
ABSTRACT
Purpose
Evaluate the type and quantity of quality information (i.e. Chemistry, Manufacturing, and Control) requested by the US FDA and EMA in queries pertaining to biosimilar applications.
Methods
Numbers/types of queries received following regulatory submissions (FDA/EMA, n = 7/n = 5) for seven biosimilars (PF-filgrastim [Nivestym], PF-rituximab [Ruxience®], PF-trastuzumab [Trazimera®], PF-bevacizumab [Zirabev®], PF-pegfilgrastim [Nyvepria®], PF-adalimumab [Abrilada™/Amsparity®], PF-infliximab [Ixifi]) from a single product portfolio were analyzed considering published regulatory authority (RA) guidance and in relation to sections/subsections of Module 3: Quality from the Common Technical Document regulatory dossier and topics based on keyword assignment.
Results
Queries were most frequently assigned (FDA/EMA %, range) to Drug Substance Manufacture (subsection 3.2.S.2; 21–35%/13–50%), Control of Drug Substance (3.2.S.4; 3–11%/5–17%), Drug Product Pharmaceutical Development (3.2.P.2; 1–12%/1–15%) and Manufacture (3.2.P.3; 17–41%/2–13%), and Analytical Similarity (3.2.R; 4–21%/4–20%). The proportion of Drug Substance and Drug Product queries was significantly different between RAs (n1 = 952, n2 = 468, p-value <0.001; two-sample proportion z-test). Topic assignments included: Control (12–27%/12–28%), Manufacturing (56–72%/34–66%), Stability (1–12%/2–24%), Biosimilarity (5–16%/5–25%), and Container Closure (0–3%/0–9%).
Conclusion
The focus of both RAs on topics related to manufacturing and controls is valuable in understanding expectations for scientific and technical content related to gaining biosimilar approval.
1. Introduction
A biosimilar is a biologic drug, which is highly similar to the authorized originator biologic or reference product (used hereafter) in terms of structure and biological activity and is expected to have the same treatment benefits and risks [Citation1]. A biosimilar is not considered a generic of a biologic molecule due to natural protein variability and the typically complex manufacturing process required for biologic medicines [Citation2].
In both the United States (US) and European Union (EU), the biosimilar RA marketing application approval follows a rigorous regulatory review process, which includes the same standards for quality, safety, and efficacy that apply to all biologic medicines [Citation3,Citation4]. Biosimilar drugs are evaluated and reviewed following established scientific guidelines requiring the manufacturer to conduct a comprehensive comparative assessment including analytical, quality, and tailored clinical studies with the reference product [Citation3–8]. The US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidance documents for marketing applications outline regulatory authority (RA) policy, and include recommendations for application holders to follow in order to facilitate authoring, reviews, and approvals [Citation3,Citation4].
Key to the approval of any biosimilar application, data must demonstrate that the biosimilar has a highly similar quality profile when compared with the reference product. The FDA and EMA guidance documents for the development of biosimilars additionally recommend applicants include the following content related to the comparative analytical assessment: expression system, manufacturing process, physiochemical properties, functional activities, biological activity, target binding, impurities, reference product, reference standards, finished product strength and stability [Citation5,Citation8].
Compared with reference biologics, biosimilars achieve regulatory approval based on the ‘totality of the evidence’ derived from a relatively truncated comparative non-clinical and clinical development pathway, while the data to support the quality submission is greatly expanded to include comparative analytical assessment () [Citation9]. Nevertheless, compared with the biologic reference product, the regulatory pathway is no less stringent overall for a biosimilar [Citation10,Citation11]. Due to the relatively condensed clinical development pathway compared with that required for the reference product, biosimilars can offer potential cost savings, and their availability can expand patients’ access to effective biologic therapy and bring considerable healthcare and patient benefits [Citation12–16].
Figure 1. Totality of the evidence that supports biosimilarity. Reproduced from: Stebbing J, Mainwaring PN, Curigliano G, et al., Understanding the Role of Comparative Clinical Studies in the Development of Oncology Biosimilars, J Clin Oncol, 2020, 38(10):1070–1080, https://ascopubs.Org/doi/10.1200/JCO.19.02953. © 2020 by American Society of Clinical Oncology. © 2020, Wolters Kluwer Health [Citation9]. Iceberg image copyright © Adike/Shutterstock.com.
![Figure 1. Totality of the evidence that supports biosimilarity. Reproduced from: Stebbing J, Mainwaring PN, Curigliano G, et al., Understanding the Role of Comparative Clinical Studies in the Development of Oncology Biosimilars, J Clin Oncol, 2020, 38(10):1070–1080, https://ascopubs.Org/doi/10.1200/JCO.19.02953. © 2020 by American Society of Clinical Oncology. © 2020, Wolters Kluwer Health [Citation9]. Iceberg image copyright © Adike/Shutterstock.com.](/cms/asset/78a4a825-199e-49f2-b4d0-b9f94b9b555d/iebt_a_2376197_f0001_oc.jpg)
Although guidelines issued by the FDA and EMA attempt to cover most scenarios related to drug manufacturing and testing, it is unrealistic to expect them to address all circumstances. Therefore, individual products must undergo an intensive review on their own merit to help guide the RA on their decision regarding authorization. The information to be provided for RA review under Module 3 (M3): Quality Section of the International Conference on Harmonisation (ICH) guideline of the Common Technical Document (CTD) [Citation17]. In addition to standard manufacturing, control, and development information, the biosimilar applicant is required to generate and demonstrate structural and functional similarity with the reference product.
Both the FDA and EMA provide precise requirements in terms of establishing the comparability of the biosimilar through considerations such as the choice of reference product (in terms of the source market and batch number), analytical methods used, and attributes [Citation5,Citation8,Citation18]. Applicants typically provide the results of this comparability analysis as an additional subsection within Module 3, resulting in the larger submission and content with respect to a reference biologic for RA review.
Information required from applicants to support biosimilar marketing applications by RAs varies. A companion analysis for the overall regulatory assessment of this portfolio of biosimilar approvals was previously published [Citation19]. However, published literature is scarce regarding the type and quantity of CMC information requested by different RAs in response to biosimilar marketing applications. An assessment of the questions on the quality module of 22 marketing authorization applications, for biosimilars submitted to the EMA up to October 2015, found that most CTD deficiencies were identified in the Drug Substance (3.2.S) and Drug Product information (3.2.P) [Citation20]. Here, we report the results of a focused analysis of the M3: Quality section of the CTD or CMC-related queries received from the US (FDA) and EU (EMA) over a 4-year period (2017–2020) of 12 biosimilar marketing applications. This was done to understand how these applications were assessed by the RAs and identify trends in the areas of review specifically for the quality sections of a biosimilar application.
2. Materials and methods
Between 2017 and 2020, 12 biosimilar applications for market authorization were submitted to the FDA (n = 7) and EMA (n = 5), comprising seven different biosimilar drug products () [Citation19]. Contemporaneous and simultaneous submissions of these applications to the FDA and EMA allowed for analysis and comparison of the RA approaches to the review. To ensure a consistent data set for analysis, the earlier biosimilar EMA marketing authorization for filgrastim (PF-filgrastim; Nivestim®), and the EMA and FDA applications for marketing authorizations for epoetin (PF-epoetin: Retacrit®) biosimilars, which were submitted prior to 2017, were excluded. This was done to retain focus on recent trends observed during biosimilar reviews from these RAs. Queries received during the review of all applications submitted to the FDA and EMA were collated, and each was assigned as outlined in retrospectively by the authors. Analysis of all queries for the RA reviews was analyzed, including those issued via assessment report, information requests, and complete responses.
Figure 2. Method of retrospective analysis of queries received from the FDA and EMA for each biosimilar. Abbreviations: CTD, Common Technical Document; DS, Drug Substance; DP, Drug Product; EMA, European Medicines Agency; FDA, US Food and Drug Administration; RA, Regulatory Authority; ICH, International Conference on Harmonisation; M3, Module 3; RS, Reference Standards; US, United States.
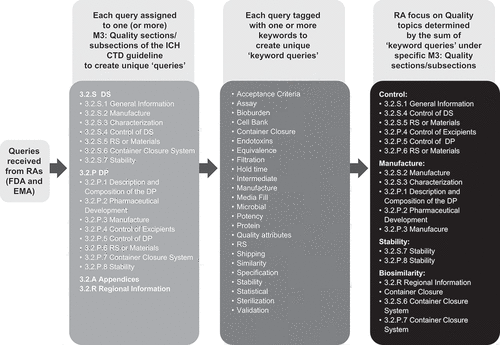
Table 1. Biosimilars and approval dates. Adapted from Analysis of the Regulatory Science Applied to a Single Portfolio of Eight Biosimilar Product Approvals by Four Key Regulatory Authorities, Pharmaceuticals (Basel), 2021, 14(4):306, © 2021 by Ingram B, Lumsden S, Radosavljevic A, et al., and licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/) [Citation19].
2.1. Total CMC queries and M3: Quality CTD section/subsection assignment
Queries received for each biosimilar were reviewed and assigned to the relevant Drug Substance (3.2.S) and Drug Product (3.2.P) subsections of the M3: Quality section of the CTD () [Citation17]. Queries relating to non-commitment Appendix Information (Section 3.2.A) and Regional Information (3.2.R), where additional data for comprehensive analytical comparability is presented, were assigned as appropriate to these sections without further subsection classification. Where a query was relevant to multiple subsections, for instance to update stability for Drug Substance as well as Drug Product, the query was counted as one relating to both Drug Substance Stability (3.2.S.7) and Drug Product Stability (3.2.P.8) subsections to generate unique ‘queries’ for each. The relative frequency of queries under each section (3.2.A/3.2.R) and subsection (3.2.S/3.2.P) as a percentage of the overall number of queries received from the FDA and EMA were determined separately for each biosimilar.
2.2. CMC quality keywords
Based on a review of all the queries received from the RAs, the authors identified a set of keywords that they considered covered the most prevalent quality topics raised by the RAs in the queries received (). Use of keyword assignment was devised to allow more detailed insight into the specific areas of interest of the RAs in relation to how each biosimilar application was reviewed.
2.3. Key words
Acceptance Criteria; Assay; Bioburden; Cell Bank; Container Closure; Endotoxins; Equivalence; Filtration; Hold time; Intermediate; Manufacture; Media Fill; Microbial; Potency; Protein; Quality Attributes; Reference Standard; Shipping; Similarity; Specification; Stability; Statistical; Sterilization; and Validation.
Following assignment of the queries to the sections/subsections of M3: Quality section of the CTD (), each query was tagged with one or more keywords based on the occurrence of terms related to each keyword heading, to create unique ‘keyword queries.’ For instance, all queries containing terms such as ‘manufacturing,’ ‘manufactured’, or ‘manufacturer(s)’ were tagged with the keyword ‘Manufacture.’ Keyword assignments were verified by the second author. For any keyword assignments where there was uncertainty or disagreement, this was resolved by the careful review of the query in its entirety. This was done to ensure that the true meaning of the query was fully understood. Each query could contain terms related to a number of different topics, as such, the query could be tagged with multiple keywords. The frequency of occurrence of each keyword query as a percentage of the overall keyword queries originating from the queries received from the FDA and EMA was determined separately for each biosimilar application.
2.4. CMC M3: quality section topics
The authors considered that the keyword queries falling under the following sections/subsections of the M3: Quality Section of the CTD would provide an indication of the RA focus on the primary Quality topics of Control, Manufacture, Stability, Biosimilarity, and Container Closure ():
Control (Sections 3.2.S.1; 3.2.S.4; 3.2.S.5; 3.2.P.4; 3.2.P.5; 3.2.P.6)
Manufacture (Sections 3.2.S.2; 3.2.S.3; 3.2.P.1; 3.2.P.2; 3.2.P.3)
Stability (Sections 3.2.S.7; 3.2.P.8)
Biosimilarity (Section 3.2.R)
Container Closure (Sections 3.2.S.6; 3.2.P.7)
The frequency of each keyword query as a percentage of all keyword queries under each Quality topic was determined separately for each biosimilar and RA (FDA and EMA).
2.5. CMC quality keywords
The compiled data for the query analysis for each subsection was then compiled into the related M3 Nodes of Drug Substance (3.2.S), Drug Product (3.2.P) non-commitment Appendix information (3.2.A), and Regional Information containing analytical similarity data (3.2.R) where additional data for comprehensive analytical comparability are presented.
2.6. Statistical analysis
Statistical analysis via two-proportion z-test was performed on the basis of M3: Quality section results as they related to each RA.
3. Results
3.1. CMC queries and M3 section/subsection assignment
The numbers of CMC-related queries received from the FDA and EMA for each of the 12 biosimilar applications are shown in , respectively. Of the applications for the five biosimilars assessed by both RAs, there was a relatively higher number of queries from the FDA than from the EMA for each of the four monoclonal antibody biosimilars, PF-trastuzumab, PF-bevacizumab, PF-rituximab, and PF-adalimumab.
Figure 3. CMC queries from the FDA and M3 Quality Sections/Subsections assignment. The graphs are offset in both directions for clarity. Abbreviations: CMC, Chemistry, Manufacturing and Controls; DP, Drug Product; DS, Drug Substance; FDA, US Food and Drug Administration; M3, Module 3; RS, Reference Standards; US, United States.
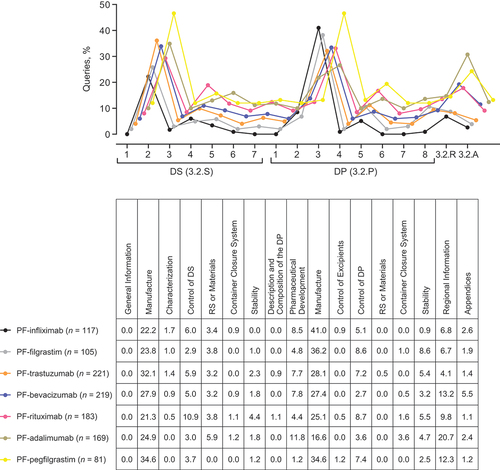
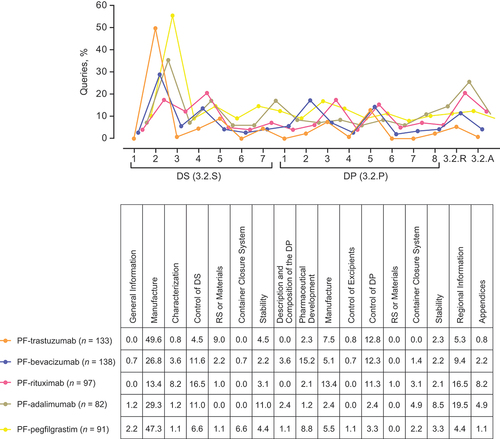
Figure 4. CMC queries from the EMA and M3 Quality Sections/Subsections assignment. The graphs are offset in both directions for clarity. Abbreviations: CMC, Chemistry, Manufacturing and Controls; EMA, European Medicines Agency; M3, Module 3.
Based on the M3: Quality section/subsection assignment, the focus of the FDA review was broadly similar across the seven biosimilars assessed, with the percentage of queries (range) most frequently associated with Drug Substance Manufacture (3.2.S.2; 21–35%), Drug Product Pharmaceutical Development (3.2.P.2; 1–12%), and Manufacture (3.2.P.3; 17–41%), and Regional Information containing analytical similarity data (3.2.R; 4–21%) ().
Drug Substance Manufacture (3.2.S.2; 13–50%), and Control of Drug Substance (3.2.S.4; 5–17%), and Drug Product Pharmaceutical Development (3.2.P.2; 1–15%) and Manufacture (3.2.P.3; 2–13%), and Regional Information containing analytical similarity data (3.2.R; 4–20%) were the subsections most frequently queried across the 5 applications assessed by the EMA ().
The compiled data for the query analysis for each subsection was summed for the related M3 Nodes Drug Substance (3.2.S) (FDA: 32–45%; EMA: 42–69%), Drug Product (3.2.P) (FDA: 40–59%; EMA: 22–41%), Regional Information containing analytical similarity data (3.2.R) (FDA: 4–21%; EMA: 4–20%) and non-commitment Appendix information (3.2.A) (FDA: 1–6%; EMA: 1–8%). This was done to evaluate the distribution of CMC-related queries. The total percentage of Drug Substance (FDA 39%, EMA 56%) and Drug Product (FDA 48%, EMA 30%) sections queries were higher than the total percentage of Regional Information containing analytical similarity data (FDA 11%, EMA 10%) and Appendix (FDA 3%, EMA 3%) queries.
3.2. CMC keywords
The keyword assessments originating from queries received from the FDA and EMA for each biosimilar are provided in Supplementary Table S1 and Supplementary Table S2, respectively. Overall, 3,543 CMC focused keywords were identified based on the queries received from the FDA (n = 2,668) and EMA (n = 875) for the 7 and 5 applications assessed, respectively.
Among the most frequently identified keyword queries were, ‘Manufacture’ (FDA 11%, EMA 10%), ‘Validation’ (FDA 8%, EMA 15%), ‘Filtration’ (FDA 9%, EMA 4%), ‘Stability’ (FDA 7%, EMA 9%), ‘Endotoxins’ (FDA 6%, EMA 1%), ‘Sterilization’ (FDA 6%, EMA 4%), ‘Assay’ (FDA 6%, EMA 9%), ‘Acceptance criteria’ (FDA 6%, EMA 11%), ‘Similarity’ (FDA 2%, EMA 4%), and ‘Specification’ (FDA 5%, EMA 7%). Overall keyword assessment found similar assessments when comparing the FDA and EMA, again focusing on Manufacturing as noted in Section/Subsection analysis.
3.3. M3: Quality section topics
The frequencies of occurrence of keyword entries under the Quality topics of Control, Manufacturing, Stability, Biosimilarity, and Container Closure for the FDA and EMA by biosimilar are shown in , respectively. For both RAs, the majority of the keyword entries fall under the sections/subsections of M3: Quality section considered to be related to Manufacture (FDA: 56–72%; EMA: 34–66%).
Figure 5. Frequency of keyword entries under Quality topics originating from the queries received from the FDA. Abbreviations: FDA, US Food and Drug Administration; US, United States.
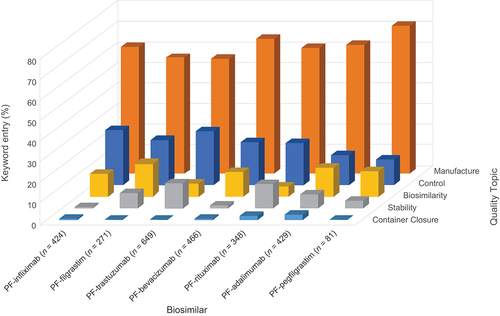
Figure 6. Frequency of keyword entries under Quality topics originating from the queries received from the EMA. Abbreviations: EMA, European Medicines Agency.
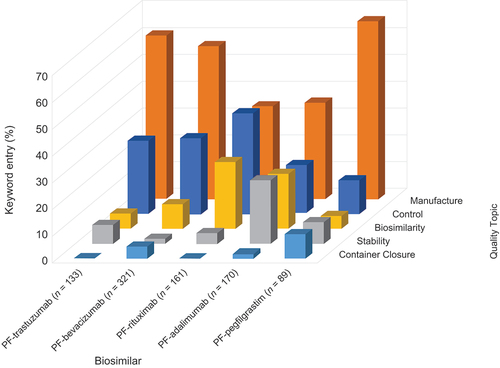
Overall, the data indicated the major focus of RA review for both the FDA and EMA is related to the manufacturing and control of both the Drug Substance and Drug Product as compared with the comprehensive analytical comparability or non-commitment Appendix information.
3.4. Statistical analysis
The proportion of Regional Information containing analytical similarity data (3.2.R) between the FDA (10.6%) and EMA (10.4%) was not significantly different based on two-sample proportion z-test (n1 = 1095, n2 = 541, p-value = 0.88). The proportion of Drug Substance (3.2.S: FDA 39.4%; EMA 56.4%) and Drug Product (3.2.P: FDA 47.6%; EMA 30.1%) queries were significantly different between the two RAs (n1 = 952, n2 = 468, p-value <0.001).
4. Discussion
Pfizer applied for and received approval for seven biosimilar applications from the FDA and for five biosimilar applications from the EMA within the time range this article is focused on. During the RA review process for all of these 12 applications, a number of queries were received and satisfactorily addressed, resulting in the successful approval of each application. The details of these queries, with a focus on the CMC sections of the applications, have been analyzed to assess the focus of the RAs during the review process. Initial findings from the analysis for all major classifications (including CMC, non-clinical, clinical, and regulatory) were previously discussed in a companion publication [Citation19], which describes the overall focus of RA review in support of the totality of regulatory analysis of biosimilar approvals.
During the preparation of the Biosimilar License Applications for submission to the FDA for evaluation and review, the extent and level of detail included in the M3: Quality sections/subsections were consistent for both the Drug Substance and Drug Product for each of the seven biosimilars reviewed here. Additionally, dossier content closely followed published guidelines available at the time of submission.
The findings from this analysis show that the most intense scrutiny of these major RAs was on the manufacturing process associated with both the Drug Substance (3.2.S) and the Drug Product (3.2.P) CTD sections. These findings are in line with those from an earlier analysis by Cilia and coauthors of biosimilar marketing applications submitted to the EMA. The most prevalent major deficiencies were identified in the Drug Product (3.2.P) and most prevalent minor findings identified in Drug Substance (3.2.S) CTD sections [Citation20]. In the current analysis, queries received from the RAs were also mainly focused on the Drug Substance (3.2.S) and the Drug Product (3.2.P) CTD sections to a greater extent than on the Regional Information (3.2.R) despite this latter section containing analytical similarity data. However, the proportion of queries received overall for DS and DP was significantly different between the FDA and EMA (p-value <0.001). This provides a valuable guide for sponsors undertaking CTD dossier development in planning for regulatory interactions.
Relevant findings from the M3: Quality Section Topic analysis found that keyword queries received from the FDA reviews were assigned to ‘Manufacturing’ almost threefold more frequently overall compared with ‘Control.’ Assignments under the remaining headings of ‘Stability,’ ‘Biosimilarity,’ and ‘Container Closure’ were fewer overall.
A similar pattern was seen in the assignment of the keyword queries received from the EMA where keywords assigned to ‘Manufacturing’ occurred around twice as often as ‘Control,’ except for PF-rituximab, where the proportions were comparable.
For approval of any biosimilar application, data must be presented that demonstrate that the biosimilar has a highly similar quality profile when compared with the reference product. The analysis performed here suggests that provided information submitted is in line with the RA (both FDA and EMA) guidelines, as only a limited number of review queries are raised relating to Regional Information containing analytical similarity data (3.2.R).
In light of the findings from this overall analysis, it should not be deemed that less regulatory attention or information be included in M3: Quality sections and subsections not regularly mentioned. Instead, if guidelines are closely followed, the RAs have shown a tendency to acknowledge the provided information as sufficient.
5. Conclusions
Analysis of CMC focused reviews performed by the FDA and EMA of biosimilars described in this article provides insight into trends that have recently been followed by these RAs. The distribution of CMC-related queries for this biosimilar portfolio may individually represent unique features associated with each molecule; however, ultimately, each biosimilar was independently reviewed in accordance with the RA regulations, resulting in relatable trends that can be used to identify areas of focus.
Overall, this analysis of CMC information reviewed by the FDA and EMA is consistent with the fundamental importance of data to support the manufacturing and testing of biosimilars and further to demonstrate similarity, as the basis for extrapolation of the biosimilar to other indications of the reference biologic. An appropriate regulatory strategy for a biosimilar marketing application should utilize both scientific advice and biosimilar guidance.
Based on the breadth of information associated with seven FDA and five EMA biosimilar applications, statistical data were generated and used to support the conclusions that RA review is independent of the quality section. In general, for the Pfizer portfolio, DS and DP manufacturing was the focus of the FDA and EMA review with greater emphasis on DS manufacturing by EMA and a balanced focus on DS and DP manufacturing by the FDA. Additionally, based on this analysis, fewer queries were received for 3.2.R Analytical Similarity than for 3.2.S or 3.2.P indicating the sponsors alignment with the published biosimilar guidance and the RAs expectations. Finally, the trend in the total number of queries received during marketing application review decreased for this portfolio over the multi-year submission period based on iterative learning from RA interactions and queries.
Although other Marketing Authorization Holders may have a slightly different experience to that outlined here, the large number of biosimilars submitted and reviewed lends credibility to the conclusions reached from this analysis. Furthermore, this query analysis may lend some valuable insight into the biosimilar development and marketing application approval process in leveraging the importance of both the FDA and EMA reviews for both DS and DP manufacturing and control with considerable influence gained from agency interactions and guidance related to analytical similarity.
6. Limitations
This study was designed to evaluate the industry perspective on RA quality assessment or review of biosimilar applications. The results are limited in scope to the two major RAs (FDA and EMA) and associated guidance documents; however, further analysis of other major global RAs and of non-biosimilar applications could be considered important for this analysis. The keyword assessment was designed by the authors to enable efficient data collection and analysis and could be considered limited in scope. Molecular complexity in relation to the queries issued by the RAs for the biosimilar applications was not assessed in this study. Furthermore, comparison of public domain information for Pfizer-developed biosimilars and other biosimilar molecules was not within the scope of this analysis and could have limited interpretation of results.
Declaration of interest
HR Hufnagel is an employee of and holds stock or options in Pfizer. SD Tennyson was an employee of and held stock or options in Pfizer at the time of the development of the manuscript. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
A reviewer on this manuscript has disclosed research, speaking and/or consulting support from Eli Lilly and Company, GlaxoSmithKline/Stiefel, AbbVie, Janssen, Alovtech, vTv Therapeutics, Bristol-Myers Squibb, Samsung, Pfizer, Boehringer Ingelheim, Amgen, Dermavant, Arcutis, Novartis, Novan, UCB, Helsinn, Sun Pharma, Almirall, Galderma, Leo Pharma, Mylan, Celgene, Ortho Dermatology, Menlo, Merck & Co, Qurient, Forte, Arena, Biocon, Accordant, Argenx, Sanofi, Regeneron, the National Biological Corporation, Caremark, Teladoc, BMS, Ono, Micreos, Eurofins, Informa, UpToDate, and the National Psoriasis Foundation. They hold stock in Causa Research and Sensal Health. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Author contributions
HR Hufnagel and SD Tennyson have contributed equally to the study design development, data extraction, data analysis, and/or interpretation. Both authors critically reviewed and revised the manuscript and approved the final version for publication.
Biosims_CMC_Publication_EOBT_Revised_Supp_Mat_25Jun24.docx
Download MS Word (22.9 KB)Acknowledgments
The authors wish to thank Jeffrey Dicker for research assistance with extraction and collation of the query responses and Peili Wang for performing the statistical analysis. Medical writing support was provided by Iain McDonald, PhD, of Envision Pharma Group and was funded by Pfizer.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14712598.2024.2376197
Additional information
Funding
References
- US Food and Drug Administration (FDA). Patient materials 2020. [cited 2020 Oct 7]. Available from: https://www.fda.gov/drugs/biosimilars/patient-materials
- Morrow T, Felcone LH. Defining the difference: what makes biologics unique. Biotechnol Healthc. 2004;1(4):24–29.
- European Medicines Agency. Guideline on similar biological medicinal products. London: EMA; 2014 [cited 2014 Oct 23]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf
- US Food and Drug Administration (FDA). Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. Silver Spring (MD): FDA; 2015. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
- European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). 2014 [cited 2014 May 22]. Available from: https://www.ema.europa.eu/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf
- US Food and Drug Administration (FDA). Quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product; guidance for industry 2015.[cited 2020 Feb27]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/quality-considerations-demonstrating-biosimilarity-therapeutic-protein-product-reference-product
- US Food and Drug Administration (FDA). Clinical pharmacology data to support a demonstration of biosimilarity to a reference product 2016. [cited 2020 May 6]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-pharmacology-data-support-demonstration-biosimilarity-reference-product
- US Food and Drug Administration (FDA). Development of Therapeutic protein biosimilars: Comparative analytical assessment and other quality-related considerations 2019. [cited 2018 Mar 2]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analytical-assessment-and-other-quality
- Stebbing J, Mainwaring PN, Curigliano G, et al. Understanding the role of comparative clinical studies in the development of oncology biosimilars. J Clin Oncol. 2020;38(10):1070–1080. doi: 10.1200/JCO.19.02953
- US Food and Drug Administration (FDA). Biosimilar development, review, and approval. 2017 [cited 2017 Oct 20]. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-development-review-and-approval
- European Medicines Agency. Biosimilar medicines: marketing authorisation. Available from: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/biosimilar-medicines-marketing-authorisation
- Chanchlani N, Mortier K, Williams LJ, et al. Use of infliximab biosimilar versus originator in a pediatric United Kingdom inflammatory bowel disease induction cohort. J Pediatr Gastroenterol Nutr. 2018;67(4):513–519. doi: 10.1097/MPG.0000000000002011
- Gulacsi L, Brodszky V, Baji P, et al. The rituximab biosimilar CT-P10 in rheumatology and cancer: a budget impact analysis in 28 European countries. Adv Ther. 2017;34(5):1128–1144. doi: 10.1007/s12325-017-0522-y
- Jha A, Upton A, Dunlop WC, et al. The Budget impact of biosimilar infliximab (Remsima®) for the treatment of autoimmune diseases in five European countries. Adv Ther. 2015;32(8):742–756. doi: 10.1007/s12325-015-0233-1
- Kim J, Ha D, Song I, et al. Estimation of cost savings between 2011 and 2014 attributed to infliximab biosimilar in the South Korean healthcare market: real-world evidence using a nationwide database. Int J Rheum Dis. 2018;21(6):1227–1236. doi: 10.1111/1756-185X.13295
- Tee M, Tee CA. Pms19 - a budget impact analysis of introducing a forced treatment pathway using the lowest priced anti-tumor necrosis factor agent for rheumatoid arthritis and ankylosing spondylitis in the philippines. Value Health. 2018;21:S290–S291. doi: 10.1016/j.jval.2018.09.1733
- ICH. ICH Guideline. The common technical document for the registration of pharmaceuticals for human use: Quality – M4Q(R1). 2002 [cited 2002 Sep 12]. Available from: https://database.ich.org/sites/default/files/M4Q_R1_Guideline.pdf
- Arato T. Japanese regulation of biosimilar products: past experience and current challenges. Br J Clin Pharmacol. 2016;82(1):30–40. doi: 10.1111/bcp.12931
- Ingram B, Lumsden RS, Radosavljevic A, et al. Analysis of the regulatory science applied to a single portfolio of eight biosimilar product approvals by four key regulatory authorities. Pharmaceuticals (Basel). 2021;14(4):306. doi: 10.3390/ph14040306
- Cilia M, Ruiz S, Richardson P, et al. Quality issues identified during the evaluation of biosimilars by the European Medicines Agency’s committee for medicinal products for human use. AAPS Pharm Sci Tech. 2018;19(2):489–511. doi: 10.1208/s12249-017-0892-0