
ABSTRACT
Background
This study compared the pharmacokinetics (PK), immunogenicity, and safety of candidate tocilizumab biosimilar, CT-P47, administered via auto-injector (CT-P47 AI) or pre-filled syringe (CT-P47 PFS), in healthy Asian adults.
Research design and methods
In this phase I, multicenter, open-label study, participants were randomized 1:1 to receive a single 162 mg/0.9 mL dose of CT-P47 via AI or PFS. Primary endpoints were area under the concentration – time curve from time zero to infinity (AUC0–inf) and maximum serum concentration (Cmax). PK equivalence was determined if 90% confidence intervals (CIs) for the ratios of geometric least-squares means (gLSMs) were within the predefined 80–125% equivalence margin. Secondary PK parameters, immunogenicity, and safety outcomes were also assessed.
Results
Of 314 participants randomized (155 CT-P47 AI; 159 CT-P47 PFS), 310 received the study drug (153 CT-P47 AI; 157 CT-P47 PFS). Primary and secondary PK results, immunogenicity and safety were similar between groups. Ninety percent CIs for the ratio of gLSMs were within the predefined equivalence margin for AUC0–inf (85.87–102.94) and Cmax (82.98–98.16).
Conclusions
PK equivalence between CT-P47 AI and CT-P47 PFS was demonstrated in healthy Asian adults, with comparable immunogenicity and safety between the two devices.
Trial registration
ClinicalTrials.gov: NCT05617183.
Plain Language Summary
Tocilizumab is a biologic medicine used to treat inflammatory diseases, such as rheumatoid arthritis. A biosimilar is a drug that is an almost identical copy of an approved original (‘reference’) biologic medicine; it has identical efficacy and safety to the original medicine but is typically less expensive. CT‑P47 is in development as a possible tocilizumab biosimilar.
Some patients prefer injections using an auto-injector (AI) rather than a pre-filled syringe (PFS), for reasons including ease of use and convenience. With an AI, medicine is delivered automatically by firmly pressing the device against the skin, whereas, with a PFS, a needle is inserted into the skin and medicine delivered by depressing the plunger. The injection of CT‑P47 using a PFS has shown comparable pharmacokinetics (i.e., the uptake, metabolism and excretion of the drug by the body) and safety to tocilizumab. Therefore, if the pharmacokinetics and safety of CT‑P47 administered via AI and PFS were shown to be similar, this might expand the choice of administration devices available to patients.
In this study, 310 healthy adults received a single injection of CT‑P47 via AI or PFS. Blood samples were taken over 43 days to analyze pharmacokinetics. The uptake, metabolism and elimination of CT‑P47 by the body was similar when administered by each device, suggesting that CT‑P47 can be administered by either AI or PFS.
1. Introduction
Tocilizumab is a recombinant, humanized, monoclonal antibody, targeting the interleukin-6 receptor (IL-6R) [Citation1]. Tocilizumab binds to both soluble and membrane-bound IL-6R, thereby inhibiting the dysregulated IL-6-mediated effects underlying several immune-mediated inflammatory diseases and certain lymphoproliferative disorders [Citation1]. The intravenous (IV) formulation of tocilizumab received regulatory approval from the European Medicines Agency (EMA) in 2009 [Citation2] and the US Food and Drug Administration (FDA) in 2010 [Citation3], followed by later approval of a subcutaneous (SC) formulation [Citation4,Citation5]. Indications for the SC formulation are rheumatoid arthritis (RA), giant cell arteritis, systemic juvenile idiopathic arthritis, polyarticular juvenile idiopathic arthritis and, in the United States only, systemic sclerosis‑associated interstitial lung disease [Citation4,Citation5].
Only two tocilizumab biosimilars have been approved by the EMA [Citation6] or FDA [Citation7] to date. CT‑P47 is one of the candidate biosimilars of reference tocilizumab in development [Citation8,Citation9]. Consistent with the SC administration devices available for reference tocilizumab [Citation4,Citation5], auto-injector (AI), and pre-filled syringe (PFS) dosage forms are being developed for CT-P47.
Patients have expressed satisfaction with, and preference for, AI over PFS administration [Citation10–15]. Specifically in patients with RA, the TOUCH trial found that most patients preferred AI to PFS administration of adalimumab for reasons including ease of use, convenience, faster injection, safety protection features, and less pain [Citation11]. Correspondingly, higher rates of self-administration have been observed in patients with RA receiving methotrexate via AI than via PFS [Citation14]. Importantly, patients with RA, including those with severe hand disability, can successfully self-administer injections using AIs [Citation16,Citation17]. For instance, a human factor study investigating the usability and patient acceptance of an AI in patients with RA found that disease severity had no significant impact on compliance with device instructions for use (compliance by degree of hand disability: lower, 83.9%; higher, 88.8%) [Citation16]. Moreover, in a study measuring the performance of simulated self-administered AI in patients with RA, hand disability, as measured by the Cochin scale [Citation18], had no impact on injection performance and patients with severe hand disability demonstrated a comparable level of injection performance to healthy individuals [Citation17]. Additionally, in a real-life study of patients with RA, their caregivers and healthcare professionals, successful administration of reference tocilizumab via AI was achieved by 92.3% of users at first assessment and by 98.1% at second assessment; in addition, there were no new safety concerns [Citation19].
Following the demonstration of bioequivalence of CT-P47 via PFS to European Union–approved tocilizumab (EU-tocilizumab) in the CT-P47 1.1 study [Citation9], and in light of patient preference for AI and potential benefits offered with AI over PFS [Citation10–14], the purpose of the current study was to develop the totality of evidence, per the FDA guidelines for developing biosimilars [Citation20], by investigating CT-P47 administered SC via AI. Specifically, this phase I, randomized, multicenter, open-label, two-arm, parallel-group, single-dose study aimed to compare the pharmacokinetics (PK), immunogenicity and safety of CT-P47 administered SC via AI or PFS in healthy Asian adults.
2. Participants and methods
2.1. Study design and procedures
This randomized, multicenter, open-label, two-arm, parallel-group phase I study was conducted at four study centers in South Korea (Table S1 in the online supplement). Screening was conducted from Day − 28 to Day − 2. Participants were randomized (1:1) to receive a single dose of CT-P47 AI or CT-P47 PFS followed by assessment of PKs, safety, and immunogenicity over the 6-week study period (until the end-of-study [EOS] visit at Day 43). A computer-generated randomization list was developed by a statistician before the study began using an interactive web response system. Using the randomization list, unique randomization numbers were assigned to participants in sequence. Randomization was balanced using randomly permuted blocks (sizes: two and four) and stratified by Day − 1 body weight (<70 vs 70–<90 vs ≥90 kg), sex (male vs female) and study center.
Participants received a single 162 mg/0.9 mL dose of CT-P47 via AI or PFS on Day 1, administered by study center staff into the outer upper arm area. Participants fasted for ≥8 h before and ≥4 h after study drug administration. Water was permitted until 1 h before and 1 h after study drug administration. Participants remained at the study center until 24-h post-dose assessments were completed, with subsequent visits conducted on an outpatient basis.
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and the International Council for Harmonisation Good Clinical Practice guideline. All national, state, and local laws and regulations were followed. The study protocol, informed consent form, advertisements used for the recruitment of participants and any other written materials provided to participants were approved by the relevant institutional review board(s) (Table S1) prior to study initiation. Participants provided written informed consent prior to study enrollment. The study was pre-registered with ClinicalTrials.gov [NCT05617183] [Citation21].
2.2. Participants
Full eligibility criteria are reported in the Supplementary methods. Adults aged 19–55 years (inclusive) who were healthy (defined as having no clinically relevant abnormalities identified by a detailed medical history, full examination, and clinical laboratory tests) were eligible for enrollment. Individuals were required to have a body weight of ≥60 to ≤100 kg (males) or ≥ 50 to ≤100 kg (females), with a body mass index (BMI) of 18.5 to 28.0 kg/m2 (inclusive). Exclusion criteria comprised: a history or current presence of clinically significant atopy or hypersensitivity/allergic reactions to study drug excipients or monoclonal antibodies; a history or current presence of systemic or local infections, gastrointestinal conditions or malignancy (except adequately treated squamous or basal cell carcinoma of the skin); laboratory abnormalities in alanine aminotransferase, aspartate aminotransferase, total bilirubin, absolute neutrophil count or platelet count at screening or Day − 1; and a history of exposure to tocilizumab, candidate tocilizumab biosimilars or IL-6–targeted drugs.
2.3. Study objectives
The primary study objective was to demonstrate PK equivalence of CT-P47 AI versus CT-P47 PFS in terms of area under the concentration – time curve from time zero to infinity (AUC0–inf) and maximum serum concentration (Cmax). Additional PK parameters were evaluated as secondary objectives of the study, considering area under the concentration – time curve from time zero to the last quantifiable concentration (AUC0–last), time to Cmax (Tmax), terminal half-life (t½), percentage of AUC0–inf obtained by extrapolation (%AUCext), terminal elimination rate constant (λz), apparent total body clearance (CL/F), apparent volume of distribution during the terminal phase (Vz/F) and serum tocilizumab concentration at each time point. The immunogenicity and safety of CT-P47 AI versus CT-P47 PFS were also evaluated (immunogenicity and safety endpoints are listed in the Supplementary methods).
2.4. Study assessments
Blood samples for PK assessment were drawn on Day 1 (pre-dose and 8 h post-dose) and at all following visits. Serum tocilizumab concentrations were ascertained using an electrochemiluminescence (ECL) method (Meso Scale Discovery, Meso Scale Diagnostics, Rockville, MD, U.S.A.). Concentrations below the lower limit of quantification (BLQ; 0.08 μg/mL) before study drug administration (or soon after administration, before the first measurable concentration) were set to zero. Post-dose concentrations that were BLQ, measurable concentrations after consecutive BLQ values and all other incidences of BLQ values were set to missing. PK parameters were computed by noncompartmental analysis using Phoenix WinNonlin version 8.3 (Pharsight, St Louis, MO, U.S.A.).
The immunogenicity of CT-P47 was assessed pre-dose at Day 1 and at Days 13 and 43. Anti‑drug antibodies (ADAs) were evaluated using a validated ECL bridging assay (sensitivity 3.021 ng/mL) that employed a three-tiered approach (screening, confirmatory, and titer). Confirmed ADA-positive samples were further analyzed for titer determination using a titer assay that serially dilutes samples. The neutralizing activity (i.e. neutralizing antibodies; NAbs) of ADA-positive samples was characterized using a validated ECL assay with affinity capture elution (sensitivity 148.787 ng/mL).
Safety assessments performed throughout included adverse event (AE) monitoring, clinical laboratory testing, and other evaluations. Treatment-emergent adverse events (TEAEs) were defined as AEs that were not present before or worsened after study drug administration. TEAEs were coded using the Medical Dictionary for Regulatory Affairs version 25.1 and severity was graded in line with the Common Terminology Criteria for AEs version 5.0. TEAEs of special interest (TEAESIs) were infection, hypersensitivity including anaphylaxis, injection site reaction (ISR), hepatic event, hemorrhage, gastrointestinal perforation, malignancy, and demyelinating disorder (capture logic can be found in the Supplementary methods). Clinical laboratory tests were conducted during screening and at Days − 1, 2, 6, 13, 29 and 43. Local injection site pain was measured by a 100-mm visual analog scale (VAS) within 15 min post-dose.
2.5. Statistical analysis
Categorical variables were summarized as n (%) of participants; continuous variables were summarized using descriptive statistics. An analysis of covariance (ANCOVA) was run to analyze the log-transformed primary PK endpoints (AUC0–inf and Cmax). Treatment was coded a fixed effect, whilst Day − 1 body weight, sex, and study center were coded covariates. For each primary PK endpoint, the difference in least-squares mean (LSM) between CT-P47 AI and CT-P47 PFS and associated 90% confidence interval (CI) were back-transformed to provide the ratio of geometric LSMs (gLSMs) and corresponding 90% CIs. The equivalence of PK (AUC0–inf and Cmax) between CT-P47 AI and CT-P47 PFS was determined if the 90% CI was within the predefined 80–125% equivalence margin.
Power calculations revealed that a sample size of 278 (139 per group) would provide 90% power to demonstrate equivalence of PKs (CT-P47 AI and CT-P47 PFS), using a 90% CI approach based on the predefined equivalence margin and assuming a coefficient of variation (CV%) of 61% and the expected geometric mean ratio of 1.0. With an expected 10% drop-out rate, approximately 310 participants (155 per group) needed to be randomized.
The analysis sets are described in the Supplementary methods. PK parameters were analyzed in the PK set. Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, U.S.A.) and missing data were not imputed.
3. Results
3.1. Participant disposition
The first participant was randomized on 15 November 2022; the final participant’s final visit was on 24 February 2023. Overall, 314 individuals were randomized (155 CT-P47 AI; 159 CT-P47 PFS) (). Four (1.3%) participants, two in each group, withdrew consent prior to the study drug administration. Overall, 300 (95.5%) participants completed the study. Ten (3.2%) participants discontinued after the study drug administration, all due to withdrawal by subject.
Figure 1. Participant flow diagram.
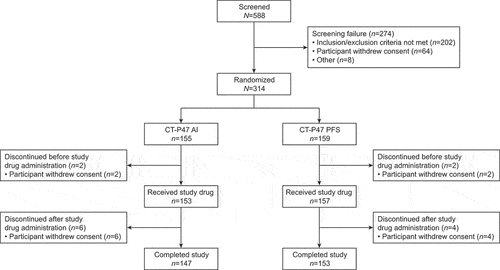
Participant demographics and baseline characteristics were similar between groups (). Overall, 215 (68.5%) participants were male and all participants were Asian. At screening, median (range) body weight was 69.3 (50.3–92.5) kg and 69.2 (50.5–100.0) kg in the CT-P47 AI and CT-P47 PFS groups, respectively. Correspondingly, median (range) BMI was 23.9 (19.0–28.0) kg/m2 and 23.8 (18.9–27.9) kg/m2.
Table 1. Participant demographics and baseline characteristics (intent-to-treat).
3.2. Pharmacokinetics
Mean serum concentrations were similar between groups until 43 days post-dose ( and Table S2). Primary () and secondary () PK variables were comparable between groups. PK equivalence between CT-P47 AI and CT-P47 PFS was demonstrated for the primary PK endpoints, AUC0–inf and Cmax (). In each case, the 90% CIs of the gLSMs ratios for CT-P47 AI versus CT-P47 PFS were within the predefined 80–125% equivalence margin.
Figure 2. Mean (SD) serum concentrations of CT-P47 for CT-P47 AI and CT-P47 PFS (PK set).
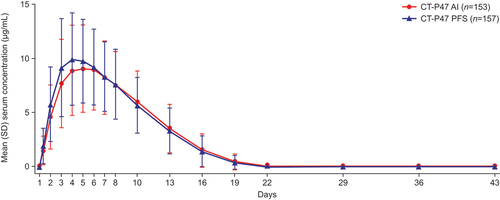
Figure 3. Summary and statistical analysis of the primary pharmacokinetic endpoints (PK set).
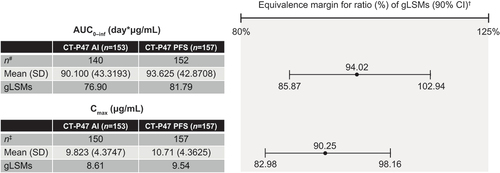
Table 2. Secondary pharmacokinetic results (PK set).
3.3. Immunogenicity
At baseline, 3 (2.0%) participants in the CT-P47 AI group and no participants in the CT-P47 PFS group were ADA positive. ADA positivity increased during the study and the proportion of participants with at least one positive post-baseline ADA result was comparable between groups (CT-P47 AI, 26 [17.0%]; CT-P47 PFS, 32 [20.4%]) (Table S3). At baseline, no participants were NAb positive. A similar proportion of participants in each group had at least one positive post-baseline NAb result (CT-P47 AI, 12 [7.8%]; CT-P47 PFS, 14 [8.9%]). At EOS visit, mean ADA titers were higher in the CT-P47 PFS group compared with the CT-P47-AI group because two participants showed higher titers of 12,480 and 24,960 relative to other participants; mean (standard deviation [SD]) titers were 933.75 (1005.78) and 2090.16 (4826.38) in the CT-P47 AI and CT-P47 PFS groups, respectively.
Serum concentrations (Figure S1) and primary and secondary PK results (Table S4) were generally comparable between the treatment groups in each ADA subset and between the ADA subsets in each treatment group. It is considered that there was no distinct impact of ADA status on serum concentration or PK results.
3.4. Safety
The full dose of the study drug was successfully received by 153 (100.0%) and 157 (100.0%) participants in the CT-P47 AI and CT-P47 PFS groups, respectively. Eighty-two (53.6%) and 77 (49.0%) participants reported at least one TEAE in the CT-P47 AI and CT-P47 PFS groups, respectively (). Correspondingly, TEAEs considered to be study drug related were reported by 64 (41.8%) and 60 (38.2%) participants. Investigations was the most common type of TEAE by system organ class (CT-P47 AI, 40 [26.1%]; CT-P47 PFS, 32 [20.4%]) (Table S5). Neutrophil count decreased was the most commonly reported TEAE overall and was reported by a higher proportion of participants in the CT-P47 AI than the CT-P47 PFS group (CT-P47 AI, 23 [15.0%]; CT-P47 PFS, 14 [8.9%]) (Table S6). All TEAEs classified as neutrophil count decreased recovered without treatment. No TEAEs led to study drug discontinuation or death and no treatment-emergent serious adverse events (TESAEs) were reported.
Table 3. Adverse events (safety set).
Infections were the most frequently reported TEAESI (CT-P47 AI, 18 [11.8%]; CT-P47 PFS, 15 [9.6%]) (). The most frequently reported TEAESIs classified as infection were upper respiratory tract infection (CT-P47 AI, 7 [4.6%]; CT-P47 PFS, 4 [2.5%]) and COVID-19 (CT-P47 AI, 5 [3.3%]; CT-P47 PFS, 3 [1.9%]). Similar proportions of participants in each group experienced TEAESIs of hypersensitivity, hepatic event and hemorrhage. Four (2.6%) and two (1.3%) participants experienced at least one TEAESI of hypersensitivity in the CT-P47 AI and CT-P47 PFS groups, respectively. Among participants reporting hypersensitivity, each sign or symptom classified as hypersensitivity was reported by one or two individuals. All participants recovered without treatment, except one participant in the CT-P47 PFS group. No anaphylaxis was identified according to Sampson criteria [Citation22]. Four participants in each group (2.6% CT-P47 AI; 2.5% CT-P47 PFS) experienced at least one TEAESI of hepatic event; all participants recovered without treatment. Two (1.3%) and three (1.9%) participants experienced at least one TEAESI of hemorrhage in the CT-P47 AI and CT-P47 PFS groups, respectively. All TEAESIs classified as hemorrhage were not considered as medically significant bleeding events and all participants recovered without treatment.
Rates of ISRs were relatively low in both groups, but higher in the CT-P47 AI versus the CT-P47 PFS group (CT-P47 AI, 14 [9.2%]; CT-P47 PFS, 5 [3.2%]). The most frequently reported signs or symptoms of ISR were injection site erythema (CT-P47 AI, 10 [6.5%]; CT-P47 PFS, 3 [1.9%]) and swelling (CT-P47 AI, 5 [3.3%]; CT-P47 PFS, 1 [0.6%]). Other signs and symptoms of ISR included, injection site bruising, induration, pain, pruritus, and warmth, and were reported for one participant in either group. All participants recovered from ISRs without any treatment within 1 day, except one participant in each group. No TEAESIs of gastrointestinal perforation, malignancy, or demyelinating disorder were reported. All TEAESIs were Grade 1–2 in intensity, other than two Grade 3 COVID-19 infections in the CT-P47 AI group considered unrelated to the study drug. All participants recovered from the event.
Hematology and clinical chemistry findings were generally comparable between groups, except for creatine kinase, which was higher in the CT-P47 PFS group in a few participants at Days 13 and 29. A detailed explanation is provided in the Supplementary results, including Grade ≥3 laboratory parameters (Table S7). There were no clinically notable differences in hypersensitivity monitoring, vital sign measurements, electrocardiogram results, physical examination findings, or clinical laboratory results between the groups. No cases of pregnancy in female participants or female partners of male participants were reported during the study. No participant displayed any signs or symptoms at any time point during the study that were indicative of active tuberculosis. Comparable mean (SD) injection site pain scores assessed by 100-mm VAS were observed between groups (CT-P47 AI, 10.44 [17.34]; CT-P47 PFS, 10.17 [16.71]).
4. Discussion
This study demonstrated the equivalence of PK results (AUC0–inf and Cmax) between CT-P47 AI and CT-P47 PFS following the administration of a single dose to healthy adults, thereby meeting the primary objective of this study. Additionally, the secondary PK results (AUC0–last, Tmax, t½, %AUCext, λz, CL/F, and Vz/F), immunogenicity, and safety were similar between the groups. The equivalence of the primary (AUC0–inf and Cmax) and comparability of the secondary PK results for CT-P47 AI and CT-P47 PFS is consistent with an existing clinical trial investigating a single dose (162 mg/0.9 mL) of reference tocilizumab administered via AI or PFS in healthy individuals [Citation19].
With regard to immunogenicity, a similar number of participants had at least one positive post-baseline ADA result to the CT-P47 1.1 study [Citation9]. The proportion of ADA/NAb positivity was similar for the administration of CT-P47 via AI and PFS. Given that a similar proportion of ADA/NAb positivity has been observed between CT-P47 PFS and EU-tocilizumab PFS [Citation9], this suggests that ADA/NAb positivity with CT-P47 via AI is similar to that with EU-tocilizumab via PFS. To date, no study has compared the immunogenicity of AI and PFS administration of reference or candidate tocilizumab [Citation19,Citation23]. Considering immunogenicity by ADA status, there was no distinct impact of ADA status on serum concentration or PK results, though, comparisons between ADA status are limited by the small number of ADA-positive participants and should therefore be interpreted with caution.
In this study, a single dose of CT-P47 administered via AI or PFS was well tolerated, with a comparable overall safety profile between groups. The majority of TEAEs reported were Grade 1–2 in intensity. All Grade 4 TEAEs were laboratory abnormalities that resolved by EOS without treatment. The comparable incidence of TEAEs for CT-P47 administered via AI versus PFS is in line with findings for reference tocilizumab administered via AI or PFS in healthy individuals [Citation19]. Overall, neutrophil count decreased was the most commonly reported TEAE. This finding is unsurprising, given that previous investigations of reference tocilizumab have detailed low neutrophil counts as marked laboratory abnormalities [Citation24,Citation25]. Notably, the EU-tocilizumab prescribing information lists low neutrophil counts as a consideration for dosing [Citation4].
In keeping with the CT-P47 1.1 study [Citation9], infection was the most frequently reported TEAESI. The proportions of participants experiencing TEAESIs of hypersensitivity, hepatic event and hemorrhage were low and similar between groups. ISRs occurred in a lower proportion of participants (6.1%) than observed in a phase I study evaluating reference tocilizumab delivered via AI or PFS (19.1%) [Citation19]. A higher rate of ISRs was reported in the CT-P47 AI relative to the CT-P47 PFS group, consistent with some reports from other clinical trials comparing the two administration devices [Citation12,Citation14,Citation26–29]. This finding might be explained by the relatively higher pressure applied with an AI against the skin compared with PFS [Citation4].
This study had a few limitations. As sample size was calculated for the demonstration of PK equivalence; the study was not powered for safety, thus we cannot exclude the possibility that differences in rates of TEAEs may become apparent with larger sample sizes. Moreover, this study included male and female participants, which could result in additional variability in PK findings over and above that accounted for by differences in body weight [Citation9]; however, randomization was stratified by sex. Furthermore, this study had an open-label design due to the visible differences in AI and PFS administration devices that cannot be concealed. However, PK equivalence was not anticipated to be impacted by participant or investigator knowledge.
The CT-P47 1.1 study was a phase I, randomized, double-blind study that compared the PK, immunogenicity and safety of a single dose of CT-P47 with EU-tocilizumab administered SC via a PFS in healthy adults [Citation9]. In this study, CT-P47 demonstrated PK equivalence and comparable immunogenicity and safety with EU-tocilizumab in 289 healthy Asian adults [Citation9]. The current study demonstrated bioequivalence of AI and PFS versions of CT-P47 in healthy Asian adults. Therefore, the evidence suggests that CT-P47 via AI is bioequivalent to EU-tocilizumab via PFS, at least in healthy individuals. Both the CT-P47 1.1 study [Citation9] and the current study were conducted in healthy adults, who provide the most sensitive population to detect differences in PK profiles. As a next step, PKs are being evaluated in the CT-P47 3.1 study, an ongoing phase III study comparing the efficacy and safety of CT-P47 IV and EU-tocilizumab administered in patients with moderate-to-severe RA who may have confounding factors and variable disease characteristics [Citation30]. Moreover, beyond the demonstration of PK equivalence between AI and PFS delivery, FDA recommendations for the development of biosimilars [Citation31] require real-life assessment of patient handling experience to assess self‑administration device performance. Therefore, the usability of CT-P47 AI is being assessed in the ongoing CT-P47 3.2 study in patients with moderate-to-severe active RA [Citation32].
5. Conclusions
The present study demonstrated equivalence of PK results for CT-P47 AI and CT-P47 PFS in healthy Asian adults. Immunogenicity and safety were comparable between CT-P47 administered via AI and PFS. These findings suggest that CT-P47 can be administered via AI or PFS, thereby potentially expanding the choice of administration devices available to patients, caregivers, and healthcare professionals.
Abbreviations
| %AUCext | = | Percentage of AUC0–inf obtained by extrapolation |
| ADA | = | Anti‑drug antibody |
| AE | = | Adverse event |
| AI | = | Auto-injector |
| ANCOVA | = | Analysis of covariance |
| AUC0–inf | = | Area under the concentration – time curve from time zero to infinity |
| AUC0–last | = | Area under the concentration – time curve from time zero to the last quantifiable concentration |
| BLQ | = | Below the lower limit of quantification |
| BMI | = | Body mass index |
| CI | = | Confidence interval |
| CL/F | = | Apparent total body clearance |
| Cmax | = | Maximum serum concentration |
| CV% | = | Coefficient of variation |
| ECL | = | Electrochemiluminescence |
| EMA | = | European Medicines Agency |
| EOS | = | End-of-study |
| EU | = | European Union |
| FDA | = | Food and Drug Administration |
| gLSM | = | Geometric least-squares mean |
| IL-6R | = | Interleukin-6 receptor |
| ISR | = | Injection site reaction |
| IV | = | Intravenous |
| LSM | = | Least-squares mean |
| NAb | = | Neutralizing antibody |
| PFS | = | Pre-filled syringe |
| PK | = | Pharmacokinetic |
| RA | = | Rheumatoid arthritis |
| SC | = | Subcutaneous |
| SD | = | Standard deviation |
| t½ | = | Terminal half-life |
| TEAE | = | Treatment-emergent adverse event |
| TEAESI | = | Treatment-emergent adverse event of special interest |
| TESAE | = | Treatment-emergent serious adverse event |
| Tmax | = | Time to maximum serum concentration |
| VAS | = | Visual analog scale |
| Vz/F | = | Apparent volume of distribution during the terminal phase |
| λz | = | Terminal elimination rate constant |
Declaration of interest
KS Yu, H Ryu, D Shin, MK Park, JG Hwang, SJ Moon, and MG Kim have nothing to disclose, other than sponsorship with respect to the current study by Celltrion, Inc. E Keystone has had consulting agreements and/or held advisory board membership for AbbVie, Celltrion, GSK, Lilly, Pfizer, Sandoz, and Samsung Bioepsis, and has received honoraria for speaking agreements with AbbVie, Celltrion, GSK, Lilly, Pfizer, and Sandoz. JS Smolen has received grants for his institution from AbbVie, AstraZeneca, Janssen, Lilly, Novartis, and Roche, and has received honoraria for consulting or speaking engagements from AbbVie, Amgen, Astro, BMS, Celgene, Celltrion, Chugai, Gilead, ILTOO, Janssen, Lilly, MSD, Novartis-Sandoz, Pfizer, Roche, Samsung, Sanofi, and UCB. SH Kim, YJ Bae, DB Jeon, JY Jang, GE Yang, JH Bae and JY Lee are employees of Celltrion, Inc. GR Burmester has received honoraria for consulting and lectures from AbbVie, Chugai, Galapagos, Lilly, Pfizer, Roche, and Sanofi. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
All authors contributed to study design and data analysis or interpretation. KS Yu, H Ryu, D Shin, MK Park, JG Hwang, SJ Moon, MG Kim, SH Kim, YJ Bae, DB Jeon, JY Jang, GE Yang, JH Bae, and JY Lee contributed to data collection. All authors critically reviewed and critically revised the manuscript, approved the final version for publication and agree to be accountable for the accuracy and integrity of the work.
Ethical approval
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and the International Council for Harmonisation Good Clinical Practice guideline. All national, state, and local laws and regulations were followed. The study protocol, informed consent form, advertisements used for the recruitment of participants and any other written materials provided to participants were approved by the relevant institutional review board(s) (Table S1) prior to study initiation. Participants provided written informed consent prior to study enrollment.
Supplemental Material
Download MS Word (250.5 KB)Acknowledgments
We thank all participants and investigators involved in the study. Medical writing support, including development of a draft outline and subsequent drafts in consultation with the authors, collating author comments, copyediting, fact checking, and referencing, was provided by Samantha Booth, at Aspire Scientific Limited (Bollington, UK). Funding for medical writing support for this article was provided by Celltrion, Inc. (Incheon, Republic of Korea). This study was pre-registered with ClinicalTrials.gov [NCT05617183].
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14712598.2024.2321360.
Additional information
Funding
References
- Tanaka T, Narazaki M, Kishimoto T. Interleukin (Il-6) immunotherapy. Cold Spring Harb Perspect Biol. 2018;10(8):a028456. doi: 10.1101/cshperspect.a028456
- European Medicines Agency. Assessment report for RoActemra [Internet]. 2009 [cited 2023 Jul 20]. Available from: https://www.ema.europa.eu/en/documents/assessment-report/roactemra-epar-public-assessment-report_en.pdf
- US Food and Drug Administration. Highlights of prescribing information: ACTEMRA (tocilizumab) [Internet]. 2010 [cited 2023 Jul 20]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/125276lbl.pdf
- European Medicines Agency. RoActemra summary of product characteristics [Internet]. 2023 [cited 2023 Jul 14]. Available from: https://www.ema.europa.eu/en/documents/product-information/roactemra-epar-product-information_en.pdf
- US Food and Drug Administration. Highlights of prescribing information: ACTEMRA (tocilizumab) [Internet]. 2022 [cited 2023 Oct 17]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/125472s049lbl.pdf
- European Medicines Agency. Tyenne summary of product characteristics [Internet]. 2023 [cited 2023 Oct 17]. Available from: https://www.ema.europa.eu/en/documents/product-information/tyenne-epar-product-information_en.pdf
- US Food and Drug Administration. Drugs@FDA: FDA-approved drugs [Internet]. 2023 [cited 2023 Oct 17]. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=761354
- Mehr S. Tocilizumab biosimilars approaching: developments on three fronts [Internet]. 2022 [cited 2023 Jul 14]. Available from: https://biosimilarsrr.com/2022/08/18/tocilizumab-biosimilars-approaching-developments-on-three-fronts/
- Yu KS, Kim B, Shin D, et al. Pharmacokinetics and safety of candidate tocilizumab biosimilar CT-P47 versus reference tocilizumab: a randomized, double-blind, single-dose phase I study. Expert Opin Investig Drugs. 2023;32(5):429–439. doi: 10.1080/13543784.2023.2212155
- Hsiao B, Fraenkel L. Patient preferences for rheumatoid arthritis treatment. Curr Opin Rheumatol. 2019;31(3):256–263. doi: 10.1097/BOR.0000000000000591
- Kivitz A, Cohen S, Dowd JE, et al. Clinical assessment of pain, tolerability, and preference of an autoinjection pen versus a prefilled syringe for patient self-administration of the fully human, monoclonal antibody adalimumab: the TOUCH trial. Clin Ther. 2006;28(10):1619–1629. doi: 10.1016/j.clinthera.2006.10.006
- Rho YH, Rychlewska-Hanczewska A, Sliwowska B, et al. Usability of prefilled syringe and autoinjector for sb4 (an etanercept biosimilar) in patients with rheumatoid arthritis. Adv Ther. 2019;36(9):2287–2295. doi: 10.1007/s12325-019-01027-z
- Roszkiewicz J, Swacha Z, Smolewska E. Prefilled pen versus prefilled syringe: a pilot study evaluating two different methods of methotrexate subcutaneous injection in patients with JIA. Pediatr Rheumatol Online J. 2020;18(1):64. doi: 10.1186/s12969-020-00455-4
- Saraux A, Hudry C, Zinovieva E, et al. Use of auto-injector for methotrexate subcutaneous self-injections: high satisfaction level and good compliance in self-i study, a randomized, open-label, parallel group study. Rheumatol Ther. 2019;6(1):47–60. doi: 10.1007/s40744-018-0134-2
- Kivitz A, Baret-Cormel L, van Hoogstraten H, et al. Usability and patient preference phase 3 study of the sarilumab pen in patients with active moderate-to-severe rheumatoid arthritis. Rheumatol Ther. 2018;5(1):231–242. doi: 10.1007/s40744-017-0090-2
- Schwarzenbach F, Dao Trong M, Grange L, et al. Results of a human factors experiment of the usability and patient acceptance of a new autoinjector in patients with rheumatoid arthritis. Patient Prefer Adherence. 2014;8:199–209. doi: 10.2147/PPA.S50583
- Xiao X, Li W, Clawson C, et al. Evaluation of performance, acceptance, and compliance of an auto-injector in healthy and rheumatoid arthritic subjects measured by a motion capture system. Patient Prefer Adherence. 2018;12:515–526. doi: 10.2147/PPA.S160394
- Duruoz MT, Poiraudeau S, Fermanian J, et al. Development and validation of a rheumatoid hand functional disability scale that assesses functional handicap. J Rheumatol. 1996;23:1167–1172.
- Fettner S, Mela C, Wildenhahn F, et al. Evidence of bioequivalence and positive patient user handling of a tocilizumab autoinjector. Expert Opin Drug Deliv. 2019;16(5):551–561. doi: 10.1080/17425247.2019.1604678.
- US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product [Internet]. 2015 [cited 2023 Jul 17]. Available from: https://www.fda.gov/media/82647/download
- ClinicalTrials.gov. Compare the auto-injector and pre-filled syringe of CT-P47 in healthy subjects [Internet]. 2023 [cited 2023 Jul 18]. Available from: https://www.clinicaltrials.gov/study/NCT05617183?cond=NCT05617183&rank=1
- Sampson HA, Munoz-Furlong A, Campbell RL. et al. Second symposium on the definition and management of anaphylaxis: summary report – second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. Ann Emerg Med. 2006;47:373–380. doi: 10.1016/j.annemergmed.2006.01.018
- Tomaszewska-Kiecana M, Dryja A, Ullmann M, et al. Pharmacokinetics and tolerability of prefilled syringe and auto-injector presentations of MSB11456: results of a randomized, single-dose study in healthy adults. Expert Rev Clin Immunol. 2023;19(4):447–455. doi: 10.1080/1744666X.2023.2174970
- Morcos PN, Zhang X, McIntyre C, et al. Pharmacokinetics and pharmacodynamics of single subcutaneous doses of tocilizumab administered with or without rHuPH20. Int J Clin Pharmacol Ther. 2013;51(7):537–548. doi: 10.5414/CP201847
- Zhang X, Georgy A, Rowell L. Pharmacokinetics and pharmacodynamics of tocilizumab, a humanized anti-interleukin-6 receptor monoclonal antibody, following single-dose administration by subcutaneous and intravenous routes to healthy subjects. Int J Clin Pharmacol Ther. 2013;51(6):443–455. doi: 10.5414/CP201819
- Bush J, Kawakami K, Muniz R. A phase 1, randomized, open-label, single-dose study to assess the relative bioavailability of a subcutaneous dose of FKB327 when administered using a prefilled syringe, a prefilled auto-injector, or a vial with disposable syringe in healthy subjects. BMC Pharmacol Toxicol. 2019;20(1):87. doi: 10.1186/s40360-019-0376-9
- Ramael S, Van Hoorick B, Tiessen R, et al. Similar pharmacokinetics of the adalimumab (Humira® biosimilar BI 695501 whether administered via subcutaneous autoinjector or prefilled syringe (VOLTAIRE®-AI and VOLTAIRE®-TAI): phase 1, randomized, open-label, parallel-group trials. Rheumatol Ther. 2018;5:403–421. doi: 10.1007/s40744-018-0119-1
- Cherniakov I, Cohen-Barak O, Tiver R, et al. A pharmacokinetic bioequivalence study of fremanezumab administered subcutaneously using an autoinjector and a prefilled syringe. Clin Pharmacol Drug Dev. 2021;10(9):1018–1027. doi: 10.1002/cpdd.902
- Shin D, Lee Y, Jeong D, et al. Comparative pharmacokinetics of an adalimumab biosimilar SB5 administered via autoinjector or prefilled syringe in healthy subjects. Drug Des Devel Ther. 2018;12:3799–3805. doi: 10.2147/DDDT.S169082
- ClinicalTrials.gov. A study to compare efficacy and safety of CT-P47 and RoActemra in patients with rheumatoid arthritis [Internet]. 2022 [cited 2023 Jul 17]. Available from: https://www.clinicaltrials.gov/study/NCT05489224?cond=NCT05489224&rank=1
- US Food and Drug Administration. Guidance for industry rheumatoid arthritis: developing drug products for treatment [Internet]. 2013 [cited 2023 Jul 28]. Available from: https://www.fda.gov/media/86066/download
- ClinicalTrials.gov. A study to evaluate usability of subcutaneous auto-injector of CT-P47 in patients with active rheumatoid arthritis [Internet]. 2023 [cited 2023 Jul 17]. Available from: https://www.clinicaltrials.gov/study/NCT05725434?intr=CT-P47&rank=2