
ABSTRACT
Background
This study’s objective was to demonstrate pharmacokinetic (PK) similarity and safety of denosumab biosimilar, CT‑P41, and United States–licensed reference denosumab (US-denosumab) in healthy male Asian adults, considering also pharmacodynamic (PD) outcomes.
Research design and methods
This double-blind, two-arm, parallel-group, Phase 1 study randomized (1:1) healthy males to a single (60-mg) subcutaneous dose of CT‑P41 or US-denosumab. Primary endpoints were area under the concentration – time curve (AUC) from time zero to infinity (AUC0–inf), AUC from time zero to the last quantifiable concentration (AUC0–last), and maximum serum concentration (Cmax). PK equivalence was determined if 90% confidence intervals (CIs) for ratios of geometric least-squares means (gLSMs) were within the predefined 80–125% equivalence margin. Secondary PK, PD, safety, and immunogenicity outcomes were also evaluated.
Results
Of 154 participants randomized (76 CT‑P41; 78 US-denosumab), 151 received study drug (74 CT‑P41; 77 US-denosumab). Primary and secondary PK results, PD results, safety, and immunogenicity were comparable between groups. Ninety percent CIs for ratios of gLSMs were within the predefined equivalence margin for AUC0–inf (100.4–114.7), AUC0–last (99.9–114.3), and Cmax (95.2–107.3).
Conclusions
Following a single dose in healthy males, CT‑P41 demonstrated PK equivalence with US-denosumab.
Trial registration
ClinicalTrials.gov: NCT06037395
1. Introduction
The human monoclonal antibody, denosumab, targets the receptor activator of nuclear factor kappa-B ligand (RANKL), preventing activation of its receptor (receptor activator of nuclear factor kappa-B [RANK]) on the osteoclast surface [Citation1]. This inhibits osteoclast formation and function, consequently decreasing bone resorption and increasing bone mass and bone strength [Citation1]. Reference denosumab (Prolia® [Amgen Inc., Thousand Oaks, CA, U.S.A.] 60 mg/mL) received regulatory approval in Europe and the U.S.A. in 2010 and is indicated at a dose of 60 mg for the treatment of postmenopausal women with osteoporosis, and to increase bone mass in other patient populations at high risk of fracture [Citation2,Citation3]. Reference denosumab is recommended by the International Osteoporosis Foundation and the European Society for Clinical and Economic Aspects of Osteoporosis, Osteoarthritis and Musculoskeletal Disorders as an initial treatment in patients at high risk of osteoporotic fracture and as a major component of the sequential treatment therapy suggested for patients with severe osteoporosis at very high risk of fracture [Citation4–6].
The high cost of biologic therapies can limit widespread patient access to treatment, particularly considering that prolonged therapy may be required to treat chronic conditions such as osteoporosis [Citation7]. Biosimilars can improve global patient access to effective treatments and significantly reduce the associated costs for healthcare systems [Citation7]. For instance, the introduction of infliximab and etanercept biosimilars gave rise to cost savings of £38.8 billion over a 2-year period [Citation7]. CT‑P41 is a candidate denosumab biosimilar currently in development.
This randomized, double-blind, two-arm, parallel-group, single-dose, Phase 1 study was conducted in healthy male participants with the primary objective of demonstrating the pharmacokinetic (PK) similarity of CT‑P41 versus US-licensed reference denosumab (US-denosumab). The study also evaluated the pharmacodynamics (PD), safety, and immunogenicity of CT‑P41.
2. Participants and methods
2.1. Study design and procedures
This randomized, double-blind, two-arm, parallel-group, single-dose, Phase 1 study (ClinicalTrials.gov: NCT06037395) was conducted at two study centers in the Republic of Korea (Table S1). Screening occurred from Day − 28 to Day − 2. Participants were admitted to the study center on Day − 1 and randomized (1:1) to receive CT‑P41 or US-denosumab. A randomization schedule was prepared by unblinded statisticians prior to the start of the study using an interactive web response system. Each participant was allocated a unique randomization number sequentially, based on the predetermined randomization list and according to their chronological order of inclusion in the study. Randomly mixed permuted blocks of two or four sizes were used to balance randomization. Randomization was stratified by body weight (<80 kg vs ≥80 kg) measured at Day − 1 and study center.
On Day 1, participants received a single 60 mg (in 1 mL of solution) dose of CT‑P41 or US‑denosumab via a single subcutaneous injection using a pre-filled syringe, injected into the outer upper arm on the participant’s non-dominant side by trained clinical staff. Participants were blinded to study drug assignment using a blindfold, screen, or similar method during drug administration. Participants and investigators remained blinded until data collection was completed and the database locked. However, given that the two study drugs were not identical in visual appearance, clinical staff who administered the study drug were not blinded but were not involved in any planned evaluations forming part of the blinded protocol.
Participants could take daily supplementation of vitamin D with doses between 400 IU and 1,000 IU, at the discretion of the investigator, to prevent risk of hypocalcemia and vitamin D deficiency. Individual vitamin D maintenance dose and continuation of co-administration of vitamin D were evaluated throughout the study by the investigator with continuous monitoring of participant’s vitamin D level. Calcium was permitted to treat adverse events (e.g. hypocalcemia).
Participants remained at the study center overnight until completion of the 24-hour post-dose assessments. All follow-up visits were completed on an outpatient basis (follow-up visits: Days 3, 4, 6, 8, 11, 15, 22, 29, 43, 57, 71, 85, 99, 113, 141, 169, 197, and 253 [end of study (EOS)]).
The study was conducted in accordance with the Declaration of Helsinki, the International Council for Harmonisation Guideline for Good Clinical Practice, and all applicable laws or regulations. The study protocol, informed consent form, and any other written materials provided to participants were approved by institutional review boards at each study center prior to study initiation (Table S1). Written informed consent was obtained from each participant prior to the study.
2.2. Participants
Full eligibility criteria can be found in the Supplementary methods. In brief, adult males aged 28–55 years (inclusive) who were healthy (defined as having no clinically relevant abnormalities identified by a detailed medical history, physical examination, and clinical laboratory tests) were eligible for enrollment. Individuals needed to have a body weight of ≥50.0–≤99.9 kg, a body mass index (BMI) of 18.5–29.9 kg/m2 (inclusive), total serum calcium ≥8.5 mg/dL, and serum 25-hydroxy vitamin D ≥20 ng/mL. Key exclusion criteria included: hypersensitivity to any component of the denosumab product (denosumab or excipients); receipt of any biologic agent within the longer of 90 days or five half-lives prior to study drug administration, or any investigational product in a clinical trial within the longer of 180 days or five half-lives; receipt of any therapy that might significantly affect bone metabolism; and a medical history of and/or current medical condition including known risk factors for hypocalcemia and known intolerance to calcium and vitamin D supplements.
2.3. Study endpoints
The primary study endpoints were area under the concentration – time curve (AUC) from time zero to infinity (AUC0–inf), AUC from time zero to the last quantifiable concentration (AUC0–last), and maximum serum concentration (Cmax). Additional PK parameters evaluated as secondary endpoints were partial AUC from time zero to Week 16 (pAUC0–W16), partial AUC from Week 16 to infinity (pAUCW16–inf), time to Cmax (Tmax), terminal half-life (t½), percentage of AUC0–inf obtained by extrapolation (%AUCext), terminal elimination rate constant (λz), apparent total body clearance (CL/F), apparent volume of distribution during the terminal phase (Vz/F), and mean residence time.
PD evaluation comprised assessment of the bone turnover markers serum type 1 C‑telopeptide (s-CTX) and procollagen type 1 N-terminal propeptide (P1NP). PD endpoints were percent change from baseline in s-CTX and P1NP and area under the effect curve (AUEC) for s-CTX and P1NP. The effect used in the AUEC was based on the percent change from baseline (reported as percentage inhibition). Safety and immunogenicity were also assessed throughout the study (safety and immunogenicity endpoints are listed in the Supplementary methods).
2.4. Study assessments
Serum samples for PK assessments were taken on Day 1 within 30 minutes prior to dosing, at 6 hours and 12 hours post-dose, and at all following visits. Serum CT‑P41 and US-denosumab concentrations were determined using a Meso Scale Discovery electrochemiluminescence assay (MSD-ECL; Meso Scale Diagnostics, Rockville, MD, U.S.A.). Serum concentrations below the lower limit of quantification (BLQ) that occurred prior to and after study drug administration were treated as zero. For PK parameter estimation, concentrations BLQ that occurred prior to study drug administration were treated as zero, while post-dose concentrations were treated as missing. Serum samples for PD assessment were collected on Day 1 at pre-dose, and at Days 2, 3, 4, 8, 15, 29, 85, 141, 197, and 253 (EOS); samples were taken in the morning, after an 8-hour overnight fast. PK and PD parameters were calculated by non-compartmental analysis using Phoenix WinNonlin version 8.3 (Certara U.S.A., Inc., Princeton, NJ, U.S.A.).
Safety assessments included monitoring adverse events, clinical laboratory tests, electrocardiogram (ECG) and vital sign measurements, hypersensitivity/allergic reaction assessments, physical examination, and assessment of local site pain (on a 100-mm visual analog scale [VAS]). A treatment-emergent adverse event (TEAE) was defined as any adverse event that was not present before exposure to study drug or any event already present that worsened in severity or frequency after study drug administration. TEAEs of special interest (TEAESI) were injection-site reactions (ISRs), drug-related hypersensitivity/allergic reaction, infection, and hypocalcemia (described in the Supplementary methods). TEAEs were coded using the Medical Dictionary for Regulatory Activities (version 25.1) and graded for severity according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE; version 5.0; described in the Supplementary methods).
Serum samples for immunogenicity assessments were collected pre-dose on Day 1 and at Days 15, 29, 57, 85, 141, and 253 (EOS). Anti-drug antibodies (ADAs) were evaluated via MSD-ECL, with a confirmatory assay sensitivity of 3.84 ng/mL in healthy human serum. Confirmed ADA-positive samples were further analyzed for neutralizing antibodies (NAbs), with a sensitivity of 56.7 ng/mL in healthy human serum.
2.5. Statistical analysis
The primary analysis assessed PK equivalence of CT‑P41 to US-denosumab, based on the primary endpoints (AUC0–inf, AUC0–last, and Cmax). An analysis of covariance (ANCOVA) model was used to analyze log-transformed primary endpoints, with treatment as a fixed effect and body weight on Day − 1 and study center as covariates. Back transformation provided the ratios of geometric least-squares means (gLSMs) and 90% confidence intervals (CIs) for these ratios. To demonstrate equivalence between CT‑P41 and US-denosumab, the 90% CIs for all three primary endpoints had to be contained within the equivalence margin of 80–125%.
For the statistical analysis of PD parameters, an ANCOVA model was used to analyze log‑transformed AUEC, with treatment as fixed effect and age and baseline value of s-CTX or P1NP as covariates. Back transformation provided the ratios of gLSMs and 95% CIs for these ratios.
A sample size of 132 participants (66 per group) was expected to provide 90% statistical power to demonstrate PK equivalence between CT‑P41 and US‑denosumab using this approach, assuming an expected geometric mean ratio of 1.0 and a percent coefficient of variation (%CV) of 40%, based on historic PK data, including a Phase 1 study of denosumab in healthy individuals [Citation8]. Accounting for a 10% dropout rate, 148 participants (74 per group) were to be enrolled.
The analysis populations are described in the Supplementary methods. Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, U.S.A.).
3. Results
3.1. Participant disposition
The first randomization date was 6 October 2021; the last participant’s last visit was on 20 October 2022. Overall, 154 participants were randomized (76 CT‑P41; 78 US-denosumab) () and included in the intent-to-treat set. Three participants discontinued prior to study drug administration; thus, 151 participants received study drug (74 CT‑P41; 77 US-denosumab) and 146 completed the study (72 CT‑P41; 74 US-denosumab).
Figure 1. Participant disposition (ITT set).
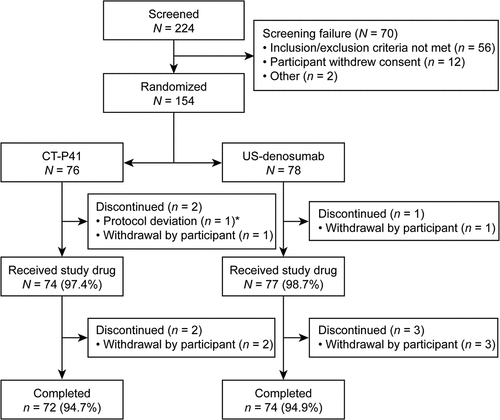
Participant demographics and baseline characteristics were similar between groups (). All participants were Asian males and the median (range) age was 38 (28–55) years. At screening, overall median (range) body weight was 74.8 (57.7–99.6) kg, and BMI was 24.5 (18.7–29.9) kg/m2. Stratification factors (Day −1 body weight [<80 kg vs ≥80 kg] and study center) were well balanced between groups.
Table 1. Demographics and baseline characteristics (ITT set).
3.2. Pharmacokinetics
Following a single dose of study drug, mean serum drug concentrations were comparable between groups throughout the study period (). All primary () and secondary () PK endpoints were comparable between groups. PK equivalence between CT‑P41 and US-denosumab was demonstrated in terms of the primary endpoints, AUC0–inf, AUC0–last, and Cmax (). For all three primary endpoints, the 90% CIs for the ratios of gLSMs were within the predefined equivalence margin of 80–125% (ratio [90% CI]: AUC0–inf, 107.3 [100.4–114.7]; AUC0–last, 106.9 [99.9–114.3]; Cmax, 101.1 [95.2–107.3]). The mean (standard deviation [SD]) value of pAUC0–W16 was 320.1 (73.0) day•μg/mL and 294.4 (80.2) day•μg/mL for CT‑P41 and US-denosumab groups, respectively (). Median Tmax was 10.0 days for both groups and the mean (SD) t½ was 16.8 (4.7) days and 16.2 (4.6) days in the CT‑P41 and US‑denosumab groups, respectively. The mean (SD) CL/F value was 0.19 (0.065) L/day and 0.22 (0.097) L/day in the CT‑P41 and US-denosumab groups, respectively.
Figure 2. Mean (SD) serum concentrations of CT‑P41 and US-denosumab (PK set). (a) Linear scale. (b) Semi-logarithmic scale.
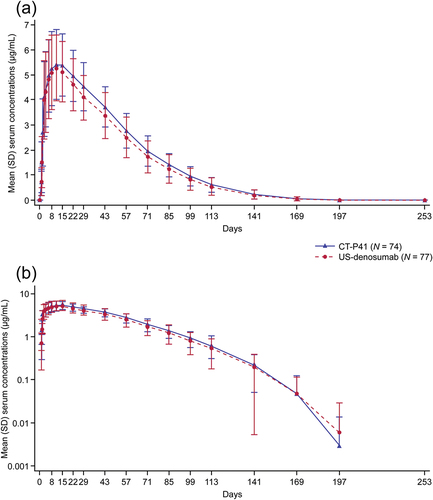
Table 2. Statistical analysis of the primary PK endpoints (PK set).
Table 3. Summary of PK parameters (PK set).
3.3. Pharmacodynamics
Overall, the trend in median percent change from baseline in s-CTX was comparable between groups (). Immediately following study drug administration, median percent change from baseline in s-CTX showed a sharp decrease until Day 8, with low levels of s-CTX persisting until Day 141 in both groups, before gradually increasing. The largest reduction in s-CTX was observed on Day 85, with median percent change from baseline at − 87.2% and − 87.4% in the CT‑P41 and US-denosumab groups, respectively.
Figure 3. Median (IQR) percent change from baseline for serum concentrations of (a) s-CTX and (b) P1NP by group (PD set).
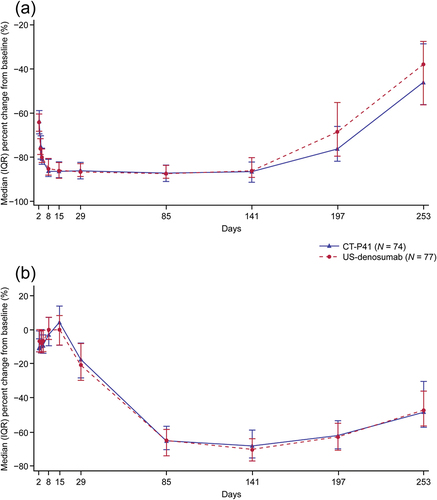
Overall trends in median percent change from baseline in P1NP were comparable between groups, with no notable differences (). Immediately following study drug administration, median percent change from baseline in P1NP was slightly decreased and remained below baseline until Day 4. From Day 4 to Day 15, median percent change from baseline in P1NP recovered above the baseline level. After Day 15, the median percent change from baseline in P1NP decreased until Day 141 (CT‑P41: −68.2%; US-denosumab: −70.3%), and then increased gradually.
The AUEC of s-CTX was comparable between the CT‑P41 and US‑denosumab groups (mean [%CV]: 19,294.9 [12.5%] and 18,955.1 [11.8%] day*% inhibition, respectively). Similarly, the AUEC of P1NP was comparable between the CT‑P41 and US-denosumab groups (mean [%CV]: 12,351.7 [24.2%] and 12,822.2 [17.3%] day*% inhibition, respectively). The gLSMs of AUEC for s-CTX and P1NP were comparable between groups. The ratios of gLSMs (95% CI) of AUEC of s-CTX and P1NP were 101.4 (97.2, 105.7) and 93.3 (83.6, 104.3), respectively.
3.4. Safety
All participants who were injected with the study drug received the total volume. Overall, 282 TEAEs were reported in 114 participants (). The majority of TEAEs were Grade 1 (mild) or Grade 2 (moderate) in intensity. All Grade 3 (severe) and 4 (life-threatening) TEAEs were abnormal laboratory test results and were considered unrelated to study drug by the investigator. In the CT‑P41 group, 55 participants (74.3%) experienced ≥1 TEAE; in the US-denosumab group, this was 59 participants (76.6%). At least one study drug-related TEAE was reported by 84 participants overall (CT‑P41: 39 participants [52.7%]; US-denosumab: 45 participants [58.4%]). The most frequently occurring TEAE was blood calcium decreased (<8.5 mg/dL: CT‑P41: 28 participants [37.8%]; US-denosumab: 35 participants [45.5%]) (Table S2). All TEAEs of blood calcium decreased were Grade 1–2 in intensity and did not require treatment. There were no treatment-emergent serious adverse events, TEAEs leading to study discontinuation, or TEAEs resulting in death in either group.
Table 4. Summary of safety findings (safety set).
In terms of TEAESI, two participants (2.7%) in the CT‑P41 group had TEAEs of ISR (with signs and symptoms of injection-site erythema and injection-site pain); both events were Grade 1 in intensity and resolved without treatment. TEAEs of infection were reported in 40 participants (CT‑P41: 18 participants [24.3%]; US‑denosumab: 22 participants [28.6%]) (Table S3). All were Grade 1–2 in intensity and resolved without sequelae. Nasopharyngitis was the most frequently reported study drug-related infection in both the CT‑P41 and US-denosumab groups (3 participants [4.1%] and 3 participants [3.9%], respectively). No TEAEs of drug-related hypersensitivity/allergic reaction or hypocalcemia were reported.
The most frequently reported CTCAE Grade 3 laboratory parameter was hypertriglyceridemia (6 participants [8.1%] and 5 participants [6.5%] in the CT‑P41 and US‑denosumab groups, respectively) (Table S4). CTCAE Grade 4 laboratory parameters were reported in six participants (5 participants [6.8%] and 1 participant [1.3%] in the CT‑P41 and US-denosumab groups, respectively). All Grade 4 cases were reported as a TEAE but were all unrelated to the study drug. Local injection-site pain scores, assessed by 100-mm VAS, were comparable between groups (mean [SD] score: CT‑P41: 4.3 [7.7] mm; US-denosumab: 4.9 [7.6] mm). There were no notable differences in ECG, vital signs (including hypersensitivity monitoring), or physical examination findings between groups.
3.5. Immunogenicity
All participants had ≥1 post-treatment positive ADA result (CT‑P41: 74 participants [100%]; US-denosumab: 77 participants [100%]; one participant [0.7%] in the CT‑P41 group had a positive ADA test prior to study drug administration) (Table S5). The proportion of participants with positive ADA results peaked on Day 85, and was similar between groups (CT‑P41: 72 participants [97.3%]; US-denosumab: 73 participants [94.8%]). Four participants (2.6%) had ≥1 positive NAb result (CT‑P41: 2 participants [2.7%]; US‑denosumab: 2 participants [2.6%]). ADA titers at the time of the NAb-positive result were all low at 100, and all participants were ADA negative on Day 253 (EOS).
4. Discussion
This study evaluated the PK equivalence of CT‑P41 and US-denosumab in a population of healthy male Asian adults. Following the administration of a single dose to healthy adult males, PK equivalence between CT‑P41 and US-denosumab was demonstrated based on AUC0–inf, AUC0–last, and Cmax, owing to the fact that the 90% CIs of the ratio of gLSMs were within the predefined 80–125% equivalence margin. PK similarity was also supported by results for the secondary PK endpoints. CT‑P41 demonstrated non-linear PKs consistent with the known PK profile of reference denosumab, whereby changes in elimination rate are observed at different concentrations over time [Citation2,Citation3,Citation9]. The t½ reported for CT‑P41 and US-denosumab (16.8 and 16.2 days, respectively) were lower than previously reported for EU-approved denosumab (26 days [Citation2]) and US-denosumab (25.4 days [Citation3]). This can potentially be explained by the duration of the study period and terminal timepoints selected for the estimation of λz, as there are changes in the elimination rate of denosumab over time due to non-linear PKs [Citation2,Citation3,Citation9].
Overall, trends in bone turnover markers were comparable between CT‑P41 and US‑denosumab in this study. The s-CTX bone resorption marker significantly decreased from baseline up to Day 8 and remained low until Day 141. The transient increase observed in the P1NP bone formation marker at early timepoints suggests a compensatory response to decreased bone resorption. Prolonged inhibition of bone resorption has been demonstrated to eventually lead to a decline in P1NP, owing to the coupling of bone formation and resorption [Citation10].
The safety profiles of CT‑P41 and US-denosumab were comparable, and there were no new safety findings with respect to previous studies of reference denosumab [Citation11–15]. The most frequent TEAE in both groups was blood calcium decreased (<8.5 mg/dL: observed in 63 participants [41.7%] overall); however, despite the relatively high frequency, there were no significant cases reported as hypocalcemia (a TEAE of interest in the study), consistent with some reports from other studies investigating reference denosumab in postmenopausal women receiving calcium and vitamin D supplements [Citation11–13,Citation15]. An absence of events reported as hypocalcemia in the current study could be due to the healthy condition of the participants and the daily supplementation (at the discretion of the investigator) of vitamin D during the study.
TEAEs of infection were reported in 40 participants (26.5%) in total, with most considered unrelated to treatment by the investigator. Nasopharyngitis was the most common infection considered related to study treatment by the investigator, in line with findings from some previous studies evaluating reference denosumab [Citation11,Citation12,Citation14].
The immunogenicity of CT‑P41 and US-denosumab was similar based on the proportions of participants with post-treatment ADAs and NAbs. All participants in the CT‑P41 and US-denosumab groups had ≥1 post-treatment ADA result, suggesting that there was no product-specific difference in ADA incidence between the two products. One participant in the CT‑P41 group had a positive ADA test at pre-dose and maintained post-treatment positive ADA results until Day 85. For this participant, ADA titers were low throughout and did not demonstrate a significant change from baseline (100 at pre-dose; 300 at post-treatment until Day 85). This suggests that the occurrence of ADAs for this participant was not treatment-boosted by the study drug. Four participants had positive NAb results that occurred on Day 85 or Day 141. The PK profiles for these participants showed no sudden decrease in serum denosumab concentration at the time of NAb positivity. Comparison of the incidence of post-treatment ADA/NAb results to other studies of denosumab products is difficult as the sensitivity, specificity, and drug tolerance of ADA/NAb detection is assay dependent [Citation16]. Moreover, three of the four participants reported nonimmune-related TEAEs only (blood calcium decreased and COVID-19), and one participant reported no TEAEs.
A strength of this study was the sampling period of 253 days, which was designed to allow PK parameters to be adequately described, including the late elimination phase where non-linear elimination is exhibited. However, unlike the single dose administered in this study, patients with chronic conditions, such as osteoporosis, are likely to receive denosumab every 6 months [Citation2,Citation3,Citation17,Citation18]. Therefore, the non-linear elimination exhibited in this study is unlikely to be observed in clinical practice, limiting the generalizability of the current findings. The study duration allowed robust profiling of bone turnover markers, which generally return to pre-treatment levels within 12 months after the last dose [Citation3].
To avoid introducing additional variability due to sex differences and to minimize the risk to pregnant women (as denosumab is contraindicated for use in pregnant women [Citation2,Citation3]), enrollment in this study was limited to male participants. All participants in this study were Asian and male and the study population was relatively young (median [range] age of 38 [28–55] years); however, the EMA and FDA prescribing information for denosumab report no impact of race, gender, or age on PKs [Citation2,Citation3,Citation17,Citation18].
Further studies to establish biosimilarity between CT‑P41 and US-denosumab include an ongoing Phase 3 multidose study comparing efficacy, PKs, PDs, and safety in patients with postmenopausal osteoporosis [Citation19].
5. Conclusions
Following the administration of a single dose to healthy male adults, PK equivalence of CT‑P41 and US-denosumab was demonstrated based on the primary endpoints of this study. Secondary PK endpoints, PDs, safety, and immunogenicity of CT‑P41 and US-denosumab were also comparable between groups, supporting further development of CT‑P41.
Abbreviations
%AUCext, percentage of area under the concentration – time curve from time zero to infinity obtained by extrapolation; %CV, percent coefficient of variation; ADA, anti-drug antibody; ANCOVA, analysis of covariance; AUC, area under the concentration – time curve; AUC0–inf, area under the concentration – time curve from time zero to infinity; AUC0–last, area under the concentration – time curve from time zero to the last quantifiable concentration; AUEC, area under the effect curve; BLQ, below the lower limit of quantification; BMI, body mass index; CI, confidence interval; CL/F, apparent total body clearance; Cmax, maximum serum concentration; CTCAE, Common Terminology Criteria for Adverse Events; ECG, electrocardiogram; ECL, electrochemiluminescence; EMA, European Medicines Agency; EOS, end of study; FDA, Food and Drug Administration; gLSM, geometric least-squares mean; ISR, injection-site reaction; ITT, intent-to-treat; MSD, Meso Scale Discovery; NAb, neutralizing antibody; P1NP, procollagen type 1 N-terminal propeptide; pAUC0–W16, partial AUC from time zero to Week 16; pAUCW16–inf, partial AUC from Week 16 to infinity; PD, pharmacodynamic; PK, pharmacokinetic; RANK, receptor activator of nuclear factor kappa-B; RANKL, receptor activator of nuclear factor kappa-B ligand; s-CTX, serum type 1 C‑telopeptide; SD, standard deviation; t½, terminal half-life; TEAE, treatment-emergent adverse event; TEAESI, treatment-emergent adverse event of special interest; Tmax, time to Cmax; US, United States; US-denosumab, United States–licensed reference denosumab; VAS, visual analog scale; Vz/F, apparent volume of distribution during the terminal phase; λz, terminal elimination rate constant.
Declaration of interest
SH Kim, YJ Bae, JH Suh, S Kim, and EK Lee are employees of Celltrion, Inc. A Kim, JH Hong, W Shin, H Yoo, and JG Jung were investigators for this study and have nothing to disclose. JY Reginster consults for Celltrion, Inc. S Silverman consults for and has received grants from Amgen, consults for and has received travel grants from Celltrion, Inc., and consults for Radius. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Ethics statement
The study was conducted in accordance with the Declaration of Helsinki, the International Council for Harmonisation Guideline for Good Clinical Practice, and all applicable laws or regulations. The study protocol, informed consent form, and any other written materials provided to participants were approved by institutional review boards at each study center prior to study initiation (Table S1). Written informed consent was obtained from each participant prior to the study.
Author contributions
A Kim, JH Hong, W Shin, H Yoo, JG Jung, SH Kim, YJ Bae, JH Suh, S Kim, and EK Lee contributed to study design. A Kim, JH Hong, W Shin, H Yoo, and JG Jung contributed to data collection. JY Reginster, SH Kim, YJ Bae, JH Suh, S Kim, EK Lee, and S Silverman contributed to data analysis and/or interpretation. All authors critically reviewed and critically revised the manuscript, approved the final version for publication, and agree to be accountable for the accuracy and integrity of the work.
CT-P41 1.2 study_supplement_revised_clean.docx
Download MS Word (67.4 KB)Acknowledgments
We thank all participants and investigators involved in the study. Medical writing support, including development of a draft outline and subsequent drafts in consultation with the authors, collating author comments, copyediting, fact checking, and referencing, was provided by Samantha Booth, PhD, at Aspire Scientific Limited (Bollington, UK). Funding for medical writing support for this article was provided by Celltrion, Inc. (Incheon, Republic of Korea). Selected results from the study were presented as a poster presentation at the American Society for Bone and Mineral Research 2023 Annual Meeting (Vancouver, BC, Canada; October 13–16, 2023).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14712598.2024.2316846
Additional information
Funding
References
- Hanley DA, Adachi JD, Bell A, et al. Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract. 2012;66(12):1139–1146. doi: 10.1111/ijcp.12022
- European Medicines Agency. Prolia summary of product characteristics [Internet]. 2023 [cited 2023 Sep 26]. Available from: https://www.ema.europa.eu/en/documents/product-information/prolia-epar-product-information_en.pdf
- US Food and Drug Administration. Prolia prescribing information [Internet]. 2023 [cited 2023 Nov 3]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/125320Orig1s211lbl.pdf
- Curtis EM, Reginster JY, Al-Daghri N, et al. Management of patients at very high risk of osteoporotic fractures through sequential treatments. Aging Clin Exp Res. 2022;34(4):695–714. doi: 10.1007/s40520-022-02100-4
- Kanis JA, Harvey NC, McCloskey E, et al. Algorithm for the management of patients at low, high and very high risk of osteoporotic fractures. Osteoporos Int. 2020;31(1):1–12. doi: 10.1007/s00198-019-05176-3
- Camacho PM, Petak SM, Binkley N, et al. American Association of Clinical Endocrinologists/American College of Endocrinology clinical practice guidelines for the diagnosis and treatment of postmenopausal osteoporosis - 2020 update. Endocr Pract. 2020;26(Suppl 1):1–46. doi: 10.4158/GL-2020-0524SUPPL
- Baumgart DC, Misery L, Naeyaert S, et al. Biological therapies in immune-mediated inflammatory diseases: can biosimilars reduce access inequities? Front Pharmacol. 2019;10:279. doi: 10.3389/fphar.2019.00279
- Chen Q, Hu C, Liu Y, et al. Pharmacokinetics, pharmacodynamics, safety, and tolerability of single-dose denosumab in healthy Chinese volunteers: a randomized, single-blind, placebo-controlled study. PLoS One. 2018;13(6):e0197984. doi: 10.1371/journal.pone.0197984
- An G. Concept of pharmacologic target-mediated drug disposition in large-molecule and small-molecule compounds. J Clin Pharmacol. 2020;60(2):149–163. doi: 10.1002/jcph.1545
- Gillett MJ, Vasikaran SD, Inderjeeth CA. The role of PINP in diagnosis and management of metabolic bone disease. Clin Biochem Rev. 2021;42(1):3–10.
- Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab. 2008;93(6):2149–2157. doi: 10.1210/jc.2007-2814
- Kendler DL, Roux C, Benhamou CL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res. 2010;25(1):72–81. doi: 10.1359/jbmr.090716
- Lewiecki EM, Miller PD, McClung MR, et al. Two-year treatment with denosumab (AMG 162) in a randomized phase 2 study of postmenopausal women with low BMD. J Bone Miner Res. 2007;22(12):1832–1841. doi: 10.1359/jbmr.070809
- Nakamura T, Matsumoto T, Sugimoto T, et al. Clinical trials express: fracture risk reduction with denosumab in Japanese postmenopausal women and men with osteoporosis: denosumab fracture intervention randomized placebo controlled trial (DIRECT). J Clin Endocrinol Metab. 2014;99(7):2599–2607. doi: 10.1210/jc.2013-4175
- Pitale S, Thomas M, Rathi G, et al. A randomized placebo-controlled trial of the efficacy of denosumab in Indian postmenopausal women with osteoporosis. Indian J Endocrinol Metab. 2015;19(1):148–154. doi: 10.4103/2230-8210.146871
- US Food and Drug Administration. Immunogenicity testing of therapeutic protein products — developing and validating assays for anti-drug antibody detection [Internet]. 2019 [cited 2023 Sep 13]. Available from: https://www.fda.gov/media/119788/download
- European Medicines Agency. Xgeva summary of product characteristics [Internet]. 2022 [cited 2023 Sep 21]. Available from: https://www.ema.europa.eu/en/documents/product-information/xgeva-epar-product-information_en.pdf
- US Food and Drug Administration. Xgeva prescribing information [Internet]. 2020 [cited 2023 Sep 21]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125320s201lbl.pdf
- ClinicalTrials.gov. A phase 3 study to compare between CT-P41 and US-licensed prolia in postmenopausal women with osteoporosis [NCT04757376] [Internet]. 2023 [cited 2023 Sep 22]. Available from: https://www.clinicaltrials.gov/study/NCT04757376