
1. Introduction
While there has been substantial progress and notable achievements in the use of adeno-associated virus (AAV) gene therapy vectors for treatment of rare diseases, setbacks related to vector toxicity and immunogenicity represent major challenges for the field. In many cases, these two issues appear to be inextricably linked[Citation1–3]. Immunogenicity of AAV vectors is thought to cause or exacerbate some of the more serious adverse events associated with AAV gene therapy, such as hepatotoxicity and thrombotic microangiopathy (TMA). Moreover, these adverse events tend to be correlated with vector dose, increasing in both prevalence and severity with higher doses [Citation1,Citation4]. Not surprisingly, high vector doses are also associated with increased immunogenicity, leading to a vicious circle when vector doses of 1E14 vg/kg or higher are required for efficacy in neuromuscular diseases [Citation1,Citation5]. These issues will likely require a multipronged approach to improve vector transduction efficiency and mitigate vector immunogenicity, and a perhaps a shift from the ‘one-and-done’ paradigm of gene therapy to a ‘slower and lower’ model of administering multiple lower doses of vector to achieve a similar therapeutic benefit.
2. Existing challenges and learnings from early clinical trials
Immunogenicity of AAV gene therapy is much more complex and nuanced than that for most protein biologic therapies, which are typically affected only by anti-drug antibodies (ADAs) [Citation2,Citation6]. ADAs also limit systemic AAV gene therapies through preexisting anti-AAV neutralizing antibodies that occur through natural exposure to wildtype AAV and which limit patient eligibility for gene therapy, and through de novo formation of neutralizing ADAs that develop after treatment and prevent the ability to re-dose AAV gene therapies. However, many of the inflammatory toxicities of AAV gene therapies appear to be driven by innate immune responses and adaptive CD8 T cell responses [Citation4].
Early clinical trials in hemophilia reported delayed elevation in serum levels of liver transaminases, which is indicative of liver inflammation and hepatoxicity. In some patients, the increase in liver enzymes correlated with a decrease in transgene expression and the appearance of circulating AAV-specific CD8 T cells [Citation4]. In many cases, the elevated liver transaminase levels and further loss of transgene expression could be successfully treated with steroids. The induction of an adaptive immune response indicates the activation of innate antigen-presenting cells (APC), such as dendritic cells, which express toll-like receptors (TLR) involved in recognition of pathogens [Citation7]. The DNA payload of AAV vectors may contain CpG motifs that bind and activate TLR9, an innate immune sensor for unmethylated CpG dinucleotides. Similarly, the capsid itself can be recognized by TLR2 resulting in activation of the APCs which can lead to the induction of adaptive immune responses. A curious aspect of the hepatoxicity associated with AAV gene therapy is the delayed onset, typically occuring 4–8 weeks after dosing. This delay suggests that AAV capsid antigens persists in some tissues and that some secondary event, or possibly cumulative hepatic stress, may cause activation of capsid-specific CD8 T cells [Citation8].
Adverse events related to elevated liver transaminases have now been widely reported in AAV gene therapy clinical trials, with increased prevalence at higher vector doses. AAV gene therapy for neuromuscular diseases has typically required doses of 1–3E14 vg/kg. Onasemnogene abeparvovec, an AAV9 therapy for spinal muscular atrophy (SMA) and the first systemic AAV gene therapy approved by the FDA, has been administered to over 1400 patients. Approximately one-third of the patients receiving a dose of 1.1E14 vg/kg have experienced at least one adverse event of hepatotoxicity [Citation1]. Three patients have been reported to experience severe hepatoxicity 7–8 weeks after treatment. Liver biopsies indicated hepatocyte degeneration and inflammatory infiltrates comprised primarily of CD8 T cells. All three patients recovered after treatment with methylprednisolone. However, tragically, three patients with X-linked myotubular myopathy (XLMTM) died 20–40 weeks after receiving a vector dose of 3.5E14 vg/kg. The patients presented with signs of severe hepatotoxicity, with bilirubin levels 35–60 times the upper limit of normal and delayed elevation in AST and ALT, and evidence of cholestasis, periportal and bile ductal reaction, and secondary fibrosis at autopsy. All three patients showed preexisting evidence of hyperbilirubinemia and intrahepatic cholestasis prior to treatment. Reports indicated a lack of inflammatory cellular infiltrates and ineffective treatment with steroids and immunosuppressants. However, an early innate immune response to the massive AAV dose could have contributed to hepatic stress resulting in exacerbation of the underlying disease [Citation8]. The clinical trial was halted and eventually restarted at a lower dose of 1.3E14 vg/kg. Unfortunately, a fourth patient died after treatment with the lower dose [Citation9].
High vector doses of AAV have also been associated with TMA, or atypical hemolytic uremic syndrome (aHUS), a syndrome characterized by hemolytic anemia, thrombocytopenia, and acute kidney injury, and associated with complement activation [Citation1,Citation10,Citation11]. Complement is a key component of the innate immune system and is comprised of plasma proteins that undergo a complex cascade of biochemical reactions that can result in direct killing of pathogens or opsonizing them for clearance, as well as chemoattraction and activation of innate immune cells and the release of vasoactive mediators. AAV can activate complement through binding of antibodies that fix complement (classical pathway) or direct binding of complement components (alternative pathway). In SMA, nine cases of TMA have been reported, including one death [Citation12]. The overall incidence is low, with over 1400 patients treated; nonetheless, one of the affected subjects had complications with persistent hypertension while another case took three months to resolve. In Duchenne muscular dystrophy (DMD), two small clinical trials involving two different AAV product candidates reported four cases of severe adverse reactions (SAEs) involving aHUS out of 15 total patients dosed [Citation11,Citation12]. One case occurred at a dose of 5E13 vg/kg, while the other three occurred at vector doses of 1–3E14 vg/kg. In addition, in September, 2021, Pfizer announced an amendment to their Phase 3 clinical trial in DMD to exclude patients with certain mutations as a result of three SAEs, including two cases of myocarditis [Citation13]. More recently, a patient death in the Pfizer Phase 1b trial was reported on 21 December 2021. No further details were available, and the clinical trial was put on hold [Citation14].
In addition to hepatotoxicity and TMA, other potential risks include hepatocellular carcinoma (HCC) and dorsal root ganglia (DRG) toxicity, which have been observed in animal models of AAV gene therapy [Citation1,Citation15–17]. HCC has been primarily associated with vector integration at high AAV doses in neonatal mice, particularly with vectors that employ a strong promoter that may result in transactivation of protooncogenes [Citation15,Citation16]. Pre-neoplastic hepatic lesions have also been reported in long-term canine studies [Citation18]. HCC has been reported in one hemophilia patient, although subsequent investigation indicate that this incidence was unlikely to be related to the AAV gene therapy vector [Citation19]. Despite the uncertain relevance to humans, a clinical program in phenylketonuria remains on clinical hold due to observations of HCC in the preclinical animal model [Citation20]. DRG toxicity has been observed in nonhuman primates (NHP) and other animal models, primarily associated with direct administration of AAV into the cerebral spinal fluid or high intravenous doses [Citation17]. DRG toxicity appears to be associated with high levels of transgene expression in DRG neurons which may cause neuronal stress [Citation21]. The clinical significance of DRG toxicity is uncertain, although ataxia and tremors has been reported in some NHP, and ataxia and proprioceptive deficits have been reported in piglets receiving an intravenous vector dose of 2E14 vg/kg [Citation17,Citation22]. Histological evaluation at autopsy revealed evidence of DRG toxicity following AAV gene therapy in a human patient with giant axonal neuropathy and a patient with familial amyotrophic lateral sclerosis [Citation1]. The latter patient reported tingling sensations and pain in the extremities after an intrathecal AAV dose of 4.2E14 vg.
The different manifestations and relative incidence of adverse reactions suggest that multiple variables may contribute to the overall risk, such as vector design, vector manufacturing and quality attributes, underlying disease, and co-morbidities. Retrospective analysis of hemophilia B gene therapy trials suggest that higher CpG content correlated with higher incidence of liver enzyme elevation, stronger CD8 T cell responses and less durable transgene expression [Citation23]. In DMD, a third clinical program, from Sarepta, has yet to announce a serious adverse event related to aHUS, with 77 patients treated as of October 2021 [Citation24]. The basis for a high incidence of aHUS in two programs but not a third is unknown. It is conceivable that different capsids would have different profiles with respect to fixation of complement or binding to preexisting antibodies. One major unknown with all AAV clinical trials is the degree of impurities from the manufacturing process, such as percentage of empty capsids or host cell impurities [Citation1,Citation5]. Since AAV doses are calculated based on determination of vector genomes (or ‘full’ capsids), the ratio of empty:full capsids could have a huge bearing on the total capsid dose.
3. Moving forward
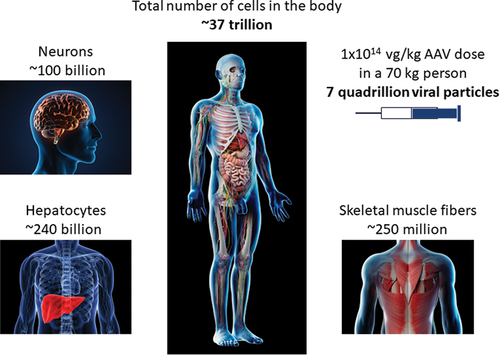
For all the notable successes and continued promise of the AAV field, there is clearly room to improve the safety and efficacy of recombinant AAV vectors, particularly for indications that currently require high vector doses. A capsid dose of 1E14 vg/kg represents a dose of over a quadrillion viral particles (not including empty capsids) in a young child, which is far greater than the total number of cells in the body (). Increasing the ratio of full:empty capsids and reducing CpG content provide an immediately actionable strategy to improve safety. Another potential strategy to increase transduction efficiency and reduce CD8 T cell responses is to introduce capsid mutations that minimize processing by the proteasome [Citation25], which results in capsid degradation and presentation of immunogenic epitopes on MHC class I molecules, or eliminating MHC-binding epitopes in capsids. Longer term strategies include sophisticated capsid engineering approaches to design or evolve capsids that show better targeting of specific tissues, such as muscle [Citation26]. Improvements in promoter and enhancer design could also improve efficacy at lower doses, although the potential risks of HCC associated with strong promoters and DRG toxicity with transgene overexpression must be considered [Citation16,Citation21]. In addition, engineering of more efficient transgenes, as exemplified by the Padua variant used in hemophilia B [Citation27], could allow for lower vector doses. Ultimately, a breakthrough in radically improving transduction efficiency may require a better understanding of the key rate limiting steps of AAV transduction, including transit through the cell from the endosome to the nucleus, viral uncoating, maintenance as a stable episome, and interaction with host cell factors [Citation26].
Figure 1. An estimate of the number of cells in the human body compared to the number of full viral particles in a high vector dose of AAV.
Both capsid engineering and pharmacological strategies are being pursued to mitigate the immunogenicity of AAV capsids. Some strategies, such as mutagenesis of capsids to remove antibody epitopes [Citation26] or use of plasmapheresis [Citation28] or IgG protease [Citation29] to eliminate anti-AAV antibodies, only address the preexisting antibody component, without addressing innate immunity or T cell responses. Indeed, the very presence of preexisting anti-AAV antibodies is indicative of prior exposure to AAV and a high likelihood of the presence of AAV-specific memory T and B cells [Citation30]. High vector doses, which can overcome low levels of neutralizing antibodies, has enabled some groups to loosen the threshold antibody titer for inclusion into clinical trials [Citation31], although preexisting memory T cells and complement fixation by preexisting low-titer antibodies could still potentially exacerbate the toxicities observed with high vector doses. Combination immunosuppressive regimens, such as rapamycin and corticosteroids or rapamycin and rituximab are being evaluated clinically for mitigation of immunogenicity [Citation32]. Rapamycin is an inhibitor of the mTOR pathway, which dosed chronically, can inhibit effector T cell activation. Rapamycin encapsulated into nanoparticles (ImmTOR) is also being evaluated for the induction of antigen-specific immune tolerance to AAV vectors. A dose of ImmTOR nanoparticles administered at the time of AAV gene therapy treatment has been shown to inhibit T and B cell responses to AAV and enable vector redosing [Citation33]. Rapamycin nanoparticles have also been shown to have hepatoprotective properties in animal models of liver inflammation [Citation34]. Notably, rapamycin has also been shown to enhance AAV vector transduction through induction of autophagy [Citation35].
4. Expert opinion
The stakes are high for mitigating the toxicity of high vector doses. The benefit of AAV gene therapy has been undeniable in SMA and hemophilia, and early data are highly promising in DMD. Even in XLMTM, those patients that tolerated therapy showed remarkable benefit. Further improvement might be achievable if the therapeutic window could be safely widened. The ultimate goal should be to provide similar efficacy at lower vector doses. This may require a combination of vector engineering and pharmacological strategies to increase vector transduction efficiency and mitigate immunogenicity. AAV gene therapy has been historically viewed as a ‘one-and-done’ therapy, meaning that a single dose of therapy was expected to provide years, if not a lifetime, of therapeutic benefit. This notion has been challenged by the waning of efficacy observed in some clinical trials and the loss of efficacy observed in patients that experience hepatotoxicity. If the barrier to vector redosing, currently limited by the formation of high titers of neutralizing antibodies after initial exposure, can be overcome, it not only opens the possibility to restore therapeutic benefit in patients that have experienced loss of transgene expression but may also provide a safer path for the treatment of neuromuscular diseases by allowing for multiple smaller doses of gene therapy rather than a single high dose of 1–3E14vg/kg. The potential of providing a therapeutic outcome by delivering multiple lower doses over a finite period of time (e.g. 1–3 months) may herald the next gene therapy paradigm that facilitates the coming wave of approved gene therapy drugs with lower toxicity profiles for unmet rare diseases – ‘one-and-done’ may be substituted with ‘lower and slower.’ One potential challenge to this approach is that vector re-dosing is likely to elicit a stronger anamnestic immune response. An immunosuppressive approach may not be sufficient to prevent the formation of memory T and B cells that would drive a secondary immune response. A strong regulatory T cell response would likely be required to maintain immune tolerance. Combination therapies may need to be employed, particularly with high vector doses.
Declaration of interest
TK Kishimoto is an employee, shareholder and executive officer of Selecta Biosciences. RJ Samulski is an employee of the University of North Carolina and a consultant of Asklepios BioPharmaceutical (AskBio). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Food and Drug Administration (FDA) Cellular, Tissue, and Gene Therapies Advisory Committee (CTGTAC) meeting #70 brief document. [cited 2022 Feb 25]. Available from: www.fdagov/media/151599/download
- Colella P, Ronzitti G, Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol Ther Methods Clin Dev. 2018 Mar 16;8:87–104. PubMed PMID: 29326962; PubMed Central PMCID: PMCPMC5758940.
- Flotte TR. Revisiting the “new” inflammatory toxicities of adeno-associated virus vectors. Hum Gene Ther. 2020;31(7–8):398–399.
- Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011 Dec 22;365(25):2357–2365. PubMed PMID: 22149959; PubMed Central PMCID: PMCPMC3265081.
- Paulk N. Gene therapy: it’s time to talk about high-dose AAV. Genet Eng Biotechnol News. 2020;40(9):14–16.
- Ronzitti G, Gross DA, Mingozzi F. Human immune responses to Adeno-Associated Virus (AAV) vectors. Front Immunol. 2020;11:670. PubMed PMID: 32362898.
- Muhuri M, Maeda Y, Ma H, et al. Overcoming innate immune barriers that impede AAV gene therapy vectors. J Clin Invest. 2021 Jan 4;131(1). PubMed PMID: 33393506. DOI: 10.1172/JCI143780.
- Watkins PB. Liver injury due to drugs and viruses: mechanistic similarities and implications for AAV gene therapy. Clin Pharmacol Ther. 2021 Dec 15; PubMed PMID: 34910298. DOI: 10.1002/cpt.2500.
- Philippidis A. Fourth boy dies in trial of Astellas gene therapy candidate. Genet Eng Biotechnol News 2021. (accessed 2022 Feb 25. https://www.genengnews.com/news/fourth-boy-dies-in-trial-of-astellas-gene-therapy-candidate/.
- Chand DH, Zaidman C, Arya K, et al. Thrombotic microangiopathy following onasemnogene abeparvovec for spinal muscular atrophy a case series. J Pediatr. 2021 Apr;231:265–268. PubMed PMID: 33259859.
- Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, et al. Current clinical applications of in vivo gene therapy with AAVs. Mol Ther. 2021 Feb 3;29(2):464–488. PubMed PMID: 33309881; PubMed Central PMCID: PMCPMC7854298.
- Mullard A. Gene therapy community grapples with toxicity issues, as pipeline matures. Nature. 2021;20:804–805.
- [cited 2022 Feb 25]. https://www.cureduchenne.org/partner-news/a-message-from-pfizer-on-our-dmd-clinical-program/
- [cited 2022 Feb 25]. https://endpts.com/one-of-pfizers-duchenne-gene-therapy-trials-put-on-hold-in-wake-of-patient-death-as-high-dose-aav-concerns-still-cloud-field/
- Donsante A, Vogler C, Muzyczka N, et al. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther. 2001Sep;817:1343–1346. PubMed PMID: 11571571.
- Chandler RJ, LaFave MC, Varshney GK, et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest. 2015Feb;1252:870–880. PubMed PMID: 25607839; PubMed Central PMCID: PMCPMC4319425.
- Hordeaux J, Buza EL, Dyer C, et al. Adeno-associated virus-induced dorsal root ganglion pathology. Hum Gene Ther. 2020Aug;3115–16:808–818. PubMed PMID: 32845779.
- Nguyen GN, Everett JK, Kafle S, et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat Biotechnol. 2021Jan;391:47–55. PubMed PMID: 33199875; PubMed Central PMCID: PMCPMC7855056.
- [cited 2022 Feb 25]. http://uniqure.com/PR_HCC%20Investigation%20Findings%20_3_29_21_FINAL.pdf
- [cited 2022 Feb 25]. https://investors.biomarin.com/2021-09-06-U-S-FDA-Placed-a-Clinical-Hold-on-BMN-307-Phearless-Phase-1-2-Gene-Therapy-Study-in-Adults-with-PKU-Based-on-Interim-Pre-clinical-Study-Findings
- Hordeaux J, Buza EL, Jeffrey B, et al. MicroRNA-mediated inhibition of transgene expression reduces dorsal root ganglion toxicity by AAV vectors in primates. Sci Transl Med. 2020 Nov 11;12(569). PubMed PMID: 33177182. DOI: 10.1126/scitranslmed.aba9188.
- Hinderer C, Katz N, Buza EL, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. 2018Mar;293:285–298. PubMed PMID: 29378426; PubMed Central PMCID: PMCPMC5865262.
- Hamilton BA, Wright JF. Challenges posed by immune responses to AAV vectors: addressing root causes. Front Immunol. 2021;12:675897. PubMed PMID: 34084173; PubMed Central PMCID: PMCPMC8168460.
- [cited 2022 Feb 25]. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-srp-9001-shows-sustained-functional
- Zhong L, Li B, Mah CS, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci U S A. 2008 Jun 3;105(22):7827–7832. PubMed PMID: 18511559; PubMed Central PMCID: PMCPMC2402387.
- Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. PubMed PMID: 32042148, Nat Rev Genet. 2020 Apr;21(4):255–272.
- George LA, Sullivan SK, Giermasz A, et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N Engl J Med. 2017 Dec 7;377(23):2215–2227. PubMed PMID: 29211678; PubMed Central PMCID: PMCPMC6029626.
- Monteilhet V, Saheb S, Boutin S, et al. A 10 patient case report on the impact of plasmapheresis upon neutralizing factors against adeno-associated virus (AAV) types 1, 2, 6, and 8. Mol Ther. 2011Nov;1911:2084–2091. PubMed PMID: 21629225; PubMed Central PMCID: PMCPMC3222518.
- Leborgne C, Barbon E, Alexander JM, et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat Med. 2020Jul;267:1096–1101. PubMed PMID: 32483358.
- Kuranda K, Jean-Alphonse P, Leborgne C, et al. Exposure to wild-type AAV drives distinct capsid immunity profiles in humans. J Clin Invest. 2018 Dec 3;128(12):5267–5279. PubMed PMID: 30352429; PubMed Central PMCID: PMCPMC6264647.
- Majowicz A, Nijmeijer B, Lampen MH, et al. Therapeutic hFIX activity achieved after single AAV5-hFIX treatment in hemophilia B patients and NHPs with pre-existing anti-AAV5 NABs. Mol Ther Methods Clin Dev. 2019 Sep 13;14:27–36. PubMed PMID: 31276009; PubMed Central PMCID: PMCPMC6586596.
- Corti M, Cleaver B, Clement N, et al. Evaluation of readministration of a recombinant adeno-associated virus vector expressing acid alpha-glucosidase in pompe disease: preclinical to clinical planning. Hum Gene Ther Clin Dev. 2015Sep;263:185–193. PubMed PMID: 26390092; PubMed Central PMCID: PMCPMC4606909.
- Meliani A, Boisgerault F, Hardet R, et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat Commun. 2018 Oct 5;9(1):4098. PubMed PMID: 30291246; PubMed Central PMCID: PMCPMC6173722.
- Ilyinskii PO, Roy CJ, LePrevost J, et al. Enhancement of the tolerogenic phenotype in the liver by ImmTOR nanoparticles. Front Immunol. 2021;12:637469. PubMed PMID: 34113339; PubMed Central PMCID: PMCPMC8186318.
- Hosel M, Huber A, Bohlen S, et al. Autophagy determines efficiency of liver-directed gene therapy with adeno-associated viral vectors. Hepatology. 2017Jul;661:252–265. PubMed PMID: 28318036; PubMed Central PMCID: PMCPMC5518300.
- Bianconi E, Piovesan A, Facchin F, et al. An estimation of the number of cells in the human body. Ann Hum Biol. 2013Nov-Dec;40(6):463–471. PubMed PMID: 23829164.