
1. Introduction
Until very recently, the management of asthma has centered around a handful of bronchodilators and corticosteroids developed empirically decades ago. The lack of therapeutic innovation is all the more surprising given the pressing clinical need: over 3000 people die of asthma a year in the United States alone, and ~50% of patients report exacerbations necessitating increased treatment in the last year.
That is all set to change. Respiratory medicine is entering a new era of biological therapies—treatments that selectively target specific inflammatory mediators and cellular pathways critical in disease pathophysiology. These treatments have already revolutionized patient care in rheumatology, gastroenterology, dermatology, and oncology. Almost all current and emerging biologic treatments for asthma target ‘type 2’ inflammation and require subcutaneous or intravenous administration. However, several pharmaceutical companies have recently developed inhibitors of prostaglandin D2 (PGD2) signaling offering an oral alternative capable of suppressing the type 2 inflammatory cascade. This editorial focuses on the rationale and efficacy of blocking PGD2 signaling in asthma.
2. Asthma pathophysiology
Most asthma is characterized by ‘type 2’ inflammation, so-called because it is thought to be mediated by type 2 helper (Th2) cells and type 2 innate lymphoid cells (ILC2s) [Citation1]. Th2 cells secrete the type 2 cytokines interleukin (IL)-4, IL-5 and IL-13, which in turn bring about the archetypal features of asthma: IgE production, mucus hypersecretion, airway hyperreactivity, and eosinophilia (). Based on this understanding of asthma immunology, monoclonal antibodies directed against these mediators have been developed as novel therapies, specifically anti-IgE (omalizumab), anti-IL-5 (mepolizumab, reslizumab), anti-IL-5Rα (benralizumab), anti-IL-13 (lebrikizumab, tralokinumab), and anti-IL-4Rα (dupilumab).
Figure 1. Asthma immunology.
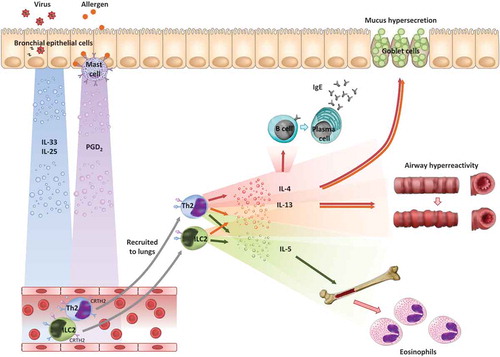
Meanwhile, attention has turned to describing the initiating events in the cascade of type 2 inflammation. Several candidates have subsequently been identified with the potential to stimulate Th2 cells and ILC2s to release type 2 cytokines, at least in vitro. These include IL-25, IL-33, thymic stromal lymphopoietin and PGD2.
3. PGD2 biology
PGD2 is a lipid inflammatory mediator produced by the sequential action of cyclooxygenases (particularly COX-2) and PGD2 synthases (PGDS)—either hematopoietic PGDS in circulating hematopoietic-derived cells, or lipocalin PGDS in the brain, heart, and adipose tissue (). Mast cells are traditionally thought to be the principal source of PGD2 as they release vast quantities in response to IgE binding [Citation2], but Th2 cells [Citation3] and macrophages [Citation4] may also produce biologically significant amounts (eosinophils and basophils can also produce PGD2, but in concentrations roughly 1000 times more dilute than mast cells).
Figure 2. Prostaglandin D2 biology.
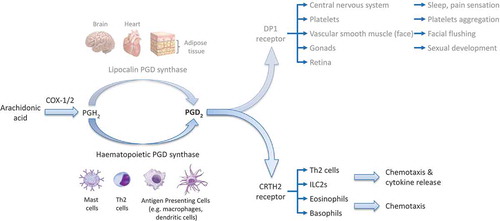
PGD2 binds the D prostanoid (DP) 1 and Chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) receptors. DP1 is found on a variety of cell types and has broadly anti-inflammatory effects. CRTH2, by contrast, is expressed selectively on immune cells, specifically eosinophils, basophils, Th2 cells, and ILC2s. Indeed, although it has long been known that PGD2 has bronchoconstricting effects when inhaled, it was only after the relatively recent discovery of the CRTH2 receptor that a pro-inflammatory role for PGD2 in allergic conditions has been described. Thus, following CRTH2 receptor binding in vitro, PGD2 triggers chemotaxis and, in the case of ILC2s and Th2 cells, the release of the type 2 cytokines IL-4, IL-5, and IL-13 [5,6]. CRTH2 binding is therefore hypothesized to be important in diseases characterized by type 2 inflammation, such as asthma, atopic dermatitis, and allergic rhinitis.
There is good evidence that the PGD2-CRTH2 pathway is upregulated in asthma. Both COX-2 [Citation7] and hematopoietic PGDS [Citation8] are more highly expressed in asthmatic lungs, suggesting a greater capacity for producing PGD2, and there are greater numbers of CRTH2+ cells [Citation9], increasing sensitivity to PGD2. These observations are more pronounced in patients with poor asthma control [Citation8].
4. The case for CRTH2 antagonists
Given the pathogenic roles of IL-4, IL-5, and IL-13 in asthma, a broader treatment that suppresses all three mediators is likely to be more effective than monoclonal antibodies targeting each individually. Of the various candidates that might trigger type 2 inflammation, PGD2-CRTH2 activation is particularly attractive as there is evidence to support it being a dominant pathway: whilst IL-25 and IL-33 can also induce type 2 cytokine release by ILC2s in vitro, their stimulatory effect is inhibited by a selective CRTH2 antagonist [Citation6].
On a practical level, selective CRTH2 antagonists are small molecules (rather than monoclonal antibodies) and can therefore be produced relatively cheaply, stored without refrigeration, and administered orally. As with all chronic disorders, non-adherence is a major problem in asthma, and an oral alternative could improve adherence and hence efficacy. There is therefore a compelling case for clinical development of selective CRTH2 antagonists.
5. Evidence to date
There have been several trials of CRTH2 antagonists in stable asthma and in the asthmatic response to an inhaled allergen challenge, a model with good positive and excellent negative predictive value for drug efficacy (). These have universally found CRTH2 antagonists to be safe and well-tolerated, even at the highest doses. The overall results in terms of efficacy, however, have been underwhelming, with inconsistent reports of statistically significant but small (and potentially not clinically significant) improvements in lung function and quality of life measures (it is worth noting that the bronchoconstriction effects of PGD2 are via the activity of its metabolite 11β-PGD2α on the thromboxane receptor, so CRTH2 blockade would not be expected to affect lung function). How do we explain the lack of clinical efficacy? Much of it may come down to two aspects of clinical trial design, selection of the appropriate population and outcome measures.
Table 1. Trials of CRTH2 antagonists in asthma.
Asthma is increasingly recognized as a heterogeneous disease with various clinical phenotypes and molecular endotypes. Given this, it is unrealistic to expect a single drug to be a panacea for all asthmatics. The type 2 inflammatory endotype, which theoretically should respond best to CRTH2 antagonism, is only one of these endotypes, albeit the most common representing around half of all asthma. However, most clinical trials of CRTH2 antagonists have selected patients on the basis of severity (e.g. use of inhaled corticosteroids or forced expiratory volume in 1 s, FEV1) rather than phenotype or endotype (see ). It makes more sense to select participants on the basis of biomarkers of type 2 inflammation, such as blood eosinophilia or exhaled nitric oxide (FeNO) levels. Indeed, when these study participants are analyzed separately, the results are more impressive. For example, there were significantly greater increases in FEV1 in subgroups with: elevated serum eosinophils [Citation10], positive skin prick tests [Citation11], and raised FeNO [Citation12].The choice of end points assessed may also be painting an unduly negative picture of CRTH2 antagonist efficacy in clinical trials of asthma. In particular, no trial to date has been powered to detect an effect on asthma exacerbations, an outcome responsible for the majority of morbidity and mortality in asthma and around half the health-care costs. Moreover, type 2 inflammation is particularly prominent during exacerbations, as demonstrated both by the marked reduction in exacerbation frequency using anti-IL-5 therapies [Citation13,Citation14] and experimentally using rhinovirus challenge models in asthma [Citation15]. Our group has recently shown that PGD2 also rises during asthma exacerbations, with levels correlating with increases in IL-5, IL-13, and measures of exacerbation severity [Citation16]. It is therefore biologically plausible that CRTH2 antagonists could be particularly effective in preventing or attenuating exacerbations, whilst simultaneously having a limited effect on stable disease. Interestingly, this appears to be the case for the emerging monoclonal antibody treatments, which produce only modest improvements in lung function and symptom scores in stable asthma, but crucially are effective in preventing ~40–50% of exacerbations (see ).
Table 2. Trials of biologicals in asthma.
6. Future directions
There are a number of clinical trials ongoing of CRTH2 antagonists in asthma that will address the shortcomings outlined earlier. These include studies that restrict inclusion to asthmatics with evidence of type 2 inflammation (NCT02560610, NCT02660489, and NCT01836471) and those powered to assess an effect on exacerbations. The latter studies include those of sufficient length and size to study naturally occurring exacerbations (NCT02563067 and NCT02555683), or precipitating exacerbations following withdrawal of oral corticosteroid maintenance therapy (NCT02560610) or experimentally following rhinovirus challenge (NCT02660489).
Should the results be positive, studies comparing CRTH2 antagonists to existing treatments and other novel monoclonal antibodies targeting type 2 pathways will be required. Their cost to health-care systems will also likely determine where they fit into existing management pathways. In addition, studies in pediatric asthma and potentially in those with other clinical phenotypes, such as aspirin-exacerbated respiratory disease, will be needed to establish benefit across the asthmatic spectrum. As is the case for other treatments targeting type 2 inflammation, the discovery of a biomarker that identifies patients most likely to benefit from CRTH2 blockade would be invaluable, particularly in those whose FeNO and serum eosinophils are suppressed by inhaled corticosteroid treatment. Nonetheless given the positive findings with CRTH2 antagonists in the subset of asthmatics with evidence of type 2 inflammation, as well as the benefit of other type 2-targeted therapies in reducing exacerbations, we believe the outstanding studies are warranted and hopeful that they yield positive results.
Declaration of interest
S Johnston is a National Institute of Health Research Senior Investigator. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Additional information
Funding
References
- Fahy JV. Type 2 inflammation in asthma–present in most, absent in many. Nat Reviews Immunol. 2014;15:57–65. [Epub 2014/Dec/24].
- Gyles SL, Xue L, Townsend ER, et al. A dominant role for chemoattractant receptor-homologous molecule expressed on T helper type 2 (Th2) cells (CRTH2) in mediating chemotaxis of CRTH2+ CD4+ Th2 lymphocytes in response to mast cell supernatants. Immunology. 2006;119:362–368. [Epub 2006/Oct/28].
- Tanaka K, Ogawa K, Sugamura K, et al. Cutting edge: differential production of prostaglandin D2 by human helper T cell subsets. J Immunol. 2000;164:2277–2280. [Epub 2000/Feb/29].
- Joo M, Kwon M, Sadikot RT, et al. Induction and function of lipocalin prostaglandin D synthase in host immunity. J Immunol. 2007;179:2565–2575. [Epub 2007/Aug/07].
- Xue L, Barrow A, Pettipher R. Interaction between prostaglandin D and chemoattractant receptor-homologous molecule expressed on Th2 cells mediates cytokine production by Th2 lymphocytes in response to activated mast cells. Clin Exp Immunol. 2009;156:126–133. [Epub 2009/Feb/18].
- Xue L, Salimi M, Panse I, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. 2014;133:1184–1194. [Epub 2014/Jan/07].
- Redington AE, Meng QH, Springall DR, et al. Increased expression of inducible nitric oxide synthase and cyclo-oxygenase-2 in the airway epithelium of asthmatic subjects and regulation by corticosteroid treatment. Thorax. 2001;56:351–357. [Epub 2001/Apr/20].
- Fajt ML, Gelhaus SL, Freeman B, et al. Prostaglandin D(2) pathway upregulation: relation to asthma severity, control, and TH2 inflammation. J Allergy Clin Immunol. 2013;131:1504–1512. [Epub 2013/Mar/20].
- Stinson SE, Amrani Y, Brightling CE. D prostanoid receptor 2 (chemoattractant receptor-homologous molecule expressed on TH2 cells) protein expression in asthmatic patients and its effects on bronchial epithelial cells. J Allergy Clin Immunol. 2015;135:395–406. [Epub 2014/Oct/15].
- Hall IP, Fowler AV, Gupta A, et al. Efficacy of BI 671800, an oral CRTH2 antagonist, in poorly controlled asthma as sole controller and in the presence of inhaled corticosteroid treatment. Pulm Pharmacol Ther. 2015;32:37–44. [Epub 2015/Apr/12].
- Pettipher R, Hunter MG, Perkins CM, et al. Heightened response of eosinophilic asthmatic patients to the CRTH2 antagonist OC000459. Allergy. 2014;69:1223–1232. [Epub 2014/May/29].
- Wenzel S, Chantry D, Eberhardt C, et al. ARRY-502, a potent, selective, oral CRTh2 antagonist reduces Th2 mediators in patients with mild to moderate Th2-driven asthma. Eur Respir J Conference: Eur Respir Soc Annu Congress. 2014;44(Suppl 58):4836.
- Pavord ID, Korn S, Howarth P, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380:651–659. [Epub 2012/Aug/21].
- Ortega HG, Yancey SW, Mayer B, et al. Severe eosinophilic asthma treated with mepolizumab stratified by baseline eosinophil thresholds: a secondary analysis of the DREAM and MENSA studies. Lancet Respir Med. 2016;4:549–556. [Epub 2016/May/15].
- Jackson DJ, Makrinioti H, Rana BM, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. 2014;190:1373–1382. [Epub 2014/Oct/29].
- Jackson D, Shamji B, Trujillo-Torralbo M, et al. Prostaglandin D2 is induced during rhinovirus-induced asthma exacerbations and related to exacerbation severity in vivo. Am J Respir Crit Care Med. 2014;189:666–673.