediately following reduction of the MDSC and TAM cell populations [Citation64]; (3) after 3–4 prodrug cycles, immunocompetent animals bearing syngeneic intracerebral gliomas were discontinued from prodrug, and continued to survive for >300 days without further treatment and without further evidence of tumor recurrence by bioluminescence imaging [Citation58]; (4) long-term surviving orthotopic tumor animals rejected rechallenge with naïve uninfected glioma cells. This rejection response was completely abrogated by prior antibody-mediated depletion of CD4+ T cells, and partially abrogated by depletion of CD8+ T cells, but not by depletion of NK cells [Citation58]; (5) T cells isolated from long-term surviving orthotopic tumor animals were analyzed by ELISpot and mixed lymphocyte-target cell reactions (MLTR). These assays showed interferon release and effector:target ratio-dependent killing in response to naïve uninfected glioma cells [Citation58]; (6) Adoptive transfer of unfractionated splenocytes and the T cell fraction of splenocytes from long-term surviving orthotopic tumor animals conferred significant survival benefit to naïve uninfected intracerebral glioma-bearing mice. In contrast, adoptive transfer of T cell–depleted splenocyte fractions did not show any benefit [Citation64].
Further detailed analyses of immunological effects of RRV prodrug activator gene therapy in orthotopic glioma models are needed. These include: more detailed immunophenotyping of effector memory T cell subsets, evaluation of dendritic cell function after bystander effect-mediated MDSC depletion, and identification of endogenous tumor antigen targets. These studies are currently ongoing using immunocompetent syngeneic intracerebral glioma models.
The immune microenvironment in gliomas in the brain is highly immunosuppressive and consists mostly of immunosuppressive MDSC, TAM, and microglia cell populations. 5-FU not only causes immunogenic cell death of infected cancer cells [Citation66] but also causes mutagenic dysregulation of nucleotide metabolism [Citation67], and selective depletion of immunosuppressive myeloid cells in the tumor microenvironment [Citation66]. However, because this chemotherapy drug is generated directly within the tumor by prodrug conversion, systemic myelotoxicity is not incurred and the immune system remains intact. Thus, tumor-associated antigen (TAA) release may occur during 5-FC treatment cycles, and is coupled with tumor-localized ‘bystander’ toxicity to immunosuppressive myeloid cells. We hypothesize that this leads to immune activation, effective TAA presentation, and lymphocytic infiltration, resulting in gradual killing and reduction of the tumor [Citation58,Citation64]. While specific TAAs that may be targeted by antitumor immune responses have not been identified, as noted above, our preclinical studies using naïve uninfected tumors for rechallenge of long-term surviving orthotopic tumor-free animals indicate that rejection is not caused by viral antigens [Citation58]. By the same token, adoptive transfer of T cells from treated animals achieved highly significant survival benefit in naïve uninfected orthotopic gliomas [Citation64], again indicating that endogenous tumor antigens and not viral antigens were targeted. This component of the mechanism of action is likely to be highly advantageous for clinical application of this technology, given the difference in size between human and mouse tumors.
Apart from size differences between patient tumors and animal tumor models, other caveats of initial preclinical studies conducted using RRV-CD are that animal tumor models frequently proliferate at a higher rate than patient tumors. Because retroviruses permanently integrate into the cancer cell genome, once the virus has established an infection, it will persist in infected cells and continue to spread whether the cells divide quickly or not. Certainly, however, the level of initial transduction and subsequent time course of replicative spread will differ depending on the proliferative index of the tumor. Another caveat is that, in particular, U-87 gliomas used in initial preclinical studies are non-invasive. However, RRV-CD/Toca 511 has also shown therapeutic efficacy in human U87-EGFRvIII, rat RG2, and murine Tu2449 gliomas [Citation35,Citation46,Citation58,Citation68,Citation69], which are all invasive tumors that more closely mimic human disease.
RRV can also be used to deliver various other prodrug activator enzymes, most of which were originally developed for use in conventional non-replicating vectors. Since RRV retain their full-length retroviral genome, additional insertion of transgene cassettes up to ~1.3 kb in size can be readily accommodated. However, larger transgenes incur genomic instability and are rapidly deleted upon serial passage of the virus. Within this size limitation, other prodrug activator genes that have shown promising results with RRV delivery in preclinical cancer models include:
Herpes simplex virus thymidine kinase (HSV-TK), which converts anti-herpetic prodrugs such as ganciclovir or valacyclovir into nucleotide analog antimetabolites by phosphorylation [Citation50,Citation70]. However, these phosphorylated nucleotide analogs are hydrophilic and do not diffuse freely across cell membranes, and hence require intercellular gap junctions to exert an efficient bystander effect.
E. coli purine nucleoside phosphorylase (PNP), which converts ribonucleoside prodrugs such as 6-methylpurine deoxyriboside or fludarabine phosphate to purine base analog antimetabolites such as 6-methylpurine and 2-fluoroadenine, respectively [Citation33,Citation37]; these free base compounds are highly hydrophobic and can readily diffuse across cell membranes. However, intestinal bacteria can also catalyze prodrug conversion and the prodrugs themselves exhibit some toxicity.
E. coli nitroreductase (NTR), which converts prodrugs such as the dinitrobenzamide CB1954 ((5-(aziridin-1-yl)-2,4-dinitro-benzamide) into a potent alkylating agent causing DNA adducts and crosslinks [Citation71]. This results in very rapid cell killing, but there is also prodrug conversion by intestinal bacteria.
Notwithstanding the above considerations, all of the above prodrug activator systems have shown therapeutic efficacy in preclinical models of glioma [Citation37,Citation50,Citation71], as well as in various other cancer models. In this context, different vector designs have been employed to maximize efficient utilization of the limited transgene capacity, including the use of alternate splice acceptors, 2A sequences, and mini-promoters [Citation22,Citation23,Citation50]. In addition, various routes of administration have been investigated in animal models, and feasibility of RRV-mediated prodrug activator gene therapy has been demonstrated for subcutaneous, intracranial, intravenous, intravesical, and intraperitoneal modes of vector injection [Citation35,Citation38,Citation47,Citation69,Citation72]. As noted above, our lead clinical candidate RRV, Toca 511, encodes an optimized version of yeast cytosine deaminase, chosen for its enhanced prodrug conversion activity and cytotoxicity, as well as its superior bystander activity [Citation21].
3. Clinical investigation of RRV for treatment of recurrent high-grade glioma and other tumors
Typically, treatment for high-grade gliomas (HGG) involves maximal safe surgical resection with combination radiotherapy and chemotherapy [Citation73]. Despite surgery, radiation, and chemotherapy, it is virtually impossible to eradicate all diffusely infiltrating glioma cells, and recurrence is almost inevitable. After initial treatment failure, patients with recurrent glioblastoma have a dismal prognosis of only ~8 months, and the 5-year overall survival after first diagnosis still remains dismal at <10% [Citation74,Citation75]. Accordingly, there remains an increased interest in developing new treatment modalities for HGG. Based on the results from our preclinical studies, we conducted clinical trials of RRV prodrug activating gene therapy in patients with recurrent primary brain tumors.
First-in-human clinical trials using RRV began in 2010 for the treatment of recurrent high-grade gliomas. Over the subsequent decade, three US-based multicenter dose escalation Phase I clinical trials (www.clinicaltrials.gov: NCT01156584, NCT01470794, NCT01985256) were conducted, evaluating different modes of vector delivery. In the first clinical trial (NCT01156584), Toca 511 (vocimagene amiretrorepvec) [Citation21] was delivered by image-guided stereotactic intratumoral injection in recurrent high-grade glioma patients. This was followed by oral administration (weekly every 4 weeks) of Toca FC, an extended-release formulation of 5-FC [Citation76]. As of the clinical data cutoff date, the median overall survival (mOS) in the efficacy evaluable population was 11.2 months (95% CI 7.1, 13.8) [Citation77]. Historical data indicate a mOS of 7.7 months in studies of subjects with recurrent GBM who did not undergo surgical resection [Citation78]. There was confirmation of vector delivery (viral RNA and transgene expression) in some tumor samples from patients who re-recurred after Toca 511 treatment and opted to undergo further resection [Citation77].
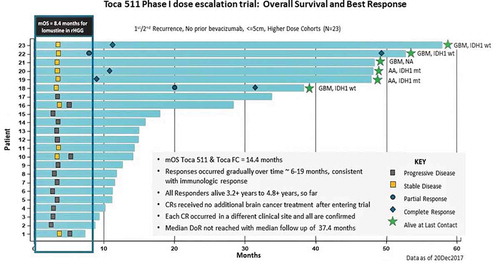
In a second Phase I dose-escalation multicenter trial (NCT01470794), Toca 511 was delivered via local injection into the tumor bed after open-flap surgical resection of recurrent high-grade glioma [Citation39,Citation40]. Both Toca 511 and Toca FC treatment were well tolerated with no dose-limiting toxicities, and the Maximum Tolerated Dose (MTD) was not reached for either agent. No treatment-related hematologic/systemic toxicities or mortality were noted [Citation39,Citation40], suggesting a superior safety profile compared to traditional alkylating chemotherapeutics [Citation79]. Survival, clinical benefit (durable objective responses and stable disease), and safety data covering >4 years for individual patients were investigated, and extensive molecular profiling was also performed. Among all efficacy-evaluable patients (n = 53), the rate of objective responses, which were all complete responses, was 11.3% (6/53) [Citation40]. Dose-dependent improvement in mOS was noted in higher-dose compared to lower dose cohorts (14.4 vs. 11.9 months), with minimal toxicity (see , ). In an ad hoc-identified patient subgroup (the ‘Phase III-eligible subgroup’, n = 23) which encompassed patients from the top three doses (109, 3.3 × 109 and 1010 TU injected), mOS for recurrent HGG was 14.8 months (). In the Phase III-eligible subgroup, the percentage of patients with objective responses was 21.7%, with five complete responders, and the stable disease rate was 21.7%, for an overall clinical benefit rate of 43.5% (, ). All responders met Phase III eligibility criteria and were treated at or within a half-log of the recommended Toca 511 Phase III dose. These five responders and one stable disease patient had long-term survival >36 months (26% of Phase III-eligible subgroup, ). One additional complete responder was in the 5-patient bevacizumab combination treatment cohort and treated with Toca FC in combination with bevacizumab for approximately 4.6 months, then continued on bevacizumab for >16.2 months. Progression-free survival was 3.2 months (95% CI, 3.0 to 3.4), and progression-free survival at 6 months was 16.3%. In tumors resected after Toca 511/Toca FC treatment, extensive necrosis and T cell infiltration was observed with only small areas of residual cancer cells, despite increased tumor signals by radiographic criteria, indicating potential pseudo-progression. Notably, all five responders in the Phase III-eligible subgroup proceeded to achieve complete responses, in some cases 2–3 years after treatment, again suggesting a delayed-onset immunotherapeutic effect [Citation40]. Genetic analyses suggest that mutations, copy-number variations, and neoantigens are linked to survival, as well as tumor-infiltrating immune cell profiles and cytokine levels in peripheral blood [Citation80].
Table 1. Survival results from Phase 1 trials of Toca 511/Toca FC therapy in recurrent high-grade glioma patients (www.clinicaltrials.gov: NCT01156584, NCT01470794, NCT01985256)
Table 2. Objective Response, durableresponse and clinical benefit rates in the Phase 1 resection trial (NCT01470794)
Figure 2. ‘Swim lane’ representation of data from the Phase 1 resection study in recurrent high-grade glioma patients, higher dose cohorts (‘Phase 3 eligible’): Toca 511 and Toca FC leads to 26% long-term survival and 22% durable complete response (CR) rate. All responses are durable CRs and associated with long-term survival (see Cloughesy et al. Citation40)
A third Phase I trial (NCT01985256), initiated in November 2013, was an ascending dose trial to assess the safety and tolerability of Toca 511 administered intravenously [Citation81,Citation82]. This clinical trial combined intravenous (IV) administration of Toca 511 as a bolus prior to surgical resection, followed by injection of Toca 511 into the tumor cavity wall after resection of accessible lesions. The efficiency of tumor transduction after intravenous RRV administration was assessed by molecular analysis of the resected tumor [Citation83]. Immunohistochemistry (IHC) was performed on resected tumor tissue after IV delivery of Toca 511 to determine if T cell infiltrates altered viral entry and/or spread. PCR analysis confirmed cytosine deaminase expression in 11 out of 17 patient samples. Based on IHC interrogation, at dose level 4 (out of five dose levels, n = 9 patients: IV injection of 14.5 ml Toca 511 per day at 3.3 × 108 TU/ml on 3 consecutive days) compared to lower single-day doses, total dose of Toca 511 trends with higher expression of cytosine deaminase protein in tumors. T cell infiltration in tumors was not limiting for cytosine deaminase expression after IV delivery of Toca 511. Among 17 patients treated, the median OS was 13.6 months (95% CI 5.8, 19.7), with stable disease observed in 3 of 17 patients (17.6%), and radiologic responses with delayed onset were observed in 2 of 17 patients following clinical progression [Citation82]. Specifically, in a patient diagnosed with anaplastic astrocytoma (IDH1 wild type) at third recurrence, a partial response was noted on routine follow-up MRI, 9 months after discontinuing Toca FC, while receiving no other anticancer therapy. Another patient (IDH1 mutant) at 1st recurrence had a complete response, which was noted 13 months after initiating Toca FC. This patient is continuing on Toca FC with no other anticancer therapy and remains in response as of last assessment. In terms of treatment-related adverse events, Toca 511 and Toca FC again appeared to be well tolerated with few Grade >3 treatment-related adverse events reported [Citation84].
The promising results from these three early-stage trials led to the initiation of a multicenter randomized Phase II trial (www.clinicaltrials.gov: NCT02414165) in 2015, which was subsequently rolled into a registrational Phase III trial (‘Toca 5 trial’, ) for recurrent high-grade glioma after FDA granted ‘Breakthrough Therapy’ designation in February 2017. Control arm patients were treated with physicians’ choice of chemotherapy (lomustine, temozolomide, or bevacizumab). This study was carried out at 67 sites globally with a total of 403 patients enrolled. Final analysis of the trial results in late 2019 showed that safety, tolerability, and adverse event profiles were favorable as seen in earlier trials (). However, the treatment failed to meet both the primary endpoint of overall survival, as well as secondary endpoints including durable response rate, durable clinical benefit rate, duration of durable response, and overall survival at 12 months [Citation85].
Figure 3. ‘TOCA 5’ Phase 2/3 clinical trial of Toca 511/Toca FC for recurrent high-grade glioma (HGG). (a) Study design of TOCA 5 trial. This was the largest randomized study conducted in the setting of recurrent HGG. GBM: glioblastoma multiforme, AA: anaplastic astrocytoma, IDH1: isocitrate dehydrogenase-1, KPS: Karnofsky Performance Score, SOC: standard of care. (b) Kaplan–Meier plot of overall survival by time from last Temozolomide treatment to randomization (ITT population). (c) Kaplan–Meier plot of overall survival by number of Toca FC cycles in the Phase 2/3 trial (intention-to-treat (ITT) population)
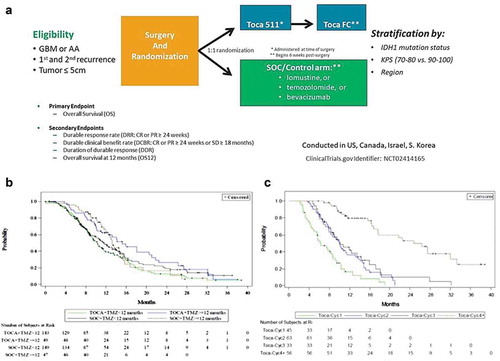
Table 3. No major safety signals with Toca 511 and Toca FC in Phase 3 trial
Additional analyses were conducted to evaluate the impact of prior temozolomide (TMZ) treatment on survival post-Toca 511 and Toca FC treatment (). Temozolomide is known to be lymphodepleting and thus has the potential to negatively impact the effectiveness of immune therapies. For patients randomized to the Toca 511 and Toca FC arm, survival was significantly longer in patients whose last TMZ treatment was more than 12 months prior to randomization in the study. The median OS was 15.2 months in patients with last TMZ at least 12 months or more, vs. 10.3 months in patients with TMZ less than 12 months prior (HR = 0.61; 95% CI: 0.41, 0.90; p = 0.0127) [Citation85]. This difference in survival based on the length of time from TMZ treatment is not observed in the control arm, whose treatments were not intended to function as immune therapies (HR = 0.86; 95% CI: 0.57, 1.30; p = 0.4830) [Citation85]. Patients at second recurrence (see below) are often more than 12 months from their prior TMZ treatment, as expected for the treatment experience of these groups where the median time from last TMZ treatment in patients at first recurrence was 2.5 months vs 17.7 months for patients at second recurrence. This observation provides insight into the role the immune system likely plays in Toca 511 and Toca FC treatment. This impact is not absolute, however, as many of the longest surviving Toca 511 and Toca FC patients were recently treated with TMZ (data on file). This most likely highlights the complexity of treating recurrent HGG where immunological health, treatment dose and several other factors are important to a patients’ overall survival.
One major factor that may have contributed to the overall failure of this trial was that, in the intent-to-treat (ITT) population, the patients had received only a median of two Toca-FC treatment cycles during the trial. We had previously shown in syngeneic mouse models that it took at least three cycles of 5-FC dosing to ensure tumor eradication [Citation35] and so we plotted the number of rounds of Toca FC against survival (). From this analysis alone, it is not possible to determine which of the two variables (doses completed or survival) is responsible for the other (i.e. it is possible that simply the longer one lives, the more chance there is to take Toca FC). Nonetheless, the plots are consistent with the idea that four cycles of 5-FC are more effective than one, two, or three courses in these cohorts. This observation could explain the differences in survival and response rate between the Phase I and Phase II/III trials. We hypothesize that this may have been in part due to physician’s decision to remove patients from treatment at the first sign of progression, despite protocol guidance that physicians treat through progression until MRI confirmation and clinical determination of progression vs. ‘pseudo-progression’.
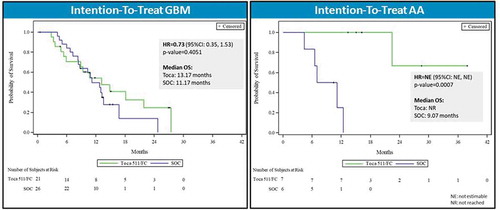
Despite the overall trial failure, analysis of the preplanned subgroups in the Phase III trial did yield interesting results. Subjects with second recurrence (n = 60) showed a 57% risk reduction for death when treated with Toca 511/Toca FC (21.82 months median OS compared to 11.14 months, HR = 0.43, p = 0.0162, ) representing an approximate doubling of survival [Citation85]. Further analysis of the second recurrence subgroup showed that patients with IDH1 mutations and anaplastic astrocytoma (AA) had the greatest survival benefit (, HR = 0.102, p = 0.009). 85 Second recurrence patients with IDH1 mutations and AA received a median of six cycles of Toca FC compared to a median of two cycles for the ITT group.
Figure 4. Overall survival in patient subgroup with two or more recurrences (intention-to-treat population). Left panel: Kaplan–Meier plot of overall survival in the second recurrence subgroup; Toca 511 and Toca FC showed 57% reduction in risk of death (~2X median survival over standard of care (SOC)). HR: hazard ratio, OS: overall survival. Right panel: Key parameters of the Toca 511/Toca FC treatment vs. SOC groups, showing good comparability. KPS: Karnofsky Performance Score, IDH: isocitrate dehydrogenase

Figure 5. Overall survival by tumor histology in patient subgroup with two or more recurrences (intention-to-treat population). Left panel: Kaplan–Meier plot of overall survival in second recurrence subgroup patients with glioblastoma multiforme (GBM). Right panel: Kaplan–Meier plot of overall survival in second recurrence subgroup patients with anaplastic astrocytoma (AA). HR: hazard ratio, OS: overall survival
Molecular data from the Toca 5 trial indicated that there were no significant arm imbalances in baseline tumor-infiltrating lymphocyte or peripheral blood mononuclear cell profiles [Citation86]. These data also showed that AA patient tumors and baseline blood have a significantly more robust immune profile compared to GBM patients. In addition, compared to IDH1 wild-type patients, IDH1 mutant tumors and baseline blood also appeared to have greater potential to generate antitumor responses based on significantly increased infiltrating lymphocytes and higher levels of CD8+ T cells. Similar to the previous two subgroups, second recurrence patient tumors and baseline blood also showed a better immune profile to potentially generate antitumor responses compared to patients at first recurrence. Overall, distinct significant differences in baseline tumor-infiltrating lymphocyte or peripheral blood mononuclear cell profiles were observed across the subgroups analyzed.
Clinical evaluation of Toca 511/Toca FC has also been pursued in other metastatic solid tumors (primarily colorectal and pancreatic cancer) in another Phase I trial (www.clinicaltrials.gov: NCT02576665) evaluating safety, vector uptake and expression, and the changes in infiltrating tumor cells pre- and post-Toca FC. The preliminary data show that (1) most patients enrolled had advanced colorectal cancer; (2) IV delivery of up to ~2.5 × 1010 TU appeared safe and well tolerated; (3) at the three highest doses, there were patients with enough vector uptake and expression, in both ‘hot’ and ‘cold’ tumor areas, to expect generation of a therapeutic response (based on mouse model data, a minimum of 3–5% local tumor infection); (4) changes were observed in infiltrating immune cells after Toca-FC treatment in some patients that would correspond to induction of anticancer immune responses [Citation87].
Plans are also in progress to evaluate this gene therapy approach in combination with the standard of care (chemotherapy and radiation) in newly diagnosed HGG patients (NRG-BN006) [Citation88].
4. Safety considerations for clinical use of RRV
The current clinical trials have demonstrated improved outcomes compared to historical controls in Phase I trials, and equivalence to standard-of-care in a Phase II/III trial with non-optimal Toca FC dosing. However, all trials so far have demonstrated an excellent safety profile for Toca 511 and Toca FC in >350 patients treated to date. Analyses of molecular fate of the viral genome have confirmed the tumor specificity of the virus in patients, the universal disappearance from the blood of viral sequences coinciding with development of neutralizing antibodies on the relatively rare (10–30%) occasions when that was observed, and the apparent lack of potentially dangerous integration sites [Citation39,Citation84].
Lack of viral dissemination within the host remains a key measure of safety: as noted, innate and adaptive immune mechanisms are active in normal cells and tissues but defective or suppressed in tumors, which is key to the tumor-selectivity of this virus (as well as most oncolytic viruses) and its lack of systemic dissemination. Indeed, in preclinical studies, vector biodistribution occurred over time in lymphoid tissues of athymic nude mice lacking T cell-mediated adaptive immunity, and in permissive BALB/c mice which are the natural hosts for MLV due to their genetic defects in innate anti-retroviral immunity. However, RRV infection and spread was found to be restricted in fully immune-competent mouse strains [Citation35,Citation58] and in rats [Citation44]. While amphotropic MLV can enter human lymphocytes and integrate into the host genome, vector spread has been shown to be inefficient in primary lymphocytes in culture [Citation2,Citation89]. Any infected lymphocytes prevent further virus replication through innate antiretroviral defense mechanisms (e.g. APOBEC3-F and -G, tetherin), and are eventually cleared through adaptive immune responses recognizing viral peptides presented by MHC on the cell surface. Accordingly, it seems unlikely that Toca 511 dissemination could happen by means of viral spread from infected lymphoid cells.
Moreover, the lack of a nuclear localization signal in RRV restricts viral replication to actively dividing cells, thereby imparting an additional degree of selectivity for cancer cells. Furthermore, the incorporation of a prodrug-activating enzyme adds a safety switch to the therapy, in that any replicating normal cells that might be infected by the vector will also be killed upon pro-drug administration. Also as noted above, the favorable safety profile of the current clinical vector in human subjects, plus preclinical efficacy data with Toca 511 administered intravenously [Citation69], allowed for the initiation of Phase I trials investigating the efficacy of tumor targeting following IV delivery of the vector, in patients either undergoing re-resection after recurrence of HGG (NCT01985256, n = 17 patients) or suffering from metastatic solid tumors (NCT02576665, see above, n = 21 patients).
5. Potential pitfalls and barriers to success
The failure of the Toca 511 Phase III trial was certainly a disappointment overall, and a number of potential pitfalls and barriers to success may have contributed to this failure. These include: the efficiency of initial virus delivery to tumors, potential physical and biological barriers to virus replication, lack of adherence to the multi-cycle prodrug regimen, and inability to induce an adequate antitumor immune response.
Mode of initial virus delivery: In the Phase III trial, virus was injected into post-resection tumor cavity walls. However, once resected, it is difficult to know exactly where pockets of infiltrating unresected tumor may be located. Hence, even with multiple injections at different sites, this runs the risk that not all residual tumor in the resection margins will be injected. However, all three Phase I dose escalation trials evaluating different routes of virus delivery (i.e. injection into post-resection tumor cavity walls, MRI-guided stereotactic injection directly into tumors in situ, and systemic intravenous infusion) showed similar median overall survival outcomes () that exceeded historical benchmarks, with complete responses in some patients. This suggests that the mode of delivery may not have had substantial impact on the Phase III trial outcome.
Physical barriers to virus replication: Diffusely infiltrating glioma cells may be difficult to reach for virus released from infected cells in the body of the tumor, as intervening quiescent normal brain tissue represents a physical barrier to infecting these cells. Similarly, necrotic areas of the tumor also represent a physical barrier for virus particles, which must traverse these areas by simple diffusion before they can reach a cancer cell substrate for further infection and replication. However, as retroviruses permanently integrate into the cancer cell genome, infected cancer cells as well as their progeny cells constitutively produce virus. As long as these cells also infiltratively migrate alongside uninfected cells, they can transmit the virus through direct cell-cell contact whenever the uninfected cells enter mitosis.
Biological barriers to virus replication: Possible biological barriers to virus replication include possible downregulation of PiT-2, the inorganic phosphate transporter protein which serves as the cellular receptor for amphotropic RRV. However, cancer cells require high levels of phosphate for nucleotide synthesis and cellular metabolism. Another potential biological barrier is the possible upregulation of innate antiviral defense mechanisms activated by interferon signaling, such as APOBEC3F/G, SAM-HD1, and tetherin. However, interferon signaling pathways and innate antiviral defenses are frequently defective in cancer cells. In fact, as noted above, this is one of the fundamental mechanisms underlying tumor selectivity of viruses generally, including RRV as well as oncolytic viruses.
It is possible that some of these innate antiviral defense mechanisms are further downregulated as tumors progress to second or third recurrence. In addition, the biology of anaplastic astrocytoma and IDH-mutant tumors may similarly predispose to virus permissivity, as suggested by significant survival benefit of Toca 511/Toca FC treatment observed in these patient subgroups as compared to control treatments. These possibilities will be explored in further clinical trials focused on these patient subgroups, to be conducted by a new biotech sponsor, Denovo Biopharma.
It is also possible that, as virus replication proceeds, cancer cells with residual interferon signaling may activate antiviral defenses, and these cells would preferentially survive. Since retroviruses are not intrinsically cytolytic, such selection pressure would not be applied until prodrug administration, so this is presumably less of a potential issue as compared to oncolytic viruses. However, after multiple prodrug cycles that eliminate the most permissive cells first, there may be selective survival of more resistant cells, thereby impeding further virus replication.
As noted above, RRV will continue to spread progressively whether cancer cells divide quickly or not, but the level of initial transduction and subsequent time course of replicative spread will differ depending on the proliferative index of the tumor. Also as noted, entry into quiescence has been reported to be a mechanism by which cancer cells escape 5-FU inhibition of thymidylate synthase. However, retroviruses permanently integrate into the cancer cell genome. Accordingly, once the virus has established an infection, the integrated provirus will persist in infected cells and their progeny cells, even if they go into quiescence and escape cytotoxicity from intracellular 5-FC prodrug conversion to 5-FU. Therefore, once infected, even quiescent cells will continue to produce virus constitutively. This enables RRV released from these cells to continue spreading whenever quiescent uninfected cancer cells resume mitosis, which eventually must occur if the tumor is to continue growth. Also, eventual resumption of mitosis again renders infected quiescent cells susceptible to ‘thymineless death’ from prodrug conversion upon the next administration of prodrug. This enables multiple cycles of prodrug to achieve continued efficacy.
Lack of adherence to multi-cycle prodrug regimen: In this context, inadequacy of prodrug dosing was likely a major problem. As noted above and shown in , the median number of prodrug cycles administered to patients in the Phase III trial was only two cycles [Citation85]. In contrast, 3–4 prodrug cycles were required for antitumor immune responses to be fully activated in preclinical models [Citation35,Citation58], and a highly positive correlation was observed between the number of prodrug cycles administered and survival outcomes in early phase clinical trials [Citation39,Citation40]. Similarly, in the Phase III trial, improved survival was associated with increasing number of prodrug cycles, with 1–3 cycles showing no long-term survival benefit, but 4+ cycles showing a plateau of ~25% long-term survivors, which is almost identical to the results achieved in the Phase I dose escalation trials (). As noted, this could be interpreted as better prognosis (AA, IDH mutant) patients simply being alive longer to take more cycles of prodrug, but the randomized matched control patients in these subgroups did not live as long, and this difference was highly statistically significant (, ). Thus, a critical factor for success in future clinical trials is likely to be maintaining adherence to continuing prodrug cycles.
Table 4. Median number of drug treatments for the intention-to-treat group (Toca FC) and the standard-of-care (SOC) control arm in the Phase 3 trial
Inability to induce an adequate antitumor immune response: As noted above, an inadequate number of prodrug cycles was associated with failure to fully eradicate orthotopic gliomas in syngeneic immunocompetent animal models, and reduced survival in the Phase III clinical trial (). Presumably this is because initial tumor transduction and vector replication at early time points are restricted to areas immediately surrounding injection sites. Accordingly, the extent of direct tumor cell killing (and hence tumor antigen release) and local bystander toxicity to MDSC and TAM populations (and hence alleviation of immunosuppression) are initially low, but increase with each successive cycle of RRV replication from viral ‘reservoir’ cells and subsequent prodrug treatment. Hence, without an adequate number of prodrug cycles, antitumor immunity may not be fully activated. Nonetheless, there are certainly other immunological parameters that may influence the ability of Toca 511/Toca FC treatment to induce a robust antitumor immune response. These may include: paucity of immunogenic tumor antigens, loss or impaired function of antigen-presenting cells, inhibition of T cell activation or induction of T cell anergy by non-myeloid cells (e.g. T regulatory cells) or signaling pathways (e.g. TGF-β). Further detailed characterization of antitumor immune responses that develop after Toca 511/Toca FC treatment, especially within the microenvironment of patient gliomas, is needed.
6. Future directions
Despite the potential pitfalls and barriers, promising evidence of highly statistically significant survival benefit has been observed in prespecified patient subgroups treated with Toca 511, as compared to their respective randomized matched control groups. Accordingly, clinical investigation of Toca 511 in these subgroups will continue under a new sponsor, Denovo Biopharma, which acquired this technology as of June 2020, with the vector now re-designated as DB107.
Further clinical studies will also undoubtedly incorporate combination regimens to enhance therapeutic efficacy. In 2005, the Stupp study demonstrated increased survival in first line GBM patients when standard treatment modalities (TMZ and fractionated radiation) were combined, illustrating the prognostic importance of combinatorial treatments for GBM [Citation73]. Preclinical studies indicate the need to carefully evaluate regimens combining Toca 511/5-FC and TMZ, as the latter has been found to either synergistically enhance or inhibit the efficacy of prodrug activator gene therapy depending on the model used [Citation90]. Also, as noted, Phase III trial results suggested better outcomes in patients who had their last TMZ treatment more than 12 months prior to Toca 511/Toca FC [Citation85], suggesting that TMZ may inhibit subsequent antitumor immune responses due to its severe myelotoxicity.
As described above, Toca 511 converts the nontoxic antifungal 5-FC into the potent chemotherapeutic drug 5-FU within infected cancer cells, resulting in high intracellular drug concentrations. As 5-FU has been widely used to sensitize several human cancers to radiation therapy, combining Toca 511/5-FC therapy with radiation may also increase the therapeutic efficacy of the treatment. In fact, Takahashi et al. [Citation68] confirmed that intracellular levels of 5-FU produced during Toca 511/5-FC therapy could act as a radio-sensitizing agent, supporting the rationale for the combination of radiation with Toca 511/5-FC treatment as a first-line therapy for treatment of patients with brain tumors. Diffusion of 5-FU into the tumor microenvironment also kills immunosuppressive myeloid cells locally, while systemic levels of 5-FU remain trace or undetectable, avoiding systemic toxicity [Citation58,Citation64,Citation65]. This may further contribute to abscopal effects of radiation therapy in combination with Toca 511/Toca FC.
The majority of preclinical studies to date have focused on intratumoral delivery of RRV, yet there are significant clinical advantages of delivering the vector without surgery. Intravenous delivery of vector may be a noninvasive method to achieve simple repeated administrations without the added morbidity of surgery, potentially broadening treatment for patients unamenable to surgical intervention. A number of preclinical safety and efficacy studies utilizing IV delivery of Toca 511 were conducted, and this route of administration has been well tolerated [Citation32,Citation45,Citation65,Citation69]. Notably, IV administration did not result in widespread dissemination of the vector in non-permissive immune competent mice and rats, with little or no viral DNA found in blood and most of the tissues examined 180 days post-administration. Preclinical models revealed efficient vector infection and replication in tumors in both immune-competent and immunocompromised animal models, with long-term survival in immune-competent mice following IV delivery of Toca 511 in combination with 5-FC treatment [Citation32,Citation65,Citation69]. Additionally, IV delivery may allow for repeated administration of RRV in a noninvasive manner, although higher doses of Toca 511 will be required to show efficacy, as compared to intratumoral administration. As noted, a Phase 1 dose escalation study (NCT01985256) has been conducted, in which Toca511 was injected intravenously to patients with recurrent HGG for 1, 3, or 5 consecutive days. CD was expressed in tumors that were subsequently resected, with the highest level of CD protein detected following 3 days of IV injection [Citation81]. With these promising results, a clinical trial was opened in 2016 utilizing IV Toca 511 administration (NCT02576665) for systemic cancers, including pancreatic and colorectal cancer [Citation87].
In terms of developing a pipeline of clinically relevant vectors, one of the strategies that has been studied is the incorporation of immune checkpoint inhibition into therapeutic strategies involving RRV. These strategies involve both combination of Toca 511/Toca FC with existing checkpoint inhibitor antibody therapies as well as encoding an immune checkpoint inhibitor into the vector itself. Initial work focused on the incorporation of shRNA or miRNA into the vector to knock-down PD-L1 expression, with multiple configurations of RRV allowing shRNA/miRNA expression developed and tested [Citation22]. In vitro assays confirmed downregulation of PD-L1 using an RRV expressing miRNA in PD-L1+ tumor cells by restoration of immune activation (IFNγ expression). Further development of this strategy has resulted in the generation of Toca 521, an RRV expressing a single-chain variable fragment (scFv) targeting PD-L1 [Citation91]. This report confirmed that the PD-L1 scFv expressed and secreted from Toca 521 infected cells competes with PD-1 binding to PD-L1, and can rescue PD-1/PD-L1-mediated immune suppression in vitro. In comparison with systemically administered anti-PD-1 or anti-PD-L1 monoclonal antibodies, Toca 521 conferred robust, durable and highly selective antitumor activity in orthotopic models of HGG and breast cancer, without incurring systemic autoimmune adverse effects.
7. Expert opinion
The initial disappointing results of cancer gene therapy studies using replication-deficient retroviral vectors were due to very limited biodistribution past the injection needle track [Citation3,Citation4]. RRV were designed to increase the extent of gene delivery to tumors in vivo. The tumor-specific non-lytic spread and persistence of RRV allow consideration of several antitumor strategies. One of these is the concept of delivering transgenes encoding prodrug activating enzymes, which allows selective timing of the induction of cell lysis by prodrug administration. This approach also appears to be well suited for inducing antitumor immune responses, at least in animal models, through ‘bystander’ effects that eliminate immunosuppressive myeloid-derived cells within the tumor. Having now moved from bench to bedside, the therapy showed promising efficacy compared to historical benchmarks in initial clinical studies, was well tolerated, and had an excellent safety profile [Citation39,Citation40,Citation84]. However, the therapy failed to meet its endpoints in a recent Phase II/III trial [Citation85]. Despite disappointment in the overall trial results, the positive outcome for certain subgroups of patients is compelling [Citation85]. In addition, it is likely that the 5-FC dosing was far from optimal [Citation85]. Whether it is the reciprocal relationship of being able to survive long enough to take sufficient rounds of Toca FC, or the ability to survive long enough to benefit from a relatively slow development of an effective anticancer immune response, or the general immune competence of patients with relatively slow-moving disease, it seems clear that this therapy has strong potential to help a wide range of cancer patients with enough survival potential to allow development of anticancer immune responses and/or four or more cycles of Toca FC treatment [Citation80,Citation85]. With the current safety data in hand [Citation84], this points toward use in earlier stage disease, where a key issue will be whether live viral therapy, which is aimed at inducing anticancer immune responses, can be positioned to be compatible with standard-of-care (which is, more often than not, highly myeloablative chemotherapy). However, given the episodic nature of many chemotherapies and the ‘rebound’ effects of many chemotherapies on tumors [Citation92] and the immune system [Citation93], this seems quite achievable in many clinical situations. Combinations with checkpoint inhibitors such as anti-CTLA4 or anti-PD-L1 show at least additivity in animal models and do not need significant workarounds, as may be required in combination with conventional chemotherapies. We expect the same kind of additivity with most immune-based therapies such as CAR-T cells. Furthermore, the now-proven ability of RRV to target tumor cells even in the brain following intravenous administration, may allow expansion of its clinical utility to a broader range of tumor types, including secondary brain tumors caused by primary malignancies from outside the brain, metastasizing to the CNS. Employing Toca 511, with its ability to spread without much hindrance from the immune system, and then ‘pulling the trigger’ by Toca FC administration leading to local killing of cancer cells and immunosuppressive myeloid cells without systemic myelotoxicity, may have distinct advantages for this type of treatment. In conclusion, Toca 511/Toca FC continues to show strong preclinical and, we believe, clinical promise and is worth pursuing in clinical situations where there is a fit with the standard-of-care that allows for four or more rounds of prodrug cycles and a sufficient runway for an antitumor immune response to develop (at least 6–8 months). We believe that HGG remains one such indication.
Article highlights
• Retroviral replicating vectors (RRV), currently under clinical investigation for the treatment of recurrent high-grade glioma, permanently integrate into the cancer cell genome, and each infected cancer cell constitutively produces new virions that bud from the cell surface and spread to adjacent cancer cells.
• The initial rationale for the use of these vectors in cancer was their selectivity for dividing cells, as the majority of normal cells in vivo are quiescent, but it is now appreciated that additional mechanisms of tumor-selectivity include cancer cell-intrinsic defects in innate immunity, and tumor microenvironment-mediated suppression of adaptive immunity, which both act to restrict retroviral replication in normal cells and tissues.
• Preclinical studies demonstrated that RRV can mediate efficient and tumor-specific delivery of transgenes encoding prodrug activating enzymes in a variety of cancer models, and that subsequent prodrug treatment leads to synchronized cell killing of infected tumor cells, as well as activation of antitumor immune responses due to ‘bystander’ killing of adjacent immunosuppressive myeloid-derived cells residing within the tumor.
• Optimization of the viral backbone and therapeutic transgene resulted in an improved clinical candidate RRV, Toca 511, for delivery of an optimized yeast cytosine deaminase prodrug activator gene, which converts the prodrug 5-fluorocytosine (5-FC) to the active drug 5-fluorouracil (5-FU). This therapy has shown highly promising results in early-stage clinical trials for recurrent high-grade glioma.
• This vector progressed to a registrational Phase III trial, but the results announced in late 2019 found that the therapy failed to meet primary and secondary endpoints. However, the median 5-FC dosing schedule was not optimal, and promising signals were observed upon analysis of predetermined subgroups. These signals are to be validated in future clinical trials.
• Additional directions for further clinical and translational development include the application of Toca 511/Toca FC to systemic malignancies such as colorectal cancer and bladder cancer in situations where there is sufficient survival expectation to allow four courses of 5-FC prodrug and induction of anticancer immune responses (a minimum of 6-8 months), and development of RRV for tumor-selective delivery of immune checkpoint inhibitors.
This box summarizes key points contained in the article.
Declaration of interest
N Kasahara was previously a consultant for Tocagen Inc., and D Ostertag and DJ Jolly are former employees of Tocagen, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
Acknowledgments
We thank all the trial subjects for volunteering to participate in the Phase I and Phase II/III trials and the clinical investigators and their staff members who recruited them and carried out the trials.
Additional information
Funding
References
- World Cancer Report: Cancer Research for Cancer Prevention. Wild, CP, Weiderpass E, Stewart, BW. World Health Organization/International Agency for Research on Cancer (IARC) Publications; Lyon, France. 2020.
- Edelstein, ML, Abedi MR, Wixon J, et al. Gene Therapy Clinical Trials Worldwide 1989-2004 - an overview. J Gene Med. 2004;6(6):597-602. DOI: https://doi.org/10.1002/jgm.619
- Rainov NG, Ren H. Clinical trials with retrovirus mediated gene therapy–what have we learned? J Neurooncol. 2003;65(3):227–236. DOI:https://doi.org/10.1023/B:NEON.0000003652.71665.f2
- Lang FF, Bruner JM, Fuller GN, et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol. 2003;21(13):2508–2518. DOI:https://doi.org/10.1200/JCO.2003.21.13.2508
- Harrington KJ, Vile RG, Melcher A, et al. Clinical trials with oncolytic reovirus: moving beyond phase I into combinations with standard therapeutics. Cytokine Growth Factor Rev. 2010;21(2-3):91–98. DOI:https://doi.org/10.1016/j.cytogfr.2010.02.006
- Gomez-Manzano C, Fueyo J. Oncolytic adenoviruses for the treatment of brain tumors. Curr Opin Mol Ther. 2010;12(5):530–537.
- Russell SJ, Peng KW. Measles virus for cancer therapy. Curr Top Microbiol Immunol. 2009;330(213–241). DOI:https://doi.org/10.1007/978-3-540-70617-5_11
- Lal R, Harris D, Postel-Vinay S, et al. Reovirus: rationale and clinical trial update. Curr Opin Mol Ther. 2009;11(5):532–539.
- Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9(1):64–71. DOI:https://doi.org/10.1038/nrc2545
- Grandi P, Peruzzi P, Reinhart B, et al. Design and application of oncolytic HSV vectors for glioblastoma therapy. Expert Rev Neurother. 2009;9(4):505–517. DOI:https://doi.org/10.1586/ern.09.9
- Lorence RM, Roberts MS, O’Neil JD, et al. Phase 1 clinical experience using intravenous administration of PV701, an oncolytic Newcastle disease virus. Curr Cancer Drug Targets. 2007;7(2):157–167. DOI:https://doi.org/10.2174/156800907780058853
- Yoon TK, Shichinohe T, Laquerre S, et al. Selectively replicating adenoviruses for oncolytic therapy. Curr Cancer Drug Targets. 2001;1(2):85–107. DOI:https://doi.org/10.2174/1568009013334223
- Dock G. Influence of complicating diseases upon leukaemia. J Med Sci. 1904;127:563–592.
- Sinkovics JG, Horvath JC. Newcastle disease virus (NDV): brief history of its oncolytic strains. J Clin Virol. 2000;16(1):1–15. DOI:https://doi.org/10.1016/S1386-6532(99)00072-4
- Russell SJ. Replicating vectors for cancer therapy: a question of strategy. Semin Cancer Biol. 1994;5(6):437–443.
- Heiber JF, Barber GN. Evaluation of innate immune signaling pathways in transformed cells. Methods Mol Biol. 2012;797:217–238. DOI:https://doi.org/10.1007/978-1-61779-340-0_15
- Naik S, Russell SJ. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther. 2009;9(9):1163–1176. DOI:https://doi.org/10.1517/14712590903170653
- Arens R. Rational design of vaccines: learning from immune evasion mechanisms of persistent viruses and tumors. Adv Immunol. 2012;114:217–243. DOI:https://doi.org/10.1016/B978-0-12-396548-6.00009-3
- Butt AQ, Mills KH. Immunosuppressive networks and checkpoints controlling antitumor immunity and their blockade in the development of cancer immunotherapeutics and vaccines. Oncogene. 2014;33(38):4623–4631. DOI:https://doi.org/10.1038/onc.2013.432
- Logg CR, Tai CK, Logg A, et al. A uniquely stable replication-competent retrovirus vector achieves efficient gene delivery in vitro and in solid tumors. Hum Gene Ther. 2001;12(8):921–932. DOI:https://doi.org/10.1089/104303401750195881
- Perez OD, Logg CR, Hiraoka K, et al. Design and Selection of Toca 511 for Clinical Use: modified Retroviral Replicating Vector With Improved Stability and Gene Expression. Mol Ther. 2012;20(9):1689-1698. DOI:https://doi.org/10.1038/mt.2012.83
- Lin AH, Twitty CG, Burnett R, et al. Retroviral replicating vector delivery of miR-PDL1 Inhibits immune checkpoint PDL1 and enhances immune responses in vitro. Mol Ther Nucleic Acids. 2017;6:221–232. DOI:https://doi.org/10.1016/j.omtn.2016.11.007
- Hofacre A, Yagiz K, Mendoza D, et al. Efficient therapeutic protein expression using retroviral replicating vector with 2A peptide in cancer models. Hum Gene Ther. 2018;29(4):437–451. DOI:https://doi.org/10.1089/hum.2017.205
- Logg CR, Kasahara N. Retrovirus-mediated gene transfer to tumors: utilizing the replicative power of viruses to achieve highly efficient tumor transduction in vivo. Methods Mol Biol. 2004;246:499–525. DOI:https://doi.org/10.1385/1-59259-650-9:499
- Dalba C, Klatzmann D, Logg CR, et al. Beyond oncolytic virotherapy: replication-competent retrovirus vectors for selective and stable transduction of tumors. Curr Gene Ther. 2005;5(6):655–667. DOI:https://doi.org/10.2174/156652305774964659
- Tai CK, Kasahara N. Replication-competent retrovirus vectors for cancer gene therapy. Front Biosci. 2008;13:3083–3095. DOI:https://doi.org/10.2741/2910
- Logg CR, Robbins JM, Jolly DJ, et al. Retroviral replicating vectors in cancer. Methods Enzymol. 2012;507:199–228. DOI:https://doi.org/10.1016/B978-0-12-386509-0.00011-9
- Lin AH, Burrascano C, Pettersson PL, et al. Blockade of type I interferon (IFN) production by retroviral replicating vectors and reduced tumor cell responses to IFN likely contribute to tumor selectivity. J Virol. 2014;88(17):10066–10077. DOI:https://doi.org/10.1128/JVI.02300-13
- Sheridan PL, Bodner M, Lynn A, et al. Generation of retroviral packaging and producer cell lines for large-scale vector production and clinical application: improved safety and high titer. Mol Ther. 2000;2(3):262–275. DOI:https://doi.org/10.1006/mthe.2000.0123
- Jolly DJ, Ibanez C. Producer Cells for Replication Competent Retroviral Vectors. United States Patent Number:US 10,316,333 B2, Assignee:Tocagen Inc. Granted: June 11, 2019.
- Lewis PF, Emerman M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J Virol. 1994;68(1):510–516. DOI:https://doi.org/10.1128/JVI.68.1.510-516.1994
- Hiraoka K, Kimura T, Logg CR, et al. Therapeutic efficacy of replication-competent retrovirus vector-mediated suicide gene therapy in a multifocal colorectal cancer metastasis model. Cancer Res. 2007;67(11):5345–5353. DOI:https://doi.org/10.1158/0008-5472.CAN-06-4673
- Kikuchi E, Menendez S, Ozu C, et al. Delivery of replication-competent retrovirus expressing Escherichia coli purine nucleoside phosphorylase increases the metabolism of the prodrug, fludarabine phosphate and suppresses the growth of bladder tumor xenografts. Cancer Gene Ther. 2007;14(3):279–286.
- Kubo S, Takagi-Kimura M, Logg CR, et al. Highly efficient tumor transduction and antitumor efficacy in experimental human malignant mesothelioma using replicating gibbon ape leukemia virus. Cancer Gene Ther. 2013;20(12):671–677. DOI:https://doi.org/10.1038/cgt.2013.67
- Ostertag D, Amundson KK, Lopez Espinoza F, et al. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro Oncol. 2012;14(2):145–159. DOI:https://doi.org/10.1093/neuonc/nor199.
- Reik W, Weiher H, Jaenisch R. Replication-competent Moloney murine leukemia virus carrying a bacterial suppressor tRNA gene: selective cloning of proviral and flanking host sequences. Proc Natl Acad Sci U S A. 1985;82(4):1141–1145. DOI:https://doi.org/10.1073/pnas.82.4.1141
- Tai CK, Wang W, Lai YH, et al. Enhanced efficiency of prodrug activation therapy by tumor-selectively replicating retrovirus vectors armed with the Escherichia coli purine nucleoside phosphorylase gene. Cancer Gene Ther. 2010;17(9):614–623. DOI:https://doi.org/10.1038/cgt.2010.17
- Tai CK, Wang WJ, Chen TC, et al. Single-shot, multicycle suicide gene therapy by replication-competent retrovirus vectors achieves long-term survival benefit in experimental glioma. Mol Ther. 2005;12(5):842–851. DOI:https://doi.org/10.1016/j.ymthe.2005.03.017.
- Cloughesy TF, Landolfi J, Hogan DJ, et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med. 2016;8(341):341ra375. DOI:https://doi.org/10.1126/scitranslmed.aad9784.
- Cloughesy TF, Landolfi J, Vogelbaum MA, et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro Oncol. 2018;20(10):1383–1392. DOI:https://doi.org/10.1093/neuonc/noy075.
- Goff S, Traktman P, Baltimore D. Isolation and properties of Moloney murine leukemia virus mutants: use of a rapid assay for release of virion reverse transcriptase. J Virol. 1981;38(1):239–248. DOI:https://doi.org/10.1128/JVI.38.1.239-248.1981
- Stuhlmann H, Jaenisch R, Mulligan RC. Construction and properties of replication-competent murine retroviral vectors encoding methotrexate resistance. Mol Cell Biol. 1989;9(1):100–108. DOI:https://doi.org/10.1128/MCB.9.1.100
- Logg CR, Logg A, Matusik RJ, et al. Tissue-specific transcriptional targeting of a replication-competent retroviral vector. J Virol. 2002;76(24):12783–12791. DOI:https://doi.org/10.1128/JVI.76.24.12783-12791.2002
- Wang W, Tai CK, Kasahara N, et al. Highly efficient and tumor-restricted gene transfer to malignant gliomas by replication-competent retrovirus vectors. Hum Gene Ther. 2003;14(2):117–127. DOI:https://doi.org/10.1089/104303403321070810
- Hiraoka K, Kimura T, Logg CR, et al. Tumor-selective gene expression in a hepatic metastasis model after locoregional delivery of a replication-competent retrovirus vector. Clin Cancer Res. 2006;12(23):7108–7116. DOI:https://doi.org/10.1158/1078-0432.CCR-06-1452
- Wang W, Tai CK, Kershaw AD, et al. Use of replication-competent retroviral vectors in an immune competent intracranial glioma model. Neurosurg Focus. 2006;20(4):E25. DOI:https://doi.org/10.3171/foc.2006.20.4.1
- Kikuchi E, Menendez S, Ozu C, et al. Highly efficient gene delivery for bladder cancers by intravesically administered replication-competent retroviral vectors. Clin Cancer Res. 2007;13(45115 Pt 1):1–4518. DOI:https://doi.org/10.1158/1078-0432.CCR-07-0151
- Kimura T, Hiraoka K, Kasahara N, et al. Optimization of enzyme-substrate pairing for bioluminescence imaging of gene transfer using Renilla and Gaussia luciferases. J Gene Med. 2010;12(6):528–537. DOI:https://doi.org/10.1002/jgm.1463
- Yin D, Zhai Y, Gruber HE, et al. Convection-enhanced delivery improves distribution and efficacy of tumor-selective retroviral replicating vectors in a rodent brain tumor model. Cancer Gene Ther. 2013;20(6):336–341. DOI:https://doi.org/10.1038/cgt.2013.25
- Solly SK, Trajcevski S, Frisen C, et al. Replicative retroviral vectors for cancer gene therapy. Cancer Gene Ther. 2003;10(1):30–39. DOI:https://doi.org/10.1038/sj.cgt.7700521
- Trajcevski S, Solly SK, Frisen C, et al. Characterization of a semi-replicative gene delivery system allowing propagation of complementary defective retroviral vectors. J Gene Med. 2005;7(3):276–287. DOI:https://doi.org/10.1002/jgm.663
- Qiao J, Moreno J, Sanchez-Perez L, et al. VSV-G pseudotyped, MuLV-based, semi-replication-competent retrovirus for cancer treatment. Gene Ther. 2006;13(20):1457–1470. DOI:https://doi.org/10.1038/sj.gt.3302782
- Duerner LJ, Schwantes A, Schneider IC, et al. Cell entry targeting restricts biodistribution of replication-competent retroviruses to tumour tissue. Gene Ther. 2008;15(22):1500–1510. DOI:https://doi.org/10.1038/gt.2008.92
- Hlavaty J, Jandl G, Liszt M, et al. Comparative evaluation of preclinical in vivo models for the assessment of replicating retroviral vectors for the treatment of glioblastoma. J Neurooncol. 2011;102(1):59–69. DOI:https://doi.org/10.1007/s11060-010-0295-5
- Inoko K, Hiraoka K, Inagaki A, et al. Therapeutic activity of retroviral replicating vector-mediated prodrug activator gene therapy for pancreatic cancer. Cancer Gene Ther. 2018;25(7-8):184-195. DOI:https://doi.org/10.1038/s41417-018-0020-7
- Twitty CG, Diago OR, Hogan DJ, et al. Retroviral replicating vectors deliver cytosine deaminase leading to targeted 5-fluorouracil-mediated cytotoxicity in multiple human cancer types. Hum Gene Ther Methods. 2016;27(1):17–31. DOI:https://doi.org/10.1089/hgtb.2015.106
- Tamura K, Tamura M, Ikenaka K, et al. Eradication of murine brain tumors by direct inoculation of concentrated high titer-recombinant retrovirus harboring the herpes simplex virus thymidine kinase gene. Gene Ther. 2001;8(3):215–222. DOI:https://doi.org/10.1038/sj.gt.3301371
- Hiraoka K, Inagaki A, Kato Y, et al. Retroviral replicating vector-mediated gene therapy achieves long-term control of tumor recurrence and leads to durable anticancer immunity. Neuro Oncol. 2017;19(7):918–929. DOI:https://doi.org/10.1093/neuonc/nox038.
- Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–338. DOI:https://doi.org/10.1038/nrc1074
- Heidelberger, C. Biochemical mechanisms of action of fluorinated pyrimidines. Exp Cell Res. 1963;24(SUPPL9):462–471. DOI:https://doi.org/10.1016/0014-4827(63)90286-6
- Sakamoto S, Kasahara N, Iwama T. Thymidine Kinase Activities in Familial Adenomatous Polyposis, Adenomatous Polyps, and Carcinoma of the Colon. in Familial Adenomatous Polyposis. Herrera L, ed. New York: A.R. Liss; 1990. p. 315–324.
- Touil Y, Igoudjil W, Corvaisier M, et al. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin Cancer Res. 2014;20(4):837–846. DOI:https://doi.org/10.1158/1078-0432.CCR-13-1854
- Brown JA, Yonekubo Y, Hanson N, et al. TGF-beta-induced quiescence mediates chemoresistance of tumor-propagating cells in squamous cell carcinoma. Cell Stem Cell. 2017;21(5):650–664.e8. DOI:https://doi.org/10.1016/j.stem.2017.10.001
- Mitchell LA, Lopez Espinoza F, Mendoza D, et al. Toca 511 gene transfer and treatment with the prodrug, 5-fluorocytosine, promotes durable antitumor immunity in a mouse glioma model. Neuro Oncol. 2017;19(7):930–939. DOI:https://doi.org/10.1093/neuonc/nox037.
- Yagiz K, Rodriguez-Aguirre ME, Lopez Espinoza F, et al. A Retroviral replicating vector encoding cytosine deaminase and 5-FC induces immune memory in metastatic colorectal cancer models. Mol Ther Oncolytics. 2017;8:14–26. DOI:https://doi.org/10.1016/j.omto.2017.12.001
- Gmeiner WH. Fluoropyrimidine modulation of the anti-tumor immune response-prospects for improved colorectal cancer treatment. Cancers (Basel). 2020;12(6):1641. DOI:https://doi.org/10.3390/cancers12061641
- Mathews CK. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat Rev Cancer. 2015;15(9):528–539. DOI:https://doi.org/10.1038/nrc3981
- Takahashi M, Valdes G, Hiraoka K, et al. Radiosensitization of gliomas by intracellular generation of 5-fluorouracil potentiates prodrug activator gene therapy with a retroviral replicating vector. Cancer Gene Ther. 2014;21(10):405–410. DOI:https://doi.org/10.1038/cgt.2014.38
- Huang TT, Parab S, Burnett R, et al. Intravenous administration of retroviral replicating vector, Toca 511, demonstrates therapeutic efficacy in orthotopic immune-competent mouse glioma model. Hum Gene Ther. 2015;26(2):82–93. DOI:https://doi.org/10.1089/hum.2014.100.
- Kubo S, Takagi-Kimura M, Tagawa M, et al. Dual-vector prodrug activator gene therapy using retroviral replicating vectors. Cancer Gene Ther. 2019;26(5-6):128–135. DOI:https://doi.org/10.1038/s41417-018-0051-0
- Chen SH, Sun JM, Chen BM, et al. Efficient prodrug activator gene therapy by retroviral replicating vectors prolongs survival in an immune-competent intracerebral glioma model. Int J Mol Sci. 2020;21(4):1433. DOI:https://doi.org/10.3390/ijms21041433
- Collins SA, Kamath P, Matsuura S, et al. Therapeutic efficacy of vocimagene amiretrorepvec (Toca 511) prodrug activator gene therapy in peritoneal carcinomatosis models of ovarian cancer. Gynecol Oncol. 2019;154(1):E29–E30. DOI:https://doi.org/10.1016/j.ygyno.2019.03.231.
- Stupp R, Mason WP, Van Den Bent MJ, et al., European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups, National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. DOI:https://doi.org/10.1056/NEJMoa043330.
- Gorlia T, Stupp R, Brandes AA, et al. New prognostic factors and calculators for outcome prediction in patients with recurrent glioblastoma: a pooled analysis of EORTC Brain Tumour Group phase I and II clinical trials. Eur J Cancer. 2012;48(8):1176–1184. DOI:https://doi.org/10.1016/j.ejca.2012.02.004
- Stupp R, Hegi ME, Mason WP, et al., European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups, National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. DOI:https://doi.org/10.1016/S1470-2045(09)70025-7.
- Aghi M, Vogelbaum MA, Jolly DJ, et al. ST-001 Phase 1 Trial of a retroviral replicating vector (Toca 511) in recurrent high grade glioma patients demonstrates the importance of real-time MRI-guided delivery for dose-related evaluation of safety and efficacy. Neuro Oncol. 2013;15(suppl 3):iii217–iii225. DOI:https://doi.org/10.1093/neuonc/not191
- Aghi M, Vogelbaum MA, Kesari S, et al. AT-02 Intratumoral delivery of the retroviral replicating vector (RRV) Toca 511 in subjects with recurrent high grade glioma: interim report of a Phase I study (NCT 01156584). Neuro Oncol. 2014;16(suppl 5):v8. DOI:https://doi.org/10.1093/neuonc/nou237.2.
- Clarke JL, Ennis MM, Yung WKA, et al., North American Brain Tumor Consortium. Is surgery at progression a prognostic marker for improved 6-month progression-free survival or overall survival for patients with recurrent glioblastoma? Neuro Oncol. 2011;13(10):1118–1124. DOI:https://doi.org/10.1093/neuonc/nor110.
- Wick W, Puduvalli VK, Chamberlain MC, et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol. 2010;28(7):1168–1174. DOI:https://doi.org/10.1200/JCO.2009.23.2595.
- Accomando WP, Rao AR, Hogan DJ, et al. Molecular and immunologic signatures are related to clinical benefit from treatment with vocimagene amiretrorepvec (Toca 511) and 5-fluorocytosine (Toca FC) in patients with glioma. Clin Cancer Res. 2020;26(23):6176–6186. DOI:https://doi.org/10.1158/1078-0432.CCR-20-0536.
- Walbert T, Bota D, Vogelbaum M, et al. ATIM-21. Intravenous delivery of Toca 511 in patients with high grade glioma results in quantifiable expression of cytosine deaminase in tumor tissue. Neuro Oncol. 2017;19(suppl_6):vi30–vi31. DOI:https://doi.org/10.1093/neuonc/nox168.116.
- Cloughesy T, Walbert T, Bota D, et al. ATIM-02. Successful cancer-selective gene delivery following intravenous Toca 511 delivery in patients with recurrent high grade glioma (HGG). Neuro Oncol. 2016;18(suppl_6):vi17. DOI:https://doi.org/10.1093/neuonc/now212.067
- Kalkanis S, Jolly DJ, Pertschuk D, et al. AT-29. Intravenous administration of Toca 511 in patients with recurrent glioblastoma. Neuro Oncol. 2014;16(suppl 5):v15. DOI:https://doi.org/10.1093/neuonc/nou237.29.
- Hogan DJ, Zhu JJ, Diago OR, et al. Molecular analyses support the safety and activity of retroviral replicating vector Toca 511 in patients. Clin Cancer Res. 2018;24(19):4680–4693. DOI:https://doi.org/10.1158/1078-0432.CCR-18-0619
- Cloughesy TF, Petrecca K, Walbert T, et al. Effect of vocimagene amiretrorepvec in combination with flucytosine vs standard of care on survival following tumor resection in patients with recurrent high-grade glioma: a randomized clinical trial. JAMA Oncol. 2020;6(12):1939–1946. DOI:https://doi.org/10.1001/jamaoncol.2020.3161.
- Cloughesy TF, Petrecca K, Walbert T, et al. LTBK-08. Toca 511 & Toca FC versus standard of care in patients with recurrent high grade glioma. Neuro Oncol. 2019;21(Suppl_6):vi284. DOI:https://doi.org/10.1093/neuonc/noz219.1199.
- Falchook GS, Rodon Ahnert J, Venkat S, et al. Immune modulation after Toca 511 and Toca FC treatment of colorectal cancer patients. J Clin Oncol. 2020;38(4_suppl):186. DOI:https://doi.org/10.1200/JCO.2020.38.4_suppl.186
- Ahluwalia M, Pugh S, Ellingson B, et al. RBTT-11. NRG Oncology NRG-BN006: a Phase II/III randomized, open-label study of Toca 511 and Toca FC with standard of care compared to standard of care in patients with newly diagnosed glioblastoma. Neuro Oncol. 2019;21(Suppl_6):vi220–vi221. DOI:https://doi.org/10.1093/neuonc/noz175.922.
- Cornetta K, Morgan RA, Gillio A, et al. No retroviremia or pathology in long-term follow-up of monkeys exposed to a murine amphotropic retrovirus. Hum Gene Ther. 1991;2(3):215–219. DOI:https://doi.org/10.1089/hum.1991.2.3-215
- Huang TT, Hlavaty J, Ostertag D, et al. Toca 511 gene transfer and 5-fluorocytosine in combination with temozolomide demonstrates synergistic therapeutic efficacy in a temozolomide-sensitive glioblastoma model. Cancer Gene Ther. 2013;20(10):544–551. DOI:https://doi.org/10.1038/cgt.2013.51
- Mitchell LA, Yagiz K, Hofacre A, et al. PD-L1 checkpoint blockade delivered by retroviral replicating vector confers anti-tumor efficacy in murine tumor models. Oncotarget. 2019;10(23):2252–2269. DOI:https://doi.org/10.18632/oncotarget.26785
- Zupanc GKH, Zupanc FB, Sipahi R. Stochastic cellular automata model of tumorous neurosphere growth: roles of developmental maturity and cell death. J Theor Biol. 2019;467:100–110. DOI:https://doi.org/10.1016/j.jtbi.2019.01.028
- Simnica D, Akyuz N, Schliffke S, et al. T cell receptor next-generation sequencing reveals cancer-associated repertoire metrics and reconstitution after chemotherapy in patients with hematological and solid tumors. Oncoimmunology. 2019;8(11):e1644110. DOI:https://doi.org/10.1080/2162402X.2019.1644110.