
ABSTRACT
Objective
VOLTAIRE-HCLF compared the relative bioavailability of citrate-free high-concentration and reference formulations of the biosimilar adalimumab-adbm (Cyltezo®), including pharmacokinetic (PK) profiles, immunogenicity, and safety profiles in healthy volunteers.
Methods
Healthy volunteers (N = 200) aged 18–55 years and with body mass index of 18.5–29.9 kg/m2 and no prior exposure to adalimumab were randomized in a 1:1 ratio to receive a single subcutaneous injection of either adalimumab-adbm 40 mg/0.4 mL (high-concentration formulation) or 40 mg/0.8 mL (reference formulation). Participants completed 13 follow-up visits over 57 days, followed by a safety follow-up period of up to 70 days.
Results
The main PK parameters were similar for the high-concentration and reference groups. For all primary endpoints, the geometric mean ratios and 90% confidence intervals of AUC0–1344, AUC0–∞, and Cmax for both groups were entirely within the standard 80–125% bioequivalence acceptance range at 101.88% (93.31–111.23%), 105.38% (95.06–116.81%), and 91.29% (84.38–98.76%), respectively. There were no differences in the proportion of anti-drug antibody-positive participants or in the distribution of anti-drug antibody titers between the two formulations at any time point after drug dosing. Participants who were given the high-concentration formulation of adalimumab-adbm experienced a lower incidence of adverse events and local reactions than those who were given the reference formulation.
Conclusions
Overall, the high-concentration and reference adalimumab-adbm formulations had highly similar PK and immunogenicity profiles and were safe and well tolerated.
Clinical trial registration
NCT05203289
1. Introduction
Adalimumab-adbm (Cyltezo®) is a recombinant IgG1 human monoclonal antibody tumor necrosis factor blocker used in the treatment of immune-mediated inflammatory diseases including rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, chronic plaque psoriasis, hidradenitis suppurativa, and uveitis [Citation1]. Adalimumab-adbm was approved by the U.S. Food and Drug Administration (FDA) as a biosimilar to the adalimumab reference product (Humira®) [Citation2]. In October 2021, adalimumab-adbm became the first monoclonal antibody to be approved as an interchangeable biosimilar, meaning that where U.S. state laws permit, a pharmacist can substitute it for the adalimumab reference product without requiring prior authorization from the prescriber. Phase III studies (VOLTAIRE-RA, VOLTAIRE-CD, VOLTAIRE-PsO) demonstrated that adalimumab-adbm has similar efficacy, safety, and immunogenicity to the adalimumab reference product across several disease indications [Citation3–5].
Adalimumab-adbm has been developed in two different citrate-free formulations: at 50 mg/mL as a 0.8 mL solution for injection (reference formulation) and a lower-volume formulation at 100 mg/mL as a 0.4 mL solution for injection (high-concentration formulation), with both formulations containing 40 mg of active drug. Notably, larger subcutaneous injection volumes are typically associated with increased patient discomfort and pain at the injection site, both of which may contribute to poor patient outcomes and medication adherence [Citation6]. While conflicting evidence exists regarding the impact of faster injection speed on injection site pain, the combined effect of faster injections with lower drug volume has the advantage of increased tolerability and quickening the injection process for patient self-administration [Citation7,Citation8].
The main objective of the VOLTAIRE-HCLF Phase I study was to investigate the relative bioavailability of 40 mg high-concentration adalimumab-adbm (100 mg/mL, 0.4 mL) compared with 40 mg adalimumab-adbm reference formulation (50 mg/mL, 0.8 mL) as a single-dose subcutaneous administration in healthy male and female volunteers. The study also aimed to compare the safety and immunogenicity profiles of the new high-concentration formulation with the current reference adalimumab-adbm formulation.
2. Methods
2.1. Study participants
Healthy male and female volunteers with an age range of 18–55 years and body mass index of 18.5–29.9 kg/m2 were enrolled in the study. Participants were excluded from the study if they had any prior exposure to adalimumab or adalimumab biosimilar drugs, any concomitant disease, or any abnormalities in medical examination findings or laboratory values. All participants provided written informed consent before study enrollment.
2.2. Study design
VOLTAIRE-HCLF was a 14-week, double-blind, randomized, single-dose, parallel-arm Phase I trial to compare the pharmacokinetics (PK), safety, tolerability, and immunogenicity of two formulations of adalimumab-adbm conducted at two sites in the United States from February 2022 to August 2022. Due to the long half-life of adalimumab-adbm, a parallel-group design was selected for this trial. A total of 200 participants were randomized in a 1:1 ratio to receive either the high-concentration formulation (adalimumab-adbm 40 mg/0.4 mL) or the reference formulation (adalimumab-adbm 40 mg/0.8 mL) ().
Figure 1. Summary of participant disposition.
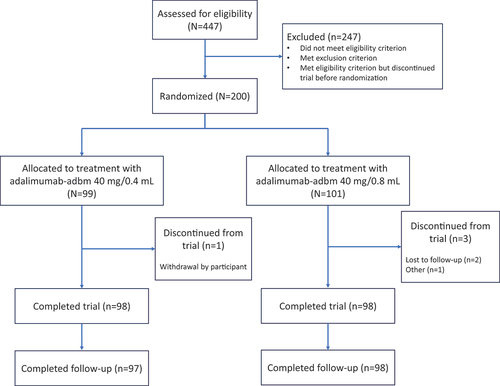
Randomization was performed by GCTS Medical Coding & Randomization Team (BI Pharma GmbH & Co. KG, Biberach, Germany). The generated randomization code was approved by a trial-independent statistician and inaccessible to all personnel directly involved in the conduct of the trial. To maintain blinding, the trial medication was administered by third-party trial personnel not involved in any other trial assessments or procedures.
Study participants received a single dose of adalimumab-adbm administered as a subcutaneous injection on Day 1 and remained in the clinic until Day 2 for monitoring. Following discharge from the trial site, participants returned to the clinic for assessment over 13 visits on Days 3, 4, 5, 6, 7, 8, 10, 15, 22, 29, 36, 43, and 57 (end of the observation period), with a safety follow-up period of up to 10 weeks (70 days from date of administration).
The study protocol was approved by the ADVARRA Institutional Review Board (Columbia, MD, USA) on 4 February 2022. The study was conducted in accordance with the International Council for Harmonisation Tripartite Guideline for Good Clinical Practice and the Declaration of Helsinki. All national, state, and local laws or regulations were followed.
2.3. Study assessments
2.3.1. Pharmacokinetic assessment
The primary endpoints to evaluate the relative bioavailability of the high-concentration and reference formulations were AUC0–1344 (area under the concentration–time curve of the analyte in plasma from 0 to 1344 hours after dose administration), AUC0–∞ (area under the concentration– time curve of the analyte in plasma from dosing extrapolated to infinity), and Cmax (maximum measured concentration of the analyte in plasma). Blood samples (3 mL) for PK assessment were collected for the preparation of K2EDTA plasma on Day 1 prior to drug administration and at 4, 12, and 24 hours following administration, and during each of the 13 follow-up visits from Days 3–57 of this study. PK parameters were determined by non-compartmental analysis using Phoenix WinNonlin version 8.1 (Certara, St. Louis, MO, USA).
2.3.2. Immunogenicity assessment
The occurrence and titer of anti-drug antibodies (ADAs) and neutralizing antibodies (nAbs) were assessed in plasma samples at baseline, Day 15, Day 29, and Day 57 using validated methods.
2.3.3. Safety and tolerability
Safety evaluations were performed by assessing vital signs, physical examinations, 12-lead electrocardiogram analysis, clinical laboratory tests, and assessment of local tolerability (i.e. swelling, induration, heat, redness, pain, and other findings). Local tolerability findings that were assessed as clinically relevant were recorded as adverse events (AEs). AEs were reported as treatment-emergent adverse events (TEAEs), AEs that constituted adverse events of special interest (AESIs), serious adverse events (SAEs), treatment-emergent infections, and AEs of injection site reactions. All AEs were collected until 10 weeks (70 days) after the administration of the trial medication. AEs that were not fully resolved at the safety follow-up visit were then followed until recovery, or in case of persistency, until sufficient improvement was achieved.
2.3.4. Statistical methods
An enrollment of 200 study participants was determined to be a sufficient sample size to assess the primary objective of this trial and achieve precision, i.e. a ratio of upper 90% confidence interval (CI) limit to the geometric mean ratio (GMR), of at least 1.13 in estimating the relative bioavailability.
The primary PK endpoints were log-transformed and subsequently analyzed using an analysis of covariance (ANCOVA) model with fixed effects for treatment, location of trial medication injection, and baseline body weight (continuous) to provide the GMRs and 90% CI for the primary endpoints across both formulations. The 90% CI was calculated based on the residual error from the ANCOVA. Descriptive statistics were calculated for all endpoints. The two formulations were considered pharmacokinetically similar if all the 90% CI of the respective GMR were within the standard bioequivalence acceptance range of 80–125%. All statistical analyses were conducted using SAS version 9.4 (SAS Institute, Inc., Cary, NC, USA).
3. Results
A total of 447 volunteers were screened and 200 participants were randomized between the two treatment groups, of whom 196 completed the trial per protocol between 21 February 2022 and 29 August 2022. Four participants prematurely discontinued the trial; two participants were lost to follow-up, one participant withdrew consent, and one participant was discontinued due to personal reasons. A summary of demographics and baseline characteristics is shown in .
Table 1. Participant demographics.
3.1. Pharmacokinetic assessment
As shown in , the mean plasma concentration–time profiles were similar in both treatment groups over the observation period, indicating that the new high-concentration formulation and the current reference formulation of adalimumab-adbm had similar PK characteristics in study participants. Plasma concentration rapidly increased over the first 48 hours for all participants and continued to rise at a more gradual rate until a median tmax of approximately 5–6 days (120–143 hours). Thereafter, plasma concentration slowly declined until Day 42 (1008 hours). Plasma concentrations were still measurable at the last blood sampling time point (1344 hours) for ~ 62% and ~ 46% of participants in the high-concentration and reference arms, respectively. Total and peak exposure of adalimumab-adbm, based on the adjusted geometric mean of AUC0–∞, AUC0–1344, and Cmax, were similar between the high-concentration and reference formulations ().
Figure 2. Mean plasma concentration–time profile between adalimumab-adbm 40 mg/0.4 mL and 40 mg/0.8 mL.
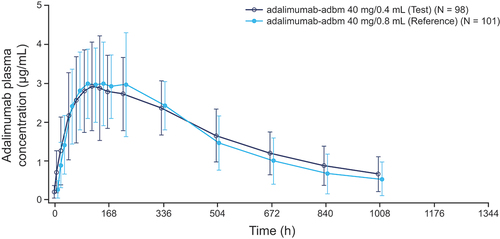
Table 2. Inferential statistical analysis of PK parameters for adalimumab-adbm 40 mg/0.4 mL and 40 mg/0.8 mL.
For all primary endpoints, the GMR and 90% CI of AUC0–1344, AUC0–∞, and Cmax for the high-concentration vs reference groups were entirely within the standard 80–125% bioequivalence acceptance range at 101.88% (93.31–111.23%), 105.38% (95.06–116.81%), and 91.29% (84.38–98.76%), respectively. Inter-individual variability in primary PK parameters was found to be moderate for both formulations. When the primary PK parameters were statistically adjusted based on the ANCOVA model on a logarithmic scale, the gCV for inter-individual variability was found to range from 34–45%.
3.2. Immunogenicity assessment
At baseline, 19 participants (19.2%) in the high-concentration treatment group and 11 participants (10.9%) in the reference formulation group were positive for ADAs, with a median titer of 4 (range: 1–32) and 8 (range: 1–128), respectively. Over the duration of the study, the percentage of ADA-positive individuals increased after administration of adalimumab-adbm, with no apparent difference in ADA percentages or ADA titer distribution observed between the two formulations at any time point after drug dosing (). Overall, the presence of preexisting ADAs in participants at baseline did not affect the PK exposure in either treatment arm.
Figure 3. Time course of (a) ADA development and (b) nAb development over time for all treatments.
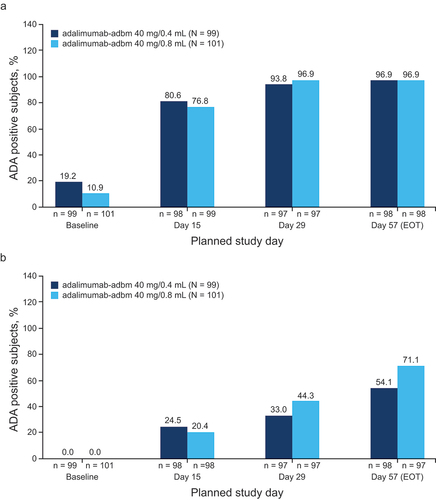
An analysis of nAb development over the course of the study showed slight variation between the two formulations of adalimumab-adbm (). The percentage of nAb-positive participants was slightly lower for the high-concentration formulation than in the reference formulation on Days 29 and 57, but the difference was not considered clinically relevant, as nAb titer distribution, PK, and safety were similar in both treatment groups.
3.3. Safety
At least one TEAE was reported for 52/200 participants (26.0%), including 18 participants (18.2%) in the high-concentration group and 34 participants (33.7%) in the reference group (). Most TEAEs were defined as mild, with no SAEs or AE-related deaths reported. The most frequently reported TEAEs were headaches, reported for 10 participants (5.0%) across both treatment groups. TEAEs of moderate intensity were reported by one participant in the high-concentration group (back pain and neck pain) and four participants (hypersensitivity, headache, dizziness, and arthralgia) in the reference group. No TEAEs of severe intensity were reported. The most frequently reported drug-related TEAEs were injection site erythema with one participant (1.0%) in the high-concentration group and eight participants (7.9%) in the reference group, followed by headache with four (4.0%) participants in each treatment group.
Table 3. Summary of frequency and incidence of subjects with AEs.
An investigator-defined AESI was reported for only one participant (1.0%) in the reference group who experienced hypersensitivity after 9 days’ drug administration that lasted 12 days. The event was moderate in intensity and resolved by the end of the observation period. No clinically meaningful changes from baseline were observed in the mean values for serum chemistry and hematology laboratory tests on Day 57. The safety laboratory results for chemistry, hematology, and urinalysis parameters showed no meaningful differences between treatment groups. There was no clinically relevant variance in vital signs, including mean systolic or diastolic blood pressure, pulse rate, or body temperature values within the treatment groups. A total of 17 treatment-related injection site reaction AEs were reported, with one event in the high-concentration formulation and 16 events across nine participants in the reference formulation. All injection site reactions were non-serious, mild in intensity, and resolved in all study participants.
4. Discussion
Bioavailability of the adalimumab-adbm high-concentration formulation was highly similar to that of the reference formulation after single subcutaneous administration in healthy volunteers. All 90% CI for total and peak exposure were entirely within the standard bioequivalence acceptance range of 80–125%. The immunogenicity profiles of the high-concentration and reference formulations of adalimumab-adbm were highly similar. Treatment with both the high-concentration and reference formulations was well tolerated and safe in these trial participants, and most AEs resolved within hours and were mild in nature. While both doses were considered to have a satisfactory safety and tolerability profile, the high-concentration formulation showed an improved local tolerability relative to the reference formulation. These findings are consistent with evidence on improved outcomes in injection site reactions with lower injection volumes. While this trial did not evaluate outcomes in the target patient populations intended for adalimumab-adbm use, the tolerability profile of high-concentration adalimumab-adbm may improve treatment adherence and patient experiences with self-injection by mitigating the incidence of local reactions.
4.1. Limitations
There are several potential limitations associated with this study primarily relating to the limited sample size and the fact that only healthy participants aged 18–55 years were included. Although the smaller sample size of healthy volunteers was appropriate for the primary objective of PK comparison, it also provides a limited assessment of the secondary objectives of immunogenicity, safety, and tolerability. Additionally, elderly and pediatric patients, patients with and without comorbid conditions and who may be taking a wide range of comedications may use adalimumab-adbm in clinical practice. However, data from the Phase III studies (VOLTAIRE-RA, VOLTAIRE-CD, VOLTAIRE-PsO, and VOLTAIRE-X, which included elderly patients) demonstrated that adalimumab-adbm reference formulation has similar efficacy, safety, PK, and immunogenicity to the adalimumab reference product across several disease indications [3–5].
5. Conclusions
Overall, the results of this Phase I clinical trial provide strong evidence for the bioequivalence of adalimumab-adbm 40 mg/0.4 mL and 40 mg/0.8 mL formulations. Based on these results, no clinically relevant differences in efficacy or safety between the high-concentration and reference adalimumab-adbm formulations would be expected in a clinical setting.
Declarations of interest
V Moschetti, S Buschke, K Hohl, and J Bertulis are employees of Boehringer Ingelheim Pharma GmbH & Co. KG. D McCabe is an employee of Boehringer Ingelheim Pharmaceuticals Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Ethics statement
The protocol was approved by the applicable independent ethics committee or institutional review board at each participating site, and the study was performed in accordance with Good Clinical Practice and the Declaration of Helsinki. All study participants provided written informed consent prior to participation in any study procedures.
Author contributions
Study conception and design: S Buschke, K Hohl, D McCabe, V Moschetti; analysis and interpretation of data: J Bertulis, S Buschke, K Hohl, V Moschetti; drafting the manuscript or revising it critically for intellectual content: S Buschke, K Hohl, V Moschetti; clinical conduct and medical oversight: V Moschetti, D McCabe; and the final approval of the version to be published: all authors. All authors provided final approval of the manuscript and agree to be accountable for all aspects of the work.
Acknowledgments
This study was supported and funded by Boehringer Ingelheim. The authors met criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors did not receive payment related to the development of this manuscript. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations. Andy Shepherd, PhD, and Jia Gao, PharmD, of Elevate Scientific Solutions LLC, provided medical writing, editorial support, and formatting support, which were contracted and funded by Boehringer Ingelheim Pharmaceuticals, Inc.
Data availability statement
To ensure independent interpretation of clinical study results and enable authors to fulfill their role and obligations under the ICMJE criteria, Boehringer Ingelheim grants all external authors access to relevant clinical study data. In adherence with the Boehringer Ingelheim Policy on Transparency and Publication of Clinical Study Data, scientific and medical researchers can request access to clinical study data after publication of the primary manuscript in a peer-reviewed journal, providing regulatory activities are complete and other criteria are met. Researchers should use the https://vivli.org/link to request access to study data and visit: https://www.mystudywindow.com/msw/datasharing for further information.
Additional information
Funding
References
- Boehringer Ingelheim Pharmaceuticals I. Cyltezo; [package insert] 2023 [cited 2023 Sep 18]. Available from: https://content.boehringer-ingelheim.com/DAM/9d0e3bae-9a17-4979-91bc-af1e011eeb8a/cyltezo-us-pi.pdf
- U.S. FDA. FDA approves cyltezo, the first interchangeable biosimilar to humira 2021. [cited 2023 Sep 19]. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-cyltezo-first-interchangeable-biosimilar-humira
- Cohen SB, Alonso-Ruiz A, Klimiuk PA, et al. Similar efficacy, safety and immunogenicity of adalimumab biosimilar BI 695501 and Humira reference product in patients with moderately to severely active rheumatoid arthritis: results from the phase III randomised VOLTAIRE-RA equivalence study. Ann Rheum Dis. 2018 Jun;77(6):914–921. doi: 10.1136/annrheumdis-2017-212245
- Hanauer S, Liedert B, Balser S, et al. Safety and efficacy of BI 695501 versus adalimumab reference product in patients with advanced Crohn’s disease (VOLTAIRE-CD): a multicentre, randomised, double-blind, phase 3 trial. Lancet Gastroenterol Hepatol. 2021 Oct;6(10):816–825. doi: 10.1016/S2468-1253(21)00252-1
- Menter A, Arenberger P, Balser S, et al. Similar efficacy, safety, and immunogenicity of the biosimilar BI 695501 and adalimumab reference product in patients with moderate-to-severe chronic plaque psoriasis: results from the randomized phase III VOLTAIRE-PSO study. Expert Opin Biol Ther. 2021 Jan;21(1):87–96. doi: 10.1080/14712598.2021.1851362
- Allegretti JR, Brady JH, Wicker A, et al. Relevance of adalimumab product attributes to patient experience in the biosimilar era: a narrative review. Adv Ther. 2024 Mar 11;41(5):1775–1794. doi: 10.1007/s12325-024-02818-9
- Dias C, Abosaleem B, Crispino C, et al. Tolerability of high-volume subcutaneous injections of a viscous placebo buffer: a randomized, crossover study in healthy subjects. AAPS PharmSciTech. 2015 Oct;16(5):1101–1107. doi: 10.1208/s12249-015-0288-y
- Zijlstra E, Jahnke J, Fischer A, et al. Impact of injection speed, volume, and site on pain sensation. J Diabetes Sci Technol. 2018 Jan;12(1):163–168. doi: 10.1177/1932296817735121