
ABSTRACT
Introduction
Pancreatic cancer is one of the deadliest solid organ cancers. In the absence of specific warning symptoms pancreatic cancer is diagnosed notoriously late. Current systemic chemotherapy regimens extend survival by a mere few months. With the advances in genetic, proteomic, and immunological profiling there is strong rationale to test kinase inhibitors to improve outcome.
Areas covered
This review article provides a comprehensive summary of approved treatments and past, present, and future developments of kinase inhibitors in pancreatic cancer. Emerging roles of protein kinase inhibitors are discussed in the context of the unique stroma, the lack of high-prevalence therapeutic targets and rapid emergence of acquired resistance, novel immuno-oncology kinase targets, and recent medicinal chemistry advances.
Expert opinion
Due to the to-date frequent failure of protein kinase inhibitors indiscriminately administered to unselected pancreatic cancer patients, there is a shift toward the development of these agents in molecularly defined subgroups which are more likely to respond. The development of accurate biomarkers to select patients who are the best candidates based on a detailed understanding of mechanism of action, pro-survival roles, and mediation of resistance of targeted kinases will be critical for the future development of protein kinase inhibitors in this disease.
1. Background
NCI’s Surveillance, Epidemiology, and End Results (SEER) program estimates a total of 62,210 new cases of pancreatic cancer (3.2% of all cancer diagnoses in the U.S.) and 49,830 deaths (8.2% of all cancer-related deaths) in the U.S. in 2022 [Citation1]. Pancreas cancer is one of the few solid organ cancers with currently rising incidence rates [Citation2–4]. Based on projected shifts in demographics and changes in estimated incidence and death rates, pancreas cancer is projected to surpass breast, prostate, and colorectal cancer to become the second leading cause of cancer-related mortality in the U.S [Citation2]. Age-adjusted incidence rates for new pancreas cancer cases and pancreas cancer-related death rates have risen by an average of 0.5% and 0.2% per year over the last decade, and projected incidence and death rates are estimated to increase to 1.3% and 0.5% annually, respectively [Citation1,Citation2]. Based on 2017 to 2019 data, the life-time risk of men and women to be diagnosed with pancreas cancer is ~1.7% [Citation1].
Most recent 5-year survival rates have seen an incremental uptick to 11.5% [Citation1,Citation5]. This incremental increase compared to the stubbornly unchanged 5-year survival rates in the single digit range observed over several decades is attributed to a shift toward detection of pancreas cancer cases at earlier stages. More recent interventions associated with improved outcomes include the creation of high-risk clinics surveilling patients with cystic pancreatic neoplasms or a family history of pancreas cancer, better diagnostic imaging and increased awareness, and the introduction of multi-agent systemic chemotherapy regimens with improved efficacy [Citation5,Citation6].
2. Medical need
Treatment for the vast majority of pancreas cancer patients remains palliative [Citation5]. Even patients diagnosed with early, localized disease, (10 to 15% of all pancreatic ductal adenocarcinoma (PDAC) patients) who are able to undergo surgical resection, nearly inevitably recur and succumb to the disease [Citation5–7]. Median overall survival for the more than 50% of patients presenting at diagnosis with metastatic disease is generally measured in months with no cures reported [Citation1,Citation5]. An area which has recently come into greater focus is the rapid deterioration of quality of life (QOL) metrics of patients afflicted by pancreas cancer [Citation8]. Across psychological outcomes patients with pancreas cancer are reported to have worse QOL than adults with other cancers [Citation8,Citation9]. Additionally, some studies report that patients with pancreatic cancer experience worse outcomes on physical and social QOL subdomains which is discussed as a result of rapid disease progression with early development of cancer cachexia [Citation10]. Compared to other solid organ cancers pancreatic cancer has a high rate of thromboembolic events (with radiographic detection in ≥25% of patients with advanced disease) and GI dysfunction due to biliary and gastric outlet obstruction and pancreatic exocrine insufficiency. Pancreatic cancer patients benefit from a multidisciplinary approach including the early involvement of pain and palliative care specialists [Citation5,Citation11]. Of note, despite a high rate of treatment-related side effects, more effective therapies with improved disease control can both delay the onset and diminish degree of deterioration of QOL compared to less effective regimens [Citation12].
3. Existing treatments
After initial randomized clinical trials established a survival gain in patients with pancreas cancer treated with single agent gemcitabine over 5-flourouracil (5-FU), the phase III PRODIGE 4/ACCORD 11 and MPACT trials showed that combination regimens are superior to single agent gemcitabine chemotherapy () [Citation13]. Patients enrolled onto the FOLFIRINOX arm of the PRODIGE 4/ACCORD 11 trial had an objective response rate (ORR) of 31.6% and disease control rate of greater than 70% which, together with a median overall survival length of 11.1 months compared to 6.8 months in the gemcitabine control group, led despite its significant toxicity to FOLFIRINOX’s regulatory approval as first-line chemotherapy in pancreas cancer patients with good performance status [Citation12]. The MPACT trial, which did not exclude patients older than 75 and enrolled patients with lower performance status, examined the addition of nab-paclitaxel (albumin-bound paclitaxel) to gemcitabine in comparison to gemcitabine monotherapy [Citation14]. A response rate of 23% and median overall survival of 8.5 months compared to 7% and 6.7 months in the gemcitabine monotherapy group, respectively, showed the superiority of the nab-paclitaxel combination regimen and added another first-line treatment option. While there has been no formal randomized comparison of FOLFIRNOX vs nab-paclitaxel-gemcitabine in the first-line setting, the slightly higher overall survival (OS) rates of the FOLFIRINOX regimen has favored in particular in younger patients with good performance status the use of FOLFIRINOX over gemcitabine-nab-paclitaxel.
Table 1. Current treatment options with designated regulatory approval in pancreatic cancer.
Approved second-line treatment options for patients with advanced pancreas cancer are currently limited to nanoliposomal irinotecan with fluorouracil/leucovorin (nal-IRI + 5-FU/LV). The NAnoliPOsomaL Irinotecan (NAPOLI-1) study, which enrolled patients with advanced pancreas cancer who had progressed after having received first-line gemcitabine-based chemotherapy, showed a median OS of 6.1 months with manageable toxicities in the nal-IRI + 5-FU/LV group versus 4.2 months in patients treated with 5-FU/LV [Citation15]. Since currently many patients are often treated with FOLFIRINOX upfront, which includes both 5-FU and irinotecan, these patients are frequently not considered for the nal-IRI/5-FU/LV regimen. A combination regimen with marginal improvement on overall outcome is the combination of the epidermal growth factor receptor (EGFR) inhibitor erlotinib and gemcitabine. The results of the large Canadian NCIC CTG PA.3 trial found in unselected, previously not treated pancreatic cancer patients a modest, yet statistically significant improvement of OS (median 6.24 months v 5.91 months; adjusted for stratification factors) in the gemcitabine/erlotinib arm compared to the gemcitabine control arm [Citation16]. However, some reviews of ‘real world’ outcome outside the initial clinical trial setting failed to show a difference, and gemcitabine/erlotinib is infrequently used in today’s treatment landscape [Citation17].
There has been a shift in recently approved treatments for pancreas cancer toward molecular subgroups and individualized treatment decisions for patients likely to benefit from administered therapy. With FDA’s landmark decision in May 2017 to grant regulatory approval of the PD-1 immune checkpoint inhibitor pembrolizumab for the treatment of microsatellite instability high (MSI-H) cancers in a tumor histology-agnostic manner for many tumor types with defective DNA mismatch repair genes, anti-PD-1 immunotherapy became available for ~0.8 to 2.5% of MSI-H/mismatch repair-deficient pancreatic cancer patients () [Citation18–21]. The PARP inhibitor olaparib was approved for maintenance treatment of metastatic pancreatic adenocarcinoma in patients with germline BRCA mutations whose disease has not progressed after at least 16 weeks of first-line chemotherapy [Citation22]. BRCA1 and 2 variants are detected in approximately 5–7% of PDAC patients [Citation23]. Based on the results of two large basket trials, FDA most recently granted approval for larotrectinib and entrectinib for the treatment of pancreatic cancers harboring fusions of the neurotrophic receptor tyrosine kinase (NTRK) gene, an even smaller molecular subgroups than tumors occurring in MSI-H or BRCA-deficient background [Citation23–25]. In this regard, results from the international phase III POLO study which provided the rationale for FDAs approval of olaparib maintenance therapy are able to illustrate how incremental advances in small, molecularly defined subgroups of patients are unlikely to change the overall dismal outcome of pancreas cancer: after screening 3,315 patients with pancreatic cancer for BRCA germline mutations, 154 patients were ultimately assigned to treatment intervention [Citation22]. While median progression-free survival (PFS) was significantly longer in the olaparib group than in the placebo group (7.4 months vs. 3.8 months; hazard ratio (HR), 0.53, 95% CI 0.35 to 0.82; p = 0.004) there was no difference in OS or quality of life between patients receiving olaparib vs placebo. Thus, overall, systemic chemotherapy remains to-date the only available treatment option for the vast majority of PDAC patients and overall outcome frequently remains governed by inevitable progression and recurrence, lack of cure, frequent treatment-related toxicities, and rapid deterioration in quality of life.
4. Market summary
Within the last two decades regulatory agencies around the world including FDA have approved more than 100 kinase inhibitors [Citation26]. The majority of these are small molecule-based protein kinase inhibitors. The number of regulatory approvals by FDA for small molecule kinase inhibitors has hereby doubled over the last five years and small molecule kinase inhibitors comprise for ~15% of all New Drug Application (NDA) approvals [Citation26–28].
The global pancreatic cancer market is estimated to increase to USD 4.2 billion by 2025 [Citation29]. There are several reasons for this expansion: in addition to the significant medical need for more effective therapies in this disease, pancreas cancer incidences continue to rise as common risk factors like tobacco usage, obesity, or diabetes mellitus remain prevalent in developed nations [Citation2,Citation4]. Pancreas cancer rates peak in the 65 to 75 age group which is a growing population in North America, Europe, and other developed nations. Thus, these demographic factors paired with the evasiveness of early disease detection and the ongoing lack of effective treatment options will continue to drive development and commercial interest [Citation5,Citation29]. Despite these favorable factors, protein kinase inhibitors are predicted to have a drop in their market share among pancreatic cancer therapeutics as their development competes with the rapidly emerging class of biologics including both monoclonal antibody (mAb)-based immuno-oncology drugs as well as antibody drug conjugates. This estimated decrease is despite the upbeat potential that to-date only ~50 to 80 of the 535 kinases encoded in the human genome have been tested as a target within clinical trials [Citation26,Citation27]. Estimates of the fraction of kinases dysregulated in cancer as high as 70%, thus, leave significant opportunity in the kinome space for de novo drug development [Citation27]. Out of the eight major groups of kinases, nearly all clinically advanced protein kinase inhibitors target kinases of the tyrosine kinase (TK), tyrosine kinase-like (TKL), STE20, STE11, and STE7-related kinases (STE), protein kinase A, protein kinase G, and protein kinase C-related (AGC), and Cdk, MAPK, GSK, Cdk-like-related (CMGC) families [Citation26]. Members of the TK family including receptor tyrosine kinases (RTKs) are the most commonly ‘drugged’ kinase class, yet ~70% of TKs and more than 80% of TKL kinases remain unexplored. In order to harness these therapeutic opportunities, recent emerging kinase inhibitor developments include targeting transcriptional kinases which are able to function as enhancers or super-enhancers and which historically have not been considered as targets. These include targeting master transcription factor (TF) regulators like the MYC proto-oncogene, cyclin-dependent kinase 7 (CDK7), or DNA-dependent protein kinase (DNA-PK) [Citation30]. The development of immuno-regulatory kinases aims at reducing immune evasion of pancreatic tumors and the restoration of immune control, and are often combined with immune checkpoint inhibitors like the programmed death receptor PD-1 or other immuno-oncology agents. The most well-investigated and advanced candidates include inhibitors of the colony stimulating factor 1 receptor (CSF1R), Bruton tyrosine kinase (BTK), spleen tyrosine kinase (SYK), phosphatidylinositol 3-kinase delta (PI3Kdelta), or receptor interacting protein kinase 1 (RIPK1) [Citation26,Citation31]. Inhibition of focal adhesion kinase (FAK), integrin-linked kinase (ILK), Janus kinase (JAK), or transforming growth factor-beta (TGFβ) receptor signaling aims to disrupt tumor-promoting stromal cells populations like cancer-associated fibroblasts or tumor associated macrophages [Citation32–34].
Recent advances in medicinal chemistry are also supporting a positive outlook for kinase inhibitors in the market space of pancreas cancer. Proteolysis-targeting chimeras (PROTACs) link an E3-ligase-binding moiety to the kinase targeting small molecule which licenses the targeted kinase protein to the proteosome resulting in degradation of the kinase [Citation26,Citation35]. Kinase degraders, like PROTOACs or heterobifunctional molecules with ubiquitin moieties (‘molecular glues’), have the advantage of not requiring 24-hours maintenance of therapeutic exposure levels within tumors. The need for maintained target inhibition like for non-covalent small molecule kinase inhibitors on the other hand is known to be difficult to achieve for chemotypes with narrow therapeutic windows and has been shown to be a cause for therapy failure. Despite these aforementioned opportunities for an increased market share, there are also challenges ahead for this class of therapeutics; for example, the trend toward development of protein kinase inhibitors for increasingly smaller, molecularly defined subgroups is likely to continue and accelerate. The rarity of the targeted variants often present in only a few percent of pancreas cancer patients will likely reduce revenue and offset other gains in market space. Traditional kinase inhibitors also face competition from new developments within the class of small molecule inhibitors like small molecule discoveries targeting essential protein-protein interactions, oncogenic phosphatases, epigenomic regulators targeting genome organization, or recent advances against previously undruggable oncogenic drivers like KRAS [Citation30,Citation36–39]. Despite these challenges including a surprisingly high rate of off-target effects of protein kinase inhibitors in current clinical use, kinase inhibitors will continue to have a key role in drug development and occupy a significant market share of future therapeutics for pancreatic cancer [Citation28].
5. Current research goals
The overarching research goal of clinical testing of protein kinase inhibitors in patients afflicted by pancreas cancer is to establish their role in improving clinical outcome of this recalcitrant disease. Protein kinase inhibitors can be given in a rationale-design fashion to suppress activation of redundant parallel signaling pathways to prevent, or delay, the development of acquired resistance and enhance response to chemo- or cancer immunotherapy. Protein kinase inhibitors are also tested to modulate the tumor microenvironment (TME) and the extracellular matrix (ECM) to breach the stromal barrier of these desmoplastic tumors which limits perfusion and drug penetration. An emerging scientific goal is to combine protein kinase inhibitors with the rapidly enlarging arsenal of immuno-oncology and cancer immunotherapy agents to elicit clinically meaningful anti-tumor responses in these immunologically cold, T cell excluded and T cell exhausted tumors [Citation40]. In this review, we provide an overview of past and current clinical testing and development of protein kinase inhibitors in pancreatic cancer and discuss the scientific rationale of selected targets and pursued combination strategies.
6. Scientific rationale
Rather than administered in an indiscriminate fashion, protein kinase inhibitor can be tailored to individual molecular and biological subgroups based on the presence of tumor-driving oncogenic signal transduction pathways, which frequently involve a cascade of either tyrosine or serine protein kinases (). Blockade of oncogenic signal transduction aims to induce cell cycle arrest, apoptosis, or senescence in cancer cells, reduce tumor progression, and induce tumor control. Detailed genomic profiling has identified unique and considerable challenges for the development of effective kinase inhibitor treatment in pancreatic cancer. After the frequent, recurrent alterations of activating KRAS mutations (up to 90% of all PDAC) loss of the tumor suppressors TP53, SMAD4, and CDKN2A (p16), pancreatic tumors harbor an average of 63 additional, non-recurrent somatic mutations involving an average of 17 signal transduction pathways which involve a variety of protein kinases and have no to minimal overlap between patients [Citation41,Citation42].
Figure 1. Protein kinase inhibitors under development in pancreatic cancer. A. Schematic representation of key targets and pharmacological agents that inhibit EGFR-RAS-MAPK signaling proteins (left). Inhibition of SHP2 pro-tumoral role within different cell populations in the tumor microenvironment (right) reduces tumor-promoting signaling via disruption of cancer cell-stromal cell crosstalk. Investigational anti-RAS agents highlighted with * indicate isoform-specific RAS inhibitors targeting the KRASG12C mutational isoform. B. Illustration of near ubiquitous focal adhesion kinase (FAK) – Src kinase activation via multitude of receptor tyrosine kinases (left) or extracellular matrix macromolecules (right) and respective pharmacological inhibitors. A selection of downstream effector signaling of FAK – Src kinase signaling is shown. C. Protein kinases involved in double strand DNA repair with emerging small molecule inhibitors.
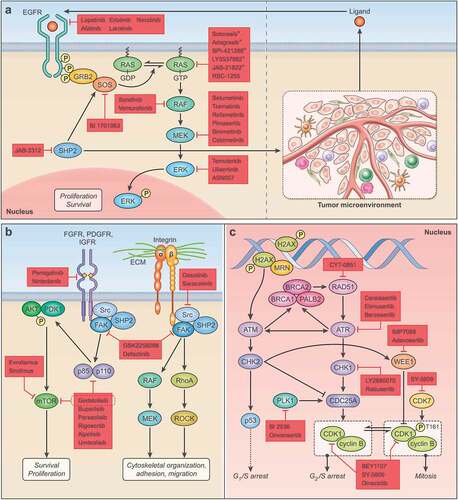
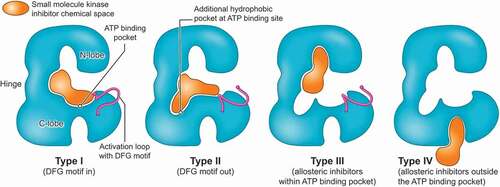
Protein kinase inhibitors aim to abolish the ability of kinases to phosphorylate its substrates. While a detailed discussion of the structural features of kinase function, and their modes of inhibition, is beyond the scope of this review, kinases consist of a smaller N- and larger C-lobe which are connected via a hinge region to form the ATP binding pocket (). The ATP molecule is anchored between the two lobes via hydrogen bonds between the hinge region and the adenine ring of the ATP [Citation26,Citation43]. The precise final position of the ATP required for hydrolysis and phosphorylation of the protein substrate is finetuned via hydrophobic interactions between highly flexible activation loops of the C-lobe and the ATP molecule. One of the activation domains includes the conserved tripeptide Asp-Phe-Gly sequence (DFG motif) which in its active conformation points toward to the phosphate groups of the ATP inside the ATP binding pocket (DFG-in conformation; active conformation) [Citation26,Citation43]. The DFG-out movement results in a rearrangement of the activation loops of the C-domain which is unfavorable for ATP hydrolysis and is indicative of the inactive state of the kinase. Type I inhibitors compete with ATP in the active state of the kinase [Citation26,Citation28]. The inhibitor mimics the hydrogen bonds of the adenine, is anchored to the hinge region, and both the DFG motif and the activation loop of the C-lobe are in an active position. Type II inhibitors exploit a hydrophobic grove opened by the DFG-out movement (). Type II inhibitors are binding to the inactive state of the targeted kinase. The vast majority of kinase inhibitors which are currently in clinical use are type I and II inhibitors [Citation28]. Type III inhibitors bind to an allosteric binding pocket within the ATP cleft and either induce a DFG-in like the clinically approved MEK inhibitors or a DFG-out conformation like FAK inhibitors currently in clinical development for pancreas cancer [Citation26,Citation44]. Type IV inhibitors engage allosteric binding sites distant from the ATP binding pocket. The improved structural and molecular knowledge on kinase function aims to accelerate kinase inhibitor designs and kinase inhibitor development to yield molecules with improved selectivity and candidates tailored to unique pharmacological needs of targeted disease indications including pancreas cancer.
Figure 2. Schematic cartoon depicting different classes of kinase inhibitors. Chemical space targeted by different classes of kinase inhibitors shown in Orange, N- and C-lobe and hinge region of protein kinase in blue. Type I inhibitors compete with ATP binding in the active state of targeted kinase, DFG motif is (shown in purple) directed toward ATP binding site (DFG-in). Type II inhibitors block ATP binding in the inactive state of the targeted kinase (DFG-out). Type III and IV are allosteric kinase inhibitors with binding within the ATP cleft (type III) or independent from the ATP binding site (type IV).
6.1. Stromal barrier, tumor microenvironment, and pharmacological considerations
Small molecule protein kinase inhibitors often have a lower affinity and target specificity compared to high molecular weight monoclonal antibody (mAb)-based therapeutics. However, this disadvantage can be offset by their higher intratumoral drug distribution. Small molecules can more easily diffuse through the tumoral stroma where large macromolecules like mAb-based therapeutics experience greater diffusion resistance by the ECM. Unique for pancreas cancer among solid organ cancers is the desmoplastic nature of the TME which forms a mechanical barrier due to an interconnected web of cellular partitions and often highly charged, large macromolecules of the ECM () [Citation45]. Long diffusion distances, opposing convectional flux, and high interstitial pressures combined with abnormal vasculature are major impediments to drug penetration and the ability of administered anti-neoplastic agents to exert their mechanism of action at the intended cell and their target [Citation45–47]. In this regard, small molecules with a low total polar surface area, which are able to cross cellular membranes to reach intracellular targets, makes them together with their ease of chemical synthesis, the availability of accurate MS/MS-based bioanalytical measurements, and lack of immunogenicity excellent clinical candidates [Citation48].
Figure 3. Impact of stromal barrier of pancreas cancer on intratumoral drug distribution. Illustration of unique desmoplastic pancreatic cancer stroma impairing effective intratumoral diffusion (top) with macromolecules of extracellular matrix (bottom left) and cellular partitions of stromal cells (bottom middle) forming barriers for drugs to reach target cells (bottom right).
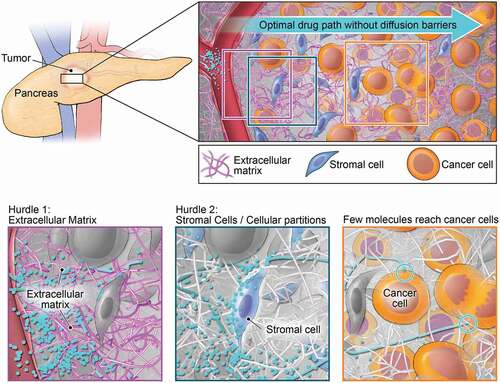
Acknowledging the detrimental impact of the desmoplastic stroma on the efficacy of administered therapeutics, pancreas cancer spearheaded the concept of combined anti-stroma, anti-cancer therapy breaking the stromal barrier to increase drug delivery and improve therapeutic responses [Citation49]. The most promising preclinical findings of anti-sonic hedgehog (SHH), anti-CTGF and TGFβ, and anti-hyaluronan via pegvorhyaluronidase alfa (PEGPH20) treatment have reached clinical testing, including phase III testing in case of PEGPH20 [Citation46,Citation47,Citation49]. Despite their initial intriguing preclinical rationale, clinical testing has not yielded improved outcomes and future development has either been discontinued (PEGPH20), is currently unclear, or still ongoing [Citation45]. The difficulties of many cancer agents to overcome poor intratumoral perfusion and achieve therapeutic levels at the target cells within the TME ask for innovative formulation strategies or chemotypes which, for instance, provide pharmacokinetic (PK) profiles characterized by high AUC tumor: plasma ratios [Citation48]. Both, the development of nab-paclitaxel, an albumin-bound, 130-nm–particle formulation of paclitaxel, and liposomal irinotecan (nal-IRI), are formulations which aim to exploit features of the TME of pancreas cancer via increased retention through enhanced permeation and retention (EPR) effects [Citation50]. While small molecule inhibitors may cover greater topographies and achieve more homogenous distribution within the TME than mAb-based therapies, these advantages might be offset by their lower target affinities, the need to cross cellular plasma membranes, or the need to maintain a minimum exposure level to ensure suppressed kinase activity [Citation48]. In this regard, well-conducted pharmacodynamic (PD) evaluations within clinical studies of MEK and AKT inhibitors have exposed some of the liabilities associated with lower target affinity and lack of target engagement as suppression of phosphorylation levels of respective kinases at phase II dose levels rarely exceeded 50% [Citation51–53]. Sustained suppression exceeding 80% is cited as required for anti-tumor response based on preclinical data [Citation54].
7. Competitive environment: a review of drugs in clinical development
Immediately after the emergence of the first molecular therapies targeting protein kinases in the first decade of the 21st century, clinical trials have evaluated kinase inhibitors in patients with pancreatic cancer. After initial studies in unselected patients with either single agent, or in combination with systemic chemotherapy, yielded disappointing results, more recent clinical testing has shifted toward biomarker-directed selection of patients and rationale-designed drug combinations who are more likely to respond.
7.1. EGFR-RAS-MAPK pathway inhibitors
With the near universal presence of activating KRAS mutations and the frequent dysregulation of EGFR/HER2 signaling there has been a keen interest in blocking canonical MAPK pathway signaling, initially at the level of EGFR and MEK and, more recently, at other signaling nodes or by targeting other regulators of the pathway [Citation55]. After release of the results of the Canadian NCIC CTG PA.3 phase III study which reported a very modest improvement of survival measured in weeks after the addition of the EGFR inhibitor erlotinib to gemcitabine first-line chemotherapy, the large phase III intergroup trial S0205 showed that the addition of the anti-EGFR monoclonal antibody cetuximab to gemcitabine did not improve outcome compared to patients treated with gemcitabine alone (6.3 versus 5.9 months; HR 1.06, CI 0.91–1.23; p = 0.23) [Citation56]. Similarly, the dual tyrosine kinase inhibitor lapatinib, which interrupts HER2 and EGFR, did not show any activity in combination with chemotherapy [Citation57–59]. In order to improve these results, a large phase II study compared outcomes of the second-generation ErbB family blocker afatinib, which covalently binds to and irreversibly blocks formation of homo- and heterodimers of EGFR-ErbB2/HER2 and ErbB4/HER4, in combination with gemcitabine to gemcitabine alone but did not show an improvement in outcome (7.3 versus 7.4 months; HR 1.06) [Citation60]. Several phase II studies examined the merit of dual, vertical EGFR-MAPK kinase pathway inhibition by combining the EGFR inhibitor erlotinib with the BRAF inhibitor sorafenib or the MEK inhibitor selumetinib, or adding a second anti-EGFR agent in the form of panitumumab [Citation61–64]. These studies have not produced an efficacy signal to warrant further investigation. A study with neratinib, a small molecule which covalently inhibits EGFR and ErbB2/HER2 and reduces expression levels of mutant KRAS in preclinical studies, is planned to be tested in combination with the repurposed histone deacetylase (HDAC) inhibitor valproate in pancreas cancer patients with RAS-mutant tumors (NCT03919292) [Citation65]. Retrospective subgroup analysis of the NCIC CTG PA.3 phase III study as well as preclinical studies in a series of pancreatic cancer patient-derived xenotransplantation (PDX) models showed that EGFR copy number or KRAS mutational status were not predictive of patients to derive benefit from anti-EGFR therapy and suggest that alternative biomarkers might identify a subgroup of patients who might derive benefit from erlotinib anti-EGFR therapy [Citation66–70].
Because 90% of PDAC harboring activating KRAS mutations, inhibitors of the downstream kinases BRAF and MEK were extensively tested in unselected pancreatic cancer patients. Randomized studies of the BRAF multikinase inhibitor sorafenib, or the MEK inhibitors trametinib or pimasertib in combination with gemcitabine versus gemcitabine alone (HRs 1.27, p = 0.26; 0.96, p = 0.45; and 1.1, p = 0.67, respectively), the MEK inhibitor selumetinib vs capecitabine 2nd-line chemotherapy (HR 1.03; p = 0.92), or selumetinib in combination with the AKT inhibitor MK-2206 compared to mFOLFOX6 chemotherapy (HR 1.37; p = 0.15) did not show any efficacy [Citation69, Citation71–74]. A study with the MEK inhibitor trametinib in combination with the mTOR inhibitor evrolismus and the CDK4/6 inhibitor ribociclib, and the MEK inhibitor binimetinib in combination with the PARP inhibitor talazoparib and the PD-L1 inhibitor avelumab was terminated because of the inability to define a recommended phase 2 dose level or because of limited efficacy [Citation75,Citation76] (NCT03637491). Of the numerous clinical investigations of MEK inhibitors in primarily unselected patients, results of refametinib in combination with gemcitabine stand out because of a ~ 23% ORR [Citation77]. The authors suggest that biomarker status including KRAS mutational status should be investigated next prior to subjecting this combination to further clinical testing [Citation77,Citation78].
Compared to protein kinase inhibitor studies targeting EGFR-RAS-MAPK pathway signaling with reported clinical findings, there has been a shift in the landscape of emerging protein kinase inhibitors which are currently under clinical evaluation and target the EGFR-RAS network for the treatment of PDAC (listed as recruiting or not yet recruiting on clinicaltrials.gov; ). This shift includes development of protein kinase inhibitors against novel effectors and regulators of the EGFR-RAS-MAPK pathway, like SHP2 or SOS1, secondly, the design of combination trials to target adaptive resistance mechanisms, and, thirdly, a significantly increased use of biomarker-driven clinical development via tumor genetic testing. For example, studies with the MEK inhibitor binimetinib (in KRAS-mutant tumors) or the ERK inhibitor ulixertinib administered in combination with the CDK4/6 inhibitor palbiciclib (NCT04870034; NCT03454035), or temuterkib administered with the SHP2 small molecule inhibitor RMC-4630 to KRAS mutant tumors (NCT04916236), or MEK and ERK inhibitor combinations with the anti-autophagy agent hydroxychloroquine, aim to overcome the often rapid development of acquired resistance observed with MEK inhibitors. Protective upregulation of autophagy following ERK inhibition was recently identified as a novel survival mechanism in KRAS-mutant PDAC, and the addition of the autophagy blocker hydroxycholoroquine (HCQ) to ERK inhibition improved outcome in preclinical models [Citation79]. Of note, pancreatic cancer harboring the KRASG12R mutation isoform (15–20% of all KRAS mutations in PDAC) are particularly sensitive to the combination of ERK and autophagy blockade and might provide a yet unexplored opportunity to enrich patients more likely to respond [Citation80]. Current clinical testing of these findings include a combination studies of the MEK inhibitor binimetinib with HCQ in KRAS-mutant pancreas cancer (NCT04132505), or the combination of the MEK inhibitor tramatinib or the ERK inhibitor temuterkib with HCQ (NCT03825289; NCT04386057, respectively). Combining the MEK inhibitor binimetinib or ERK inhibitor ulixertinib with the CDK4/6 inhibitor palbociclib, which is FDA-approved for the treatment of breast cancer, is testing preclinical findings of MAPK pathway control of cyclin D1 and CDK4/6 and synergistic responses of the two agents in preclinical PDAC models [Citation81,Citation82]. A study of ulixertinib plus nab-paclitaxel and gemcitabine in patients with pancreatic cancer on the other hand was recently terminated due to excessive toxicity (NCT02608229).
Recent pharmaceutical targeting of additional RTK effectors beyond the canonical MAPK pathway kinases BRAF, MEK, and ERK to reduce RAS activation includes the discovery of druggable structural motifs on the RAS regulator son-of-sevenless (SOS) disrupting protein-protein interactions (PPI) or the development of inhibitors against the ubiquitously expressed protein tyrosine phosphatase 2 (SHP2). Both SOS and SHP2 are essential adaptor proteins downstream of many RTKs (NCT04975256; NCT04045496) () [Citation55,Citation83,Citation84]. Preclinical findings suggest that SHP inhibitors prevent the development of adaptive resistance via upstream RTK and MAPK pathway reactivation frequently observed with MEK inhibitors [Citation84]. As vertical pathway inhibition targeting two kinases within the MAPK pathway via BRAF and MEK inhibitors has shown superior outcomes compared to single-agent therapy and received regulatory approval in BRAFV600-mutant melanoma and non-small cell lung cancer (NSCLC), SHP2 inhibitors have been described as attractive combination partners with MEK and ERK inhibitors due to their ability to prevent RTK signaling activation and are currently under investigation in KRAS-mutant cancers including pancreas cancer (NCT04916236) [Citation84–86]. Most recently, dual MAPK pathway inhibition is conducted in a biomarker-guided fashion; initially promoted by concerns of toxicity of dual SHP2 and MEK inhibition on normal cells, several early phase clinical trials are testing SHP2 inhibitors in combination with G12C mutational isoform-specific KRAS inhibitors (). This strategy has been shown in preclinical models to suppress the development of wild type RAS activation mediating resistance (NCT05288205; NCT04330664) [Citation87]. Following FDA’s approval of sotorasib for KRASG12C-mutant NSCLC, several basket trials are enrolling other KRAS-mutant histologies with low frequencies of the KRAS mutational G12C isoform including pancreas cancer. SHP2 signaling also supports an immune suppressive, pro-tumor phenotype in several cell populations of the TME. SHP2 inhibition has been shown to reprogram the immune landscape in preclinical cancer models, and the combination of SHP2 and RAS inhibitors might disrupt feed-forward signaling loops involving the MAPK pathway between the cancer cell compartment and the TME () [Citation88,Citation89]. However, considering that PDAC with KRASG12C mutations constitute 1–2% of all PDAC cases, the impact onto overall outcome of pancreas cancer will be even in the advent of remarkable activity modest. Other examples of low frequency mutations affecting MAPK signaling in PDAC where kinase inhibitors shown to be active in other solid organ malignancies might become a successful precision medicine strategy are somatic BRAFV600 mutations (frequency in PDAC of 3%; currently tested within NCT04390243 enrolling pancreatic cancer patients with somatic BRAFV600 mutation for treatment with the MEK inhibitor binimetinib and BRAF inhibitor encorafenib) [Citation90].
7.2. PI3K-mTOR pathway inhibition
The PI3K-AKT-mTOR pathway is a major oncogenic pathway fueling cancer cell metabolism, cancer cell stemness, survival, tumor growth, and metastasis in PDAC. Frequent mutations affecting effectors and regulators of the pathway include loss of the tumor suppressor PTEN (present in ~15%), activating mutations in the catalytic PI3K subunit (PI3KCA) (1%), or amplification of the AKT2 gene locus (up to 20%) [Citation42,Citation90,Citation91]. Importantly, PI3K-AKT-mTOR signaling is a major effector pathway of a variety of dysregulated RTKs including FDGFR, PDGFR, HGFR/c-MET, EGFR, ERB2/HER2, AXL, and others ().
Initial studies on PI3K-mTOR signaling inhibition with the unselective pan-PI3K inhibitor buparlisib (BKM-120) in pancreas cancer were not successful and have not been continued. Combining buparlisib with mFOLFOX showed a 76% grade 3 or higher AE rate and did not identify a tolerable dose level [Citation92]. Buparlisib in combination with the MEK inhibitor trametinib yielded no objective responses during clinical testing and PD analysis showed no suppression of the PD markers phospho-S6 or phospho-ERK [Citation53]. Similarly, rigosertib, a multikinase inhibitor with dual activity against PI3K and PLK signaling pathways, did not show an improvement in outcome when combined with gemcitabine compared to single agent gemcitabine chemotherapy alone (median OS 6.1 vs 6.4 months; HR 1.24) [Citation93]. Equally, early phase studies with the PKCβ, PI3K/AKT inhibitor enzostaurin in combination with gemcitabine or the PI3K δ-isoform-selective inhibitor parsaclisib in combination with the JAK1 blocker itacitinib did not show any efficacy [Citation94,Citation95]. A study of the GSK-3β inhibitor LY2090314 in combination with gemcitabine for patients with metastatic pancreatic cancer was halted because of slow accrual (NCT01632306).
Several phase II studies which tested the merit of mTOR inhibition, either via administration of single agent evrolismus or sirolismus, or evrolismus in combination with the MEK inhibitor trametinib or cetuximab and temsirolismus in combination with gemcitabine have not yielded improved outcomes compared to what is commonly observed after second-line therapy for PDAC (NCT00499486) [Citation75,Citation96–98]. These findings are in line with the results from the randomized phase II SWOG S1115 study which combined the AKT inhibitor MK-2206 with the MEK inhibitor selumetinib in pancreas cancer patients who were previously treated with gemcitabine and compared outcome to patients receiving oxaliplatin and fluorouracil chemotherapy [Citation74]. This study reported inferior outcomes for the combination kinase inhibitor group (median OS 3.9 vs 6.7 months; HR, 1.37; p = 0.15; median PFS 1.9 vs 2.0 months; HR, 1.61; p = 0.02) which the authors attribute, in part, to uncertainty of target inhibition and the lack of selecting patients more likely to respond.
Initial studies of dual PI3K/mTOR and pan-PI3K inhibitors across different solid organ cancers discovered a narrow therapeutic window due to the high rate of treatment-related toxicities like hyperglycemia, nausea, diarrhea, or liver dysfunction [Citation99]. With PI3K isoform-selective candidates demonstrating clinical efficacy in other types of cancer, like the FDA-approved PI3K delta-specific idelalisib for chronic lymphocytic leukemia (CLL), or the PI3K alpha-specific inhibitor alpelisib approved in combination with the anti-estrogen receptor agent fulvestrant for advanced breast cancer patients whose tumors have PI3KCA mutations and are hormone receptor positive and HER2 negative, PI3K inhibitors in clinical testing for pancreas cancer are seeing a greater use of isoform-selective agents [Citation100]. The PI3Kα isoform-specific alpelisib is currently tested in combination with the MEK inhibitor binimetinib (NCT01449058) or gemcitabine-nab-paclitaxel chemotherapy (NCT02155088). Similarly, the PI3Kδ-selective umbralisib is undergoing clinical testing in combination with gemcitabine-nab-paclitaxel (NCT02574663). Clinical testing of the pan-class I isoform PI3K/mTOR inhibitor gedatolisib in combination with the CDK4/6 kinase inhibitor palbociclib in pancreatic cancer patients (NCT03065062) is a translation of positive findings of inhibition of cyclin-dependent kinases and PI3K-AKT signaling made in PDX models of pancreas cancer [Citation82,Citation101].
7.3. FAK-Src kinase and FGFR inhibitors
Focal adhesion kinase (FAK) is an integrin-associated protein tyrosine kinase that is frequently overexpressed and activated in PDAC and has been identified as a high-merit target in PDAC due its pleiotropic tumor-promoting role [Citation102,Citation103]. Multiple stimuli from the expanded, abnormal extracellular matrix (ECM) of the tumoral stroma activate FAK at sites of cellular adhesions (). FAK activates via its catalytic kinase domain a multitude of effector pathways which are not limited to the canonical cytoskeletal activation and reorganization pathways like Rho and Ral GTPases, but also include, via Src kinase, PI3K-mTOR signaling and, via RAS, MAPK pathway activation [Citation103]. FAK is also stimulating via a scaffolding function focal adhesion turnover, cell adhesion and migration, a function which has so far been difficult to target by current anti-FAK small molecule inhibitors [Citation44,Citation103]. In addition, FAK supports the pro-tumor phenotype of several cell populations within the TME including cancer-associated fibroblasts, endothelial cells or CD4 + T regulatory cells, and targeting FAK has been shown to reprogram the TME and sensitize PDAC to immune checkpoint inhibition in preclinical animal models [Citation32]. To date, results from early phase clinical trials of the FAK inhibitor GSK2256098 administered in combination with the MEK inhibitor trametinib, or the FAK inhibitor defactinib administered within a small pilot study in combination with gemcitabine and the PD-1 immune checkpoint blocker pembrolizumab, did not match the preclinical promise and have yielded no improvement in outcome [Citation104,Citation105]. Similarly, a randomized phase II study comparing the Src inhibitor dasatinib, one of the effectors of FAK, in combination with gemcitabine to gemcitabine monotherapy did not show an improvement in outcome (HR 1.16; p = 0.57) in patients with locally advanced pancreas cancer [Citation106]. This study did not replicate the more favorable results of ORR of 29.4% or 22.7% of earlier clinical testing when dasatinib was administered as single agent or in combination with FOLFOX chemotherapy [Citation107,Citation108]. Similar negative findings were obtained with the Src kinase inhibitor saracatinib [Citation109,Citation110]. Ongoing drug development includes the combination of the FAK inhibitor defactinib and pembrolizumab as neoadjuvant or adjuvant treatment (NCT03727880), or defactinib and GSK2256098 in combination with MAPK pathway blockers (NCT03875820; NCT02428270).
Blockade of FGFR signaling, which is firmly connected to FAK activation (), is currently investigated within a precision medicine approach in pancreas cancer patients. Following FDA’s approval of the FGFR small molecule inhibitor pemigatinib for previously treated cholangiocarcinoma with FGFR2 fusions or other FGF/FGFR gene arrangements, objective responses to pemigatinib in other malignancies including in pancreatic cancer patients harboring FGF/FGFR2 alterations have been observed [Citation111]. While the overall prevalence of FGFR fusions and mutations in PDAC is ≤5%, FGFR variants are more common in adenosquamous carcinoma of the pancreas, a rare subform of pancreas cancer, and pemigatinib is currently tested in this molecular subpopulation (NCT05216120) [Citation112].
7.4. HGF/c-MET and AXL receptor inhibition
Hepatocyte growth factor (c-MET; tyrosine-protein kinase MET) signaling is frequently activated in PDAC. Whereas gene amplifications and activating mutations drive HGF/c-MET activation in NSCLC and other solid organ malignancies, secretion of the HGF ligand by cancer-associated fibroblasts and other cells of the TME as well as release of ECM-bound HGF within the stroma which carries elevated rates of turnover are the predominant mechanisms of c-MET activation in PDAC [Citation113–115]. Preclinically, c-MET activation is a well-studied mechanism of cancer cell stemness, resistance to gemcitabine, and promotes cell invasion and metastasis in pancreas cancer [Citation115,Citation116]. Cabozantinib, approved for differentiated thyroid cancer, was evaluated in combination with gemcitabine in pancreas cancer patients but a tolerable dose could not be established due to excessive toxicity [Citation117]. Current development focuses on combination with immune checkpoint inhibitors (NCT04820179; NCT05052723).
AXL is a cell surface receptor tyrosine kinase which is expressed both on cancer and stromal cells [Citation118]. It transduces signals from the ECM into the cytoplasm mediating cell plasticity, cell invasion, and tumor metastasis. Several preclinical studies linking loss of AXL signaling and AXL inhibition to improved outcome including improved response to systemic chemotherapy [Citation118,Citation119]. A phase I/II is currently evaluating the combination of the AXL inhibitor bemcentinib and gemcitabine-nab-paclitaxel (NCT03649321).
7.5. DNA damage repair (DDR) inhibitors
The POLO trial, which reported improved outcomes of PDAC patients with germline BRCA1 and 2 mutations treated with the PARP inhibitor olaparib post-platinum chemotherapy, showed that targeting DDR can have therapeutic merit in this therapy-recalcitrant disease () [Citation22]. Overall, the rate of inherited and sporadic alterations in DDR pathway genes has been reported as high as 19.7% in pancreas cancer, and mutations in DDR genes beyond BRCA1/2 like ATM, PALB2, or RAD51 are explored as molecular markers to select patients for protein kinase inhibitors targeting DDR signaling [Citation120,Citation121]. In order to extend the positive findings with olaparib in germline BRCA1 and 2-mutant pancreatic cancers to other genetic defects involving DDR pathways, two phase II studies administered olaparib to patients with pancreatic tumors with a ‘BRCAness’ phenotype [Citation122]. BRCAness was defined as a DDR genetic alteration (ATM, PALB2, ARID1A, somatic BRACA, PTEN, RAD51, CCNE, or FANCB mutation), personal or family history of BRCA-associated cancers, or ATM protein loss. Overall survival was longest in the subgroup with DDR genetic alterations with median OS exceeding 1 year (median OS 13.6 months) suggesting proof-of-concept of targeting DDR-mutant PDAC in future clinical trials [Citation122]. Upregulation of DDR mechanisms is also a hallmark of cancer stem cells (CSCs) in solid organ malignancies including pancreatic tumors [Citation123,Citation124]. CSCs frequently reside in hypoxic regions, protect their genome from environmental stress by upregulating DDR pathways, and evade eradication by current chemotherapies due to inherent increased chemoresistance [Citation125]. A randomized phase II trial comparing outcome of unselected PDAC patients treated with the CHK1 kinase inhibitor rabusertib in combination with gemcitabine to gemcitabine monotherapy tested in pancreas cancer patient the concept whether DNA damage load in the combination group can be increased beyond a tolerable threshold and increase chemosensitivity to gemcitabine in unselected patients [Citation126]. There was no difference in outcome (median OS 7.8 months rabusertib/gemcitabine vs 8.3 months gemcitabine alone) (). Single agent PLK1 inhibition via BI2536 also failed to yield an efficacy signal after phase II testing [Citation127]. Ongoing and planned clinical testing is targeting additional kinases of the DDR pathway, most frequently in combination with chemotherapy. ATR kinase inhibitors, either as monotherapy or in combination with chemotherapy, have recently shown activity in small cell lung cancer and other solid organ malignancies [Citation128,Citation129]. The ATR kinase inhibitors elimusertib and berzosertib are currently under evaluation in combination with gemcitabine or irinotecan in phase I trials enrolling patients with PDAC (NCT04514497; NCT04616534; NCT02595931). In a genotype-directed clinical approach, the ATR blocker ceralasertib is undergoing phase I evaluation in combination with the PARP inhibitor recruiting PDAC patients selected by loss of ARID1A or ATM (NCT03682289). Other kinase inhibitors, which target DDR mechanisms and are currently being tested in combination with chemotherapy and enroll pancreas cancer patients are the PLK1 inhibitor onvansertib (NCT04752696), the RAD51 blocker CYT-0851 (NCT03997968), and the CHK1 inhibitor LY2880070 (NCT02632448) ().
Table 2. Reported outcomes of pancreatic cancer patients treated with protein kinase inhibitors administered as single agent or in combination.
Table 3. Emerging protein kinase inhibitors under investigation for pancreatic cancer.
WEE1 kinase connects DDR with cell cycle checkpoint control to allow cells to repair their damaged genome (). Following DNA damage involving double strand breaks, WEE1 kinase is activated via DDR pathway members ATM, ATR, DNA-PK, or downstream CHK1 and CHK2 kinases [Citation121,Citation130]. Activated WEE1 phosphorylates CDK1 at Tyr15 and Thr14 which reduces CDK1 activity and prevents entry into mitosis, delays G2/M checkpoint transition, and prolongs the G2 phase (). WEE1 blockade aims to remove the ability of the cell properly repair double strand breaks to overwhelm the cellular genome with an intolerable amount of structural defects [Citation130]. WEE1 inhibitors in early phase clinical trials which are enrolling pancreas cancer patients include IMP7068 (NCT04768868) and adavosertib (AZD1775) in combination with gemcitabine-based chemotherapy (NCT05212025; NCT02194829).
7.6. Cyclin-dependent and cell division cycle-related kinase inhibitors
After FDA’s approval of the highly selective CDK4/6 inhibitor palbociclib in combination with the anti-estrogen receptor agent fulvestrant for the treatment of HR positive, HER2 negative breast cancer in 2016 there has been renewed interest of a possible role of CDK inhibitors in the treatment of PDAC [Citation131]. Early studies with 1st generation, ATP-competitive agents inhibiting multiple CDKs like alvociclib (flavopiridol) have shown a narrow therapeutic window and no activity [Citation131,Citation132]. Several correlative tissue observations from clinical pancreatic cancer specimens confirm an upregulation or overexpression of cyclin D1 regulating CDK4 and 6, which is associated with unfavorable clinical outcome [Citation133,Citation134]. The frequent loss of the endogenous CDK4/6 inhibitor p16 due to mutations and copy number variations including deletions and inversions of the cyclin-dependent kinase inhibitor 2A (CDKN2A) gene locus (present in up to 50% of all PDAC) together with the impaired recovery of cancer cells from cytotoxic chemotherapy when CDK4/6 inhibition is followed chemotherapy administration support CDKs as a strong drug target in pancreas cancer [Citation41,Citation135]. However, in a small phase II study of 12 pancreas cancer patients with loss or mutations in CDKN2A single agent palbociclib did not show any activity [Citation136]. These findings replicate results from earlier studies in breast cancer where CDKN2A mutation status did not predict activity of CDK4/6 inhibition [Citation137,Citation138]. Other genetic alterations of the RB pathway like amplifications of CDK4 or cyclin D1 (CCND), present in ~9% of all pancreas patients, might be molecular markers with better predictive accuracy [Citation90]. Combinations of the CDK4/6 inhibitor abemaciclib with the pan-PI3K inhibitor samotolisib or the pan-CDK inhibitor dinaciclib with the AKT inhibitor MK-2206, which aimed at dual, vertical pathway inhibition of the CDK4/6-regulating cyclin D1-PI3K/mTOR signaling axis, showed in unselected PDAC patients no activity (NCT02981342) [Citation82,Citation139]. Emerging strategies with CDK inhibitors focus on the use of selective, 2nd generation non-ATP competitive agents combined with cytotoxic chemotherapy. These include the CDK4/6 inhibitors SHR6390 or the CDK7 inhibitor SY-5609 in combination with gemcitabine nab-paclitaxel (NCT05185869; NCT04247126). Recruitment status of the phase I/II studies of BEY1107 with gemcitabine (NCT03579836) of palbociclib in combination with cisplatin (NCT02897375) is currently unknown. CDC7 is a replication checkpoint initiating DNA replication at S phase entry and cooperates with CDK1 to control G1-S transition () [Citation124,Citation140]. Rapidly dividing cancer cells display higher levels of stress including replication stress, and depletion or inhibition of CDC7 triggers cell death. These findings and the observation that CDC7 is a largely non-essential factor in normal cells makes CDC7 an attractive target [Citation140]. Results from ongoing phase I studies of LY3143921 are currently awaited (NCT03096054).
7.7. Tumor microenvironment and immuno-oncology kinase inhibition
7.7.1. JAK/STAT pathway and TGFβ receptor inhibition
Protein kinase inhibitors have been increasingly employed to disrupt the complex cancer cell-TME interface targeting pro-survival signals from dysregulated ECM components and tumor-edited stromal cells including immune cells [Citation113,Citation141]. Targeting JAK/STAT signaling, which is frequently activated in PDAC and associated with poor outcome, is expected to suppress pro-tumor cues from a multitude of stromal cells in the TME of PDAC [Citation141,Citation142]. JAK/STAT activation in PDAC is a product of activation of multiple cytokine receptors in the inflammatory tumor environment involving autocrine, paracrine signaling or the lack of sequestration of cytokine receptor ligands in the abnormal ECM characterized by an increased turnover rate [Citation141]. These include, among many other examples, interleukin 6 (IL-6)-mediated STAT3 activation which expands immune suppressive myeloid-derived suppressor cells (MDSCs) or M2-like tumor-associate macrophages, promotion of the inflammatory phenotype of CAFs, or the paclitaxel-induced TLR4 activation on stromal mononuclear cells including B cells leading to NF-kB activation and IL-6, −8, or VEGF secretion which suggest a strong rationale to use JAK/STAT inhibitors to sensitize pancreatic tumors to chemotherapy [Citation143–146]. Two large, randomized phase III studies compared the JAK1/2 inhibitor ruxolitinib in combination gemcitabine with gemcitabine alone and did not show any improvement in OS or PFS [Citation147]. While outcome with the JAK1/2 kinase inhibitors was improved in patients with serum C-reactive protein levels ≥3 mg/mL, no further development of this approach is currently undertaken. Because of these disappointing findings a study with the JAK1 inhibitor itacitinib in combination with gemcitabine nab-paclitaxel, which yielded an ORR of 28.6%, was terminated early [Citation95,Citation148]. It is possible that due to its cellular signaling pleiotropism, JAK inhibition is not universally associated with anti-tumor effects in all stromal cell populations and can induce detrimental effects in some. For example, JAK inhibition shifts the inflammatory phenotype of CAFs toward a myofibroblastic phenotype which is associated with increased TGFβ receptor signaling, tumor collagen content, and decreased tumor perfusion including perfusion with gemcitabine [Citation33,Citation47].
7.7.2. Transforming growth factor-beta signaling
A multitude of elegant preclinical studies has now established that stroma-derived TGFβ signaling is an oncogenic, tumor-promoting cytokine in the later, evolved stages of pancreas cancer [Citation149]. TGFβ signaling induces an EMT-like phenotype in pancreas cancer cells which is associated with resistance to cytotoxic chemotherapy, promotes metastasis, activates CAFs to secrete ECM components which increase interstitial pressure, expand pericytes surrounding tumor vasculature, and overall diminish tumoral perfusion and drug delivery [Citation34,Citation50,Citation113]. Pro-tumoral immune suppression mediated by TGFβ signaling includes, among many other effects, maintenance of a M2-like phenotype of tumor associated macrophages (TAMs), expansion of MDSCs, or reprogramming of CD4 T effector cells toward T regulatory cells [Citation34]. The randomized phase II study of the TGFβ receptor I inhibiting small molecule galunisertib in combination with gemcitabine did not show any improvement in outcome compared to gemcitabine alone (median OR 8.9 months vs 7.1 months; HR 0.79) [Citation150]. A follow-up study of galunisertib in combination with the PD-L1 inhibitor durvalumab equally failed to yield an efficacy signal [Citation151]. Studies with vactosertib, a small molecule with improved pharmacokinetics targeting TGFβ receptor I, have not yet started accruing or recruitment status is unknown (NCT04258072; NCT03666832). The not yet recruiting phase III study comparing chemotherapy to chemotherapy plus the anti-connective tissue growth factor (CTGF) blocker pamrevlumab (FG-3019) in locally advanced pancreas cancer will aid investigation of the role of TGFβ disruption in PDAC (NCT03941093). CTGF, which is under direct control of the TGF receptor promotor, is mediating many of TGFβ’s pro-stroma and pro-tumor effects [Citation152]. Equally, results of the phase II study of the anti-TGF-β monoclonal antibody (mAb) NIS793 with or without the PD-1 inhibitor spartalizumab in combination with nab-paclitaxel/gemcitabine (NG) versus NG alone which has started to accrue patients with metastatic PDAC will aid to clarify the role of targeting TGFβ signaling (NCT04390763). It is possible that TGFβ receptor kinase blockade via ligation of TGFβ ligands with mAbs will have different effects and show different results than TGFβ receptor kinase inhibition with small molecules.
7.7.3. Vascular endothelial & platelet-derived growth factor receptor pathway inhibition
Pancreatic tumors with their dense, desmoplastic stroma and elevated interstitial pressures are a prototype of hypoxic tumors inducing a pro-angiogenic environment characterized by activation of hypoxia-inducible factor (HIF) which activates VEGF, PDGF signaling, angiopoietin, complement as well as others [Citation153]. Despite the near universal overexpression of VEGFR2 and other angiogenic factors in pancreas cancer, multiple anti-VEGF trials including bevacizumab, axitinib, or vatalanib, either as monotherapy, or in combination with gemcitabine or erlotinib, have shown negative results () [Citation154]. Anti-VEGF/VEGFR therapy reduces endothelial fenestrations present in abnormal tumor vasculature, abrogates vascular sprouting, reduces pericyte coverage, hyperpermeability, interstitial pressures, and overall reduces vascular density and blood flow to the tumor [Citation155,Citation156]. These effects are generally short-lived and despite of reducing blood flow of up to 70% frequently not all tumor cells are killed [Citation156]. It has been postulated that surviving cancer cells further upregulate in response to hypoxia, factors like FGF, PDGF-BB, HGF, Ang-2 and others, with cancer cells converting into a more CSC-like phenotype when residing in hypoxic niches [Citation125,Citation153]. Increasing hypoxia after the initial anti-VEGF effect might shift TAMs again into a M2-like phenotype associated with immune suppression [Citation153]. Anti-VEGF agents were frequently multikinase small molecule inhibitors with an extended target profile against PDGFR, FGFR, or others and were tested either alone, or in combination with gemcitabine-based chemotherapy [Citation157–162]. Disappointingly, none of these studies produced an efficacy signal including the large, randomized phase II and III studies with vandetanib (median OS 8.83 months vs 8.95 months; HR 1.21) or axitinib (8.5 months vs 8.3 months; HR 1.014) as did a number of initial studies with the anti-VEGF mAb bevacicumab [Citation157,Citation159]. Emerging protein kinase inhibitors targeting VEGF receptor signaling are testing VEGFR inhibitors in combination with PD-1 immune checkpoint blockers (NCT04415385; NCT04887805; NCT04803851; NCT05193604). Normalization of intratumoral perfusion and normalization of hypoxia recalibrates the intratumoral immune landscape toward an anti-tumor phenotype and improves responses to immune checkpoint blockade [Citation153]. Other trials are testing VEGFR inhibitors in combination with gemcitabine nab-paclitaxel chemotherapy (NCT05168527; NCT04814485).
PDGFs are another pleiotropic class of growth factors that are able to activate tumor cells and exert tumor cell-promoting functions in a variety of stromal cells including perivascular cells, cancer associated fibroblasts (CAFs), or MDSCs [Citation163,Citation164]. Despite promising findings in preclinical models of pancreas cancer clinical results with multikinase inhibitors targeting PDGFR like regorafenib, axitinib, anlotinib, or dovitinib have been disappointing [Citation157,Citation160–162]. This includes a large phase III study if the multikinase inhibitor masitinib. Masitinib targets the adhesion kinases Lyn and Fyn kinase, PDGFR, c-KIT, Lck, FGFR3, or CSF1R and was tested in combination with gemcitabine versus gemcitabine alone in the control arm (HR 0.89; p = 0.7) [Citation165]. Subgroup analysis identified a benefit in patients with elevated baseline plasma acyl-CoA oxidase 1 (ACOS1; defined as a delta cycle threshold value of ≤3.05 of the overall pharmacogenomic population) and baseline pain intensity above 20 mm as measured on a 100 mm visual analogue scale (VAS). The phase III AB12005 follow-up study enrolling previously not treated patients with a VAS>20 and/or a 1 mg/kg/day or greater opioid analgesic dose prospectively validated the initial findings from the subgroup analysis in unresectable locally advanced pancreas patients (median OS 13.0 vs 11.2 months; HR 0.46; P = 0.0047) [Citation166]. It is currently unclear whether the modest increase is sufficient to justify further development, exploration of additional biomarkers like PDGF, FGF, or VEGF has been quoted as desirable.
7.7.4. Bruton’s tyrosine kinase inhibition
Bruton’s tyrosine kinase (BTK) small molecule inhibition targets pro-survival cues in the pancreatic cancer TME by disrupting the pro-tumorigenic impact of intratumoral B cells and the polarization of TAMs toward a M2-like phenotype which was found to be PI3Kγ-dependent [Citation31]. BTK inhibition interfering with this signaling crosstalk increased infiltration of cytotoxic CD8 T cells which was associated with improved tumor control in preclinical models of pancreas cancer [Citation31]. A phase III study of the BTK inhibitor ibrutinib in combination with nab-paclitaxel and gemcitabine did not show an improvement in outcome compared to patients treated with nab-paclitaxel-gemcitabine alone (median of 9.7 months in ibrutinib plus nab-paclitaxel/gemcitabine versus 10.8 months in nab-paclitaxel/gemcitabine group; HR 1.11; p = 0.3225) [Citation167]. Similarly, a follow-up phase IB/II study of ibrutinib and the PD-L1 inhibitor durvalumab yielded no efficacy signal as did recently released findings from early phase clinical testing of the BTK inhibitor acalabrutinib in pancreatic cancer patients (NCT02362048) [Citation168].
8. Potential developmental issues
The indiscriminate administration of protein kinase inhibitors to unselected patients with pancreatic cancer, either as single agent or in combination with systemic cytotoxic chemotherapy, has to-date been met with limited success. Targeting molecularly defined subgroups, in particular with inhibitors shown to have therapeutic merit in other malignancies, has, on the other hand, improved outcome of pancreatic cancer patients with BRCA1 and 2 mutations or tumors harboring gene fusions and aberrations of the NTRK receptor, and both olaparib and larotrectinib/entrectinib have received regulatory approval for the treatment of respective subgroups [Citation23]. Future molecular subgroups are expected to be small as such actionable variants are rare and constitute only a few percent of pancreatic cancer patients. Thus, development of some of these protein kinase inhibitors will be, in part, driven by clinical testing in other malignancies where aberrant genetic markers are more common as pancreatic cancer patients with rare actionable mutations are likely to be enrolled in basket trials which accrue patients across different histologies with the same molecular subtype. The focus toward smaller, molecularly defined subgroups compared to the larger phase II and III of unselected patients conducted previously, will overall restrict market share and carries the risk of limiting investments into future protein kinase inhibitor development in pancreatic cancer. Protein kinase inhibitors are also competing against the rapidly expanding field of immuno-oncology agents as, for example, CD40 agonism or combination studies with cancer vaccines have shown improvements in survival in late phase II clinical trials [Citation169,Citation170]. There is opportunity for synergy between protein kinase inhibitor and immuno-oncology development. While initial studies with immuno-oncology kinase inhibitors or the combination of protein kinase inhibitors and immune checkpoint inhibitors have not shown an improvement of outcome so far, selection of patients was not tailored to subsets of pancreatic cancer patients with actionable stromal or immunological dependencies. Irrespective of the selected strategy, persistent development issues for protein kinase inhibitors in pancreatic cancer which remain to be resolved include the (1) frequent lack of a comprehensive understanding of the biological role of the intended kinase target within and across tumoral and stromal cell populations, (2) overcoming the unique stromal barrier characterized by general hypoperfusion, poor drug penetration, and the uncertainty of target engagement, and (3) the need for detailed PK/PD correlations for a thorough understanding of acquired mechanisms of resistance.
9. Conclusions
A multitude of phase II and III clinical studies which examined single agent protein kinase inhibitors, or kinase inhibitors in combination with systemic cytotoxic chemotherapy, did not yield an efficacy signal in unselected pancreas cancer patients. There has been a shift toward the testing of protein kinase inhibitors in molecularly defined subtypes, in particular after the approval of larotrectinib and entrectinib for TRK fusion positive tumors or olaparib for maintenance therapy of pancreatic cancer patients with BRCA1 and 2 germline mutations. Considering the low frequency of currently actionable mutations in pancreatic cancer, some of the early phase genotype-directed clinical trials enrolling pancreatic cancers with KRASG12C mutational isoforms, FGFR genome variants, or defects in DDR pathways are carried out as basket trials which are enrolling patients across different cancer histologies. Other emerging strategies include the employment of protein kinase inhibitors to sensitize immunologically cold pancreatic tumors to immune checkpoint inhibitors or other cancer immunotherapy agents via targeting signal transduction in stromal components and pro-tumoral immune cell populations.
10. Expert opinion
The trend toward the development of protein kinase inhibitors in molecularly defined subgroups and toward greater individualization of pancreatic cancer therapy is likely going to accelerate and dominate the next decade of protein kinase inhibitor development. This includes targeting of tumors which harbor the KRAS mutational isoform G12C. Several rationale-designed combination studies with KRASG12C inhibitors aim to suppress resistance from KRASG12C inhibition and are already enrolling patients including pancreatic cancer patients. RET or ALK kinase inhibitors which have established efficacy profiles on other solid organ malignancies have shown anecdotal benefit in pancreatic cancer patients harboring ultrarare RET amplifications or ALK rearrangements (≤1% of PDAC) [Citation171,Citation172]. Thus, these and several other genetic alterations shown to be targets of kinase inhibitors in other malignancies might be low-hanging fruits for the rapid development of other genotype-directed kinase therapy options. However, considering the overall paucity of these alterations, that as of now the vast majority of KRAS mutational isoforms remain undruggable, and that the predominant common genetic alterations in this disease are tumor suppressors, the impact of these advances on overall outcome of pancreas cancer is likely to be modest.
Protein kinase inhibitors will be increasingly employed to target kinases involved in immune regulation and against targets in the TME. While none of the recent phase II and III trials evaluating the merit of anti-stroma anti-cancer combination therapies has shown any benefit to-date, the unique biology of the pancreatic cancer stroma with its multitude of pro-survival functions including its immunologic privilege, impairment of drug delivery, and mediation of resistance makes it unlikely that single-agent kinase inhibition employed solely against the cancer cell compartment will surpass current results from systemic chemotherapy. Future clinical studies on protein kinase inhibitors should be required to incorporate robust PD markers to evaluate target engagement. While recent analyses are reporting a decrease in the attrition rates of kinase inhibitors, opportunities to accelerate protein kinase inhibitor development include the incorporation of predictive biomarkers for both toxicity and efficacy, innovative dose escalation designs, and treatment selection and decisions based on individual patients’ profiles. After a target has been shown in clinical and correlative endpoints to not improve outcomes, the field should abstain from repeat testing of similar protein kinase inhibitors which offer no pharmacological or mechanism-of-action advantage. Many dysregulated kinases in cancer including in pancreatic cancer of the human kinome remain unexplored and the development of small molecule inhibitors against these novel targets is an area which holds great promise. Similarly, advances in medicinal chemistry like the design of kinase degraders which license targeted kinases to the proteosome and might carry mechanistic and pharmacological advantages over current non-covalent kinase inhibitors are exciting developments and clinical evaluation is eagerly awaited. Leveraging the already available knowledge, including recent findings form single cell sequencing studies, on the cellular, immunologic, genetic, and metabolomic make-up of these tumors will be pivotal for the effective translation and the overall success of protein kinase inhibitors in this disease.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Surveillance Epidemiology and End Results (SEER). Cancer Stat Facts: Pancreatic Cancer. (MD). cancer statistics/reports on cancer/ cancer stat facts: pancreatic cancer 2022. Bethesda: Program: U.S. Department of Health and Human Services, N.I.o.H., National Cancer Institute; 2022.
- Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921.
- Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33.
- Ma J, Jemal A. The rise and fall of cancer mortality in the USA: why does pancreatic cancer not follow the trend? Future Oncol. 2013;9(7):917–919.
- Park W, Chawla A, O’Reilly EM. Pancreatic Cancer: a Review. JAMA. 2021;326(9):851–862.
- Blackford AL, Canto MI, Klein AP, et al. Recent trends in the incidence and survival of stage 1A pancreatic cancer: a surveillance, epidemiology, and end results analysis. J Natl Cancer Inst. 2020;112(11):1162–1169.
- Kardosh A, Lichtensztajn DY, Gubens MA, et al. Long-term survivors of pancreatic cancer: a California population-based study. Pancreas. 2018;47(8):958–966.
- Bauer MR, Bright EE, MacDonald JJ, et al. Quality of life in patients with pancreatic cancer and their caregivers: a systematic review. Pancreas. 2018;47(4):368–375.
- Zabora J, BrintzenhofeSzoc K, Curbow B, et al. The prevalence of psychological distress by cancer site. Psychooncology. 2001;10(1):19–28.
- Beesley VL, Wockner LF, O‘Rourke P, et al. Risk factors for current and future unmet supportive care needs of people with pancreatic cancer. A longitudinal study. Support Care Cancer. 2016;24(8):3589–3599.
- Nakchbandi IA, Lohr JM. Coagulation, anticoagulation and pancreatic carcinoma. Nat Clin Pract Gastroenterol Hepatol. 2008;5(8):445–455.
- Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19): 1817–1825.
- Burris HA, Moore MJ, Andersen J. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 3rd, et al. 1997;15(6):2403–2413.
- Von Hoff DD, Ervin T, Arena FP, et al., Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703.
- Wang-Gillam A, Li C-P, Bodoky G, et al., Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet. 2016;387(10018): 545–557.
- Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the national cancer institute of canada clinical trials group. J Clin Oncol. 2007;25(15):1960–1966.
- Shin S, Park CM, Kwon H. Erlotinib plus gemcitabine versus gemcitabine for pancreatic cancer: real-world analysis of Korean national database. BMC Cancer. 2016;16:443.
- Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520.
- Boyiadzis MM, Kirkwood JM, Marshall JL, et al. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6(1):35.
- Marabelle A, Le DT, Ascierto PA, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: results From the Phase II KEYNOTE-158 Study. J Clin Oncol. 2020;38(1):1–10.
- Luchini C, Brosens LAA, Wood LD, et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: histology, molecular pathology and clinical implications. Gut. 2021;70(1):148–156.
- Golan T, Hammel P, Reni M, et al., Maintenance olaparib for germline BRCA -mutated metastatic pancreatic cancer. N Engl J Med. 2019;381(4):317–327.
- Fink J. Genomic testing makes inroads after first-line therapy in metastatic pancreatic cancer Targeted Therapies in Oncology. Targeted Therapies in Oncology. 2019;9(16):72.
- Drilon A, Laetsch TW, Kummar S, et al. Efficacy of Larotrectinib in TRK Fusion–Positive cancers in adults and children. N Engl J Med. 2018;378(8):731–739.
- Doebele RC, Drilon A, Paz-Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020;21(2):271–282.
- Attwood MM, Fabbro D, Sokolov AV, et al. Trends in kinase drug discovery: targets, indications and inhibitor design. Nat Rev Drug Discov. 2021;20(11):839–861.
- Wilson LJ, Linley A, Hammond DE, et al. New perspectives, opportunities, and challenges in exploring the human protein kinome. Cancer Res. 2018;78(1):15–29.
- Klaeger S, Heinzlmeir S, Wilhelm M, et al. The target landscape of clinical kinase drugs. Science. 2017;358(6367):6367.
- Pancreatic cancer treatment market size industry report 2018-2025. GVR-1-68038-774-2. San Francisco, CA: Grand View Research; 2017.
- Llombart V, Mansour MR. Therapeutic targeting of “undruggable” MYC. Vol. 75. EBioMedicine: Elsevier: 2022.103756
- Gunderson AJ, Kaneda MM, Tsujikawa T. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discovery. 2016;6(3):270–285.
- Jiang H, Hegde S, Knolhoff BL, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–860.
- Biffi G, Oni TE, Spielman B, et al. IL1-Induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9(2):282–301
- Principe DR, Timbers KE, Atia LG, et al. TGFbeta signaling in the pancreatic tumor microenvironment. Cancers (Basel). 2021;13(20):5086.
- Sun X, Gao H, Yang Y, et al. PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther. 2019;4:64.
- Lu H, Zhou Q, He J, et al. Recent advances in the development of protein-protein interactions modulators: mechanisms and clinical trials. Signal Transduct Target Ther. 2020;5(1):213.
- Zhong L, Li Y, Xiong L, et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduct Target Ther. 2021;6(1):201.
- Rosell R, Aguilar A, Pedraz C, et al. KRAS inhibitors, approved. Nat Cancer. 2021;2(12):1254–1256.
- Wang Z, Hausmann S, Lyu R, et al. SETD5-coordinated chromatin reprogramming regulates adaptive resistance to targeted pancreatic cancer therapy. Cancer Cell. 2020;37(6):834–849 e13.
- Steele NG, Carpenter ES, Kemp SB, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat Cancer. 2020;1(11):1097–1112.
- Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806.
- Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52.
- Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17(5):353–377.
- Lu Y, Sun H. Progress in the development of small molecular inhibitors of focal adhesion kinase (FAK). J Med Chem. 2020;63(23):14382–14403.
- Hakim N, Patel R, Devoe C, et al. Why HALO 301 failed and implications for treatment of pancreatic cancer. Pancreas. 2019;3(1):e1–e4.
- Neesse A, Frese KK, Bapiro TE, et al. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc Natl Acad Sci U S A. 2013;110(30):12325–12330.
- Li D, Schaub N, Guerin TM, et al. T cell-mediated antitumor immunity cooperatively induced By TGFbetaR1 antagonism and gemcitabine counteracts reformation of the stromal barrier in pancreatic cancer. Mol Cancer Ther. 2021;20(10):1926–1940.
- Zhao Z, Ukidve A, Kim J, et al. Targeting Strategies for Tissue-Specific Drug Delivery. Cell. 2020;181(1):151–167.
- Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461.
- Kano MR, Bae Y, Iwata C, et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci U S A. 2007;104(9):3460–3465.
- Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26(13):2139–2146.
- Do K, Speranza G, Bishop R, et al. Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Invest New Drugs. 2015;33(3):720–728.
- Bedard PL, Tabernero J, Janku F, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 2015;21(4):730–738.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972.
- Mustachio LM, Chelariu-Raicu A, Szekvolgyi L, et al. Targeting KRAS in cancer: promising therapeutic strategies. Cancers (Basel). 2021;13(6):1204.
- Philip PA, Benedetti J, Corless CL, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest oncology group-directed intergroup trial S0205. J Clin Oncol. 2010;28(22):3605–3610.
- Safran H, Miner T, Bahary N, et al. Lapatinib and gemcitabine for metastatic pancreatic cancer. A phase II study. Am J Clin Oncol. 2011;34(1):50–52.
- Wu Z, Gabrielson A, Hwang JJ, et al. Phase II study of lapatinib and capecitabine in second-line treatment for metastatic pancreatic cancer. Cancer Chemother Pharmacol. 2015;76(6):1309–1314.
- Cohen SJ, O’Neil BH, Berlin J, et al. A phase 1b study of erlotinib in combination with gemcitabine and nab-paclitaxel in patients with previously untreated advanced pancreatic cancer: an Academic Oncology GI Cancer Consortium study. Cancer Chemother Pharmacol. 2016;77(4):693–701.
- Haas M, Waldschmidt DT, Stahl M, et al. Afatinib plus gemcitabine versus gemcitabine alone as first-line treatment of metastatic pancreatic cancer: the randomised, open-label phase II ACCEPT study of the Arbeitsgemeinschaft Internistische Onkologie with an integrated analysis of the ‘burden of therapy’ method. Eur J Cancer. 2021;146:95–106.
- Cardin DB, Goff L, Li C-I, et al. Phase II trial of sorafenib and erlotinib in advanced pancreatic cancer. Cancer Med. 2014;3(3):572–579.
- Cohen DJ, Love LL, Ryan E, et al. Phase II study of sorafenib with gemcitabine and erlotinib (GES) in first-line advanced pancreatic cancer. J Clin Oncol. 2011;29(4):266–266.
- Ko AH, Bekaii-Saab T, Van Ziffle J, et al. A multicenter, open-label phase ii clinical trial of combined MEK plus EGFR inhibition for chemotherapy-refractory advanced pancreatic adenocarcinoma. Clin Cancer Res. 2016;22(1):61–68.
- Halfdanarson TR, Foster NR, Kim GP, et al. A phase ii randomized trial of panitumumab, erlotinib, and gemcitabine versus erlotinib and gemcitabine in patients with untreated, metastatic pancreatic adenocarcinoma: north central cancer treatment group trial N064B (Alliance). Oncologist. 2019;24(5):589–e160.
- Dent P, Booth L, Roberts JL, et al. Neratinib inhibits hippo/YAP signaling, reduces mutant K-RAS expression, and kills pancreatic and blood cancer cells. Oncogene. 2019;38(30):5890–5904.
- da Cunha Santos G, Dhani N, Tu D, et al. Molecular predictors of outcome in a phase 3 study of gemcitabine and erlotinib therapy in patients with advanced pancreatic cancer: national cancer institute of Canada clinical trials group study PA.3. Cancer. 2010;116(24):5599–5607.
- Boeck S, Jung A, Laubender RP, et al. EGFR pathway biomarkers in erlotinib-treated patients with advanced pancreatic cancer: translational results from the randomised, crossover phase 3 trial AIO-PK0104. Br J Cancer. 2013;108(2):469–476.
- Boeck S, Jung A, Laubender RP, et al. KRAS mutation status is not predictive for objective response to anti-EGFR treatment with erlotinib in patients with advanced pancreatic cancer. J Gastroenterol. 2013;48(4):544–548.
- Bodoky G, Timcheva C, Spigel DR, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012;30(3):1216–1223.
- Jimeno A, Tan AC, Coffa J, et al. Coordinated epidermal growth factor receptor pathway gene overexpression predicts epidermal growth factor receptor inhibitor sensitivity in pancreatic cancer. Cancer Res. 2008;68(8):2841–2849.
- Infante JR, Somer BG, Park JO, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014;50(12):2072–2081.
- Goncalves A, Gilabert M, François E, et al. BAYPAN study: a double-blind phase III randomized trial comparing gemcitabine plus sorafenib and gemcitabine plus placebo in patients with advanced pancreatic cancer. Ann Oncol. 2012;23(11):2799–2805.
- Van Cutsem E, Hidalgo M, Canon J-L, et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int J Cancer. 2018;143(8):2053–2064.
- Chung V, McDonough S, Philip PA, et al., Effect of Selumetinib and MK-2206 vs oxaliplatin and fluorouracil in patients with metastatic pancreatic cancer after prior therapy: SWOG S1115 study randomized clinical trial. JAMA Oncol. 2017;3(4):516–522.
- Tolcher AW, Bendell JC, Papadopoulos KP, et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann Oncol. 2015;26(1):58–64.
- LoRusso P, De Vos FYFL FM, Beck JT, et al. Phase Ib study of ribociclib (R) + trametinib (T) in patients (pts) with metastatic/advanced solid tumours. Ann Oncol. 2020;31:S484.
- Van Laethem JL, Riess H, Jassem J, et al. Phase I/II study of refametinib (BAY 86-9766) in combination with gemcitabine in advanced pancreatic cancer. Target Oncol. 2017;12(1):97–109.
- Kenney C, Kunst T, Webb S, et al. Phase II study of selumetinib, an orally active inhibitor of MEK1 and MEK2 kinases, in KRAS(G12R)-mutant pancreatic ductal adenocarcinoma. Invest New Drugs. 2021;39(3):821–828.
- Bryant KL, Stalnecker CA, Zeitouni D, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25(4):628–640.
- Hobbs GA, Baker NM, Miermont AM, et al. Atypical KRASG12R mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer Discov. 2020;10(1):104–123.
- Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014;5(15):6512–6525.
- Franco J, Balaji U, Freinkman E, et al. Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep. 2016;14(5):979–990.
- Poulikakos PI, Solit DB. Resistance to MEK inhibitors: should we co-target upstream? Sci Signal. 2011;4(166):e16.
- Fedele C, Ran H, Diskin B, et al., SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 2018;8(10): 1237–1249.
- Ruess DA, Heynen GJ, Ciecielski KJ, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 2018;24(7):954–960.
- Ahmed TA, Adamopoulos C, Karoulia Z, et al. SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Rep. 2019;26(1):65–78 e5.
- Ryan MB, Fece de la Cruz F, Phat S, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRAS(G12C) Inhibition. Clin Cancer Res. 2020;26(7):1633–1643.
- Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223.
- Fedele C, Li S, Teng KW, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med. 2021;218(1). 10.1084/jem.20201414.
- Witkiewicz AK, McMillan EA, Balaji U, et al., Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6(1): 6744.
- Ying H, Elpek KG, Vinjamoori A, et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discov. 2011;1(2):158–169.
- McRee AJ, Sanoff HK, Carlson C, et al. A phase I trial of mFOLFOX6 combined with the oral PI3K inhibitor BKM120 in patients with advanced refractory solid tumors. Invest New Drugs. 2015;33(6):1225–1231.
- O’Neil BH, Scott AJ, Ma WW, et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann Oncol. 2015;26(12):2505.
- Richards DA, Kuefler PR, Becerra C, et al. Gemcitabine plus enzastaurin or single-agent gemcitabine in locally advanced or metastatic pancreatic cancer: results of a phase II, randomized, noncomparative study. Invest New Drugs. 2011;29(1):144–153.
- Naing A, Powderly JD, Nemunaitis JJ, et al. Exploring the safety, effect on the tumor microenvironment, and efficacy of itacitinib in combination with epacadostat or parsaclisib in advanced solid tumors: a phase I study. J Immunother Cancer. 2022;10(3):e004223.
- Wolpin BM, Hezel AF, Abrams T, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol. 2009;27(2):193–198.
- Kordes S, Richel DJ, Klümpen H-J, et al. A phase I/II, non-randomized, feasibility/safety and efficacy study of the combination of everolimus, cetuximab and capecitabine in patients with advanced pancreatic cancer. Invest New Drugs. 2013;31(1):85–91.
- Karavasilis V, Samantas E, Koliou G-A, et al. Gemcitabine combined with the mTOR inhibitor temsirolimus in patients with locally advanced or metastatic pancreatic cancer. A hellenic cooperative oncology group phase I/II study. Target Oncol. 2018;13(6):715–724.
- Conway JR, Herrmann D, Evans TJ, et al. Combating pancreatic cancer with PI3K pathway inhibitors in the era of personalised medicine. Gut. 2019;68(4):742–758.
- Andre F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA -Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N Engl J Med. 2019;380(20):1929–1940.
- Hu C, Dadon T, Chenna V, et al. Combined inhibition of cyclin-dependent kinases (Dinaciclib) and AKT (MK-2206) blocks pancreatic tumor growth and metastases in patient-derived xenograft models. Mol Cancer Ther. 2015;14(7):1532–1539.
- Hochwald SN, Nyberg C, Zheng M, et al. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 2009;8(15):2435–2443.
- Murphy JM, Rodriguez YAR, Jeong K, et al. Targeting focal adhesion kinase in cancer cells and the tumor microenvironment. Exp Mol Med. 2020;52(6):877–886.
- Aung KL, Welch ME, Wang S, et al. A phase II trial of GSK2256098 and trametinib in patients with advanced pancreatic ductal adenocarcinoma (PDAC) (MOBILITY-002 Trial, NCT02428270). J Clin Oncol. 2018;36(4):409–409.
- Wang-Gillam A, Lockhart MR, Tan AC, et al. Phase I study of defactinib combined with pembrolizumab and gemcitabine in patients with advanced cancer: experiences of pancreatic ductal adenocarcinoma (PDAC) patients. Cancer Res. 2020;80(16_Supplement):CT118–CT118. abstract CT118.
- Evans TRJ, Van Cutsem E, Moore MJ, et al. Phase 2 placebo-controlled, double-blind trial of dasatinib added to gemcitabine for patients with locally-advanced pancreatic cancer. Ann Oncol. 2017;28(2):354–361.
- Chee CE, Krishnamurthi S, Nock CJ, et al. Phase II study of dasatinib (BMS-354825) in patients with metastatic adenocarcinoma of the pancreas. Oncologist. 2013;18(10):1091–1092.
- George TJ, Ali A, Wang Y, et al. Phase II study of 5-fluorouracil, oxaliplatin plus dasatinib (FOLFOX-D) in first-line metastatic pancreatic adenocarcinoma. Oncologist. 2021;26(10):825–e1674.
- Messersmith WA, Arcaroli NS, Tan J, et al. A phase II trial of saracatinib (AZD0530), an oral Src inhibitor, in previously treated metastatic pancreatic cancer. J Clin Oncol. 2010;28(15):e14515–e14515.
- Renouf DJ, Moore MJ, Hedley D, et al. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Invest New Drugs. 2012;30(2):779–786.
- Subbiah V, Iannotti NO, Gutierrez M, et al., FIGHT-101, a first-in-human study of potent and selective FGFR 1-3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann Oncol. 2022;33(5): 522–533.
- Lenkiewicz E, Malasi S, Hogenson TL, et al. Genomic and epigenomic landscaping defines new therapeutic targets for adenosquamous carcinoma of the pancreas. Cancer Res. 2020;80(20):4324–4334.
- Thomas D, Radhakrishnan P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer. 2019;18(1):14.
- Pothula SP, Xu Z, Goldstein D, et al. Targeting HGF/c-MET Axis in Pancreatic Cancer. Int J Mol Sci. 2020;21(23):9170.
- Hage C, Rausch V, Giese N, et al. The novel c-Met inhibitor cabozantinib overcomes gemcitabine resistance and stem cell signaling in pancreatic cancer. Vol. 4. London, UK: Cell Death Dis; 2013. p. e627.
- Li C, Wu J, Hynes M, et al. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology. 2011;141(6):2218–2227 e5.
- Zhen DB, Griffith KA, Ruch JM, et al. A phase I trial of cabozantinib and gemcitabine in advanced pancreatic cancer. Invest New Drugs. 2016;34(6):733–739.
- Du W, Brekken RA. Does Axl have potential as a therapeutic target in pancreatic cancer? Expert Opin Ther Targets. 2018;22(11):955–966.
- Bellomo G, Rainer C, Quaranta V, et al. Chemotherapy-induced infiltration of neutrophils promotes pancreatic cancer metastasis via Gas6/AXL signalling axis. Gut. 2022;8:228–229.
- Perkhofer L, Gout J, Roger E, et al. DNA damage repair as a target in pancreatic cancer: state-of-the-art and future perspectives. Gut. 2021;70(3):606–617.
- Crowley F, Park W, O’Reilly EM. Targeting DNA damage repair pathways in pancreas cancer. Cancer Metastasis Rev. 2021;40(3):891–908.
- Javle M, Shacham-Shmueli E, Xiao L, et al., Olaparib monotherapy for previously treated pancreatic cancer with DNA damage repair genetic alterations other than germline BRCA variants. JAMA Oncol. 2021;7(5): 693–699.
- Bian Y, Teper Y, Mathews Griner LA, et al. Target deconvolution of a multikinase inhibitor with antimetastatic properties identifies TAOK3 as a key contributor to a cancer stem cell-like phenotype. Mol Cancer Ther. 2019;18(11):2097–2110.
- Vitale I, Manic G, De Maria R, et al. DNA Damage in Stem Cells. Mol Cell. 2017;66(3):306–319.
- Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225–238.
- Laquente B, Lopez-Martin J, Richards D, et al. A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC Cancer. 2017;17(1):137.
- Mross K, Dittrich C, Aulitzky WE, et al. A randomised phase II trial of the Polo-like kinase inhibitor BI 2536 in chemo-naive patients with unresectable exocrine adenocarcinoma of the pancreas - a study within the Central European Society Anticancer Drug Research (CESAR) collaborative network. Br J Cancer. 2012;107(2):280–286.
- Thomas A, Redon CE, Sciuto L, et al. Phase I study of ATR inhibitor M6620 in combination with topotecan in patients with advanced solid tumors. J Clin Oncol. 2018;36(16):1594–1602.
- Thomas A, Takahashi N, Rajapakse VN, et al. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell. 2021;39(4):566–579 e7.
- Ghelli Luserna Di Rora A, Cerchione C, Martinelli G, et al. A WEE1 family business: regulation of mitosis, cancer progression, and therapeutic target. J Hematol Oncol. 2020;13(1):126.
- O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13(7):417–430.
- Carvajal RD, Tse A, Shah MA, et al. A phase II study of flavopiridol (Alvocidib) in combination with docetaxel in refractory, metastatic pancreatic cancer. Pancreatology. 2009;9(4):404–409.
- Gansauge S, Gansauge F, Ramadani M, et al. Overexpression of cyclin D1 in human pancreatic carcinoma is associated with poor prognosis. Cancer Res. 1997;57(9):1634–1637.
- Kornmann M, Arber N, Korc M. Inhibition of basal and mitogen-stimulated pancreatic cancer cell growth by cyclin D1 antisense is associated with loss of tumorigenicity and potentiation of cytotoxicity to cisplatinum. J Clin Invest. 1998;101(2):344–352.
- Salvador-Barbero B, Alvarez-Fernández M, Zapatero-Solana E, et al. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell. 2020;38(4):584.
- Al Baghdadi T, Halabi S, Garrett-Mayer E, et al. Palbociclib in Patients With Pancreatic and Biliary Cancer With CDKN2A Alterations: results From the Targeted Agent and Profiling Utilization Registry Study. JCO Precis Oncol. 2019;3:1–8.
- Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16(1):25–35.
- Andre F, Campone SS, Petrakova M, et al. Ribociclib + letrozole for first-line treatment of hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2-) advanced breast cancer (ABC): efficacy by baseline tumor markers. Cancer Res. 2017;77(13):CT045–CT045.
- Murphy AG, Zahurak M, Shah M, et al. A phase i study of dinaciclib in combination with MK-2206 in patients with advanced pancreatic cancer. Clin Transl Sci. 2020;13(6):1178–1188.
- Suski JM, Ratnayeke N, Braun M, et al. CDC7-independent G1/S transition revealed by targeted protein degradation. Nature. 2022;605(7909):357–365.
- Nagathihalli NS, Castellanos JA, Shi C, et al. Signal transducer and activator of transcription 3, mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic cancer. Gastroenterology. 2015;149(7):1932–1943 e9.
- Denley SM, Jamieson NB, McCall P, et al. Activation of the IL-6R/Jak/stat pathway is associated with a poor outcome in resected pancreatic ductal adenocarcinoma. J Gastrointest Surg. 2013;17(5):887–898.
- White CM, Martin BK, Lee L-F, et al. Effects of paclitaxel on cytokine synthesis by unprimed human monocytes, T lymphocytes, and breast cancer cells. Cancer Immunol Immunother. 1998;46(2):104–112.
- Rajput S, Volk-Draper LD, Ran S. TLR4 is a novel determinant of the response to paclitaxel in breast cancer. Mol Cancer Ther. 2013;12(8):1676–1687.
- Karakasheva TA, Lin EW, Tang Q, et al. IL-6 mediates cross-talk between tumor cells and activated fibroblasts in the tumor microenvironment. Cancer Res. 2018;78(17):4957–4970.
- Sheridan C. Pancreatic cancer provides testbed for first mechanotherapeutics. Nat Biotechnol. 2019;37(8):829–831.
- Hurwitz H, Van Cutsem E, Bendell J, et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Invest New Drugs. 2018;36(4):683–695.
- Beatty GL, Shahda S, Beck T, et al. A phase Ib/II study of the JAK1 Inhibitor, Itacitinib, plus nab -paclitaxel and gemcitabine in advanced solid tumors. Oncologist. 2019;24(1):14–e10.
- Principe DR, DeCant B, Mascariñas E, et al. TGFbeta signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 2016;76(9):2525–2539.
- Melisi D, Garcia-Carbonero R, Macarulla T, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer. 2018;119(10):1208–1214.
- Melisi D, Oh D-Y, Hollebecque A, et al. Safety and activity of the TGFbeta receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J Immunother Cancer. 2021;9(3):e002068.
- Chen Z, Zhang N, Chu HY, et al. Connective tissue growth factor: from molecular understandings to drug discovery. Front Cell Dev Biol. 2020;8:593269.
- Mpekris F, Voutouri C, Baish JW, et al. Combining microenvironment normalization strategies to improve cancer immunotherapy. Proc Natl Acad Sci U S A. 2020;117(7):3728–3737.
- Van Cutsem E, Vervenne WL, Bennouna J, et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol. 2009;27(13):2231–2237.
- Tong RT, Boucher Y, Kozin SV, et al. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64(11):3731–3736.
- Sitohy B, Nagy JA, Dvorak HF. Anti-VEGF/VEGFR therapy for cancer: reassessing the target. Cancer Res. 2012;72(8):1909–1914.
- Kindler HL, Ioka T, Richel DJ, et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: a double-blind randomised phase 3 study. Lancet Oncol. 2011;12(3):256–262.
- Dragovich T, Laheru D, Dayyani F, et al. Phase II trial of vatalanib in patients with advanced or metastatic pancreatic adenocarcinoma after first-line gemcitabine therapy (PCRT O4-001). Cancer Chemother Pharmacol. 2014;74(2):379–387.
- Middleton G, Palmer DH, Greenhalf W, et al. Vandetanib plus gemcitabine versus placebo plus gemcitabine in locally advanced or metastatic pancreatic carcinoma (ViP): a prospective, randomised, double-blind, multicentre phase 2 trial. Lancet Oncol. 2017;18(4):486–499.
- Ma WW, Xie H, Fetterly G, et al. A Phase Ib Study of the FGFR/VEGFR inhibitor dovitinib with gemcitabine and capecitabine in advanced solid tumor and pancreatic cancer patients. Am J Clin Oncol. 2019;42(2):184–189.
- Wu H, Zhao C HN. Outcomes of anlotinib plus nab-paclitaxel/gemcitabine as first-line treatment for patients with advanced pancreatic adenocarcinoma: a retrospective analysis in China. J Clin Oncol. 2022;40(4):772–783.
- Bozzarelli S, Rimassa L, Giordano L, et al. Regorafenib in patients with refractory metastatic pancreatic cancer: a Phase II study (RESOUND). Future Oncol. 2019;15(35):4009–4017.
- Aoto K, Ito K, Aoki S. Complex formation between platelet-derived growth factor receptor beta and transforming growth factor beta receptor regulates the differentiation of mesenchymal stem cells into cancer-associated fibroblasts. Oncotarget. 2018;9(75):34090–34102.
- Li T, Guo T, Liu H, et al. Plateletderived growth factorBB mediates pancreatic cancer malignancy via regulation of the Hippo/Yesassociated protein signaling pathway. Oncol Rep. 2021;45(1):83–94.
- Deplanque G, Demarchi M, Hebbar M, et al. A randomized, placebo-controlled phase III trial of masitinib plus gemcitabine in the treatment of advanced pancreatic cancer. Ann Oncol. 2015;26(6):1194–1200.
- Ezenfis JHO, Hermine O. Masitinib plus gemcitabine as first-line treatment of pancreatic cancer with pain: results from phase 3 study AB12005. J Clin Oncol. 2021;39(15):4018–4018.
- Tempero M, Oh D-Y, Tabernero J, et al. Ibrutinib in combination with nab-paclitaxel and gemcitabine for first-line treatment of patients with metastatic pancreatic adenocarcinoma: phase III RESOLVE study. Ann Oncol. 2021;32(5):600–608.
- Hong D, Rasco D, Veeder M, et al. A phase 1b/2 Study of the bruton tyrosine kinase inhibitor ibrutinib and the PD-L1 inhibitor durvalumab in patients with pretreated solid tumors. Oncology. 2019;97(2):102–111.
- O’Hara MH, Wolff RA ORE, O’Hara MH. Gemcitabine (Gem) and nab-paclitaxel (NP) ± nivolumab (nivo) ± CD40 agonistic monoclonal antibody APX005M (sotigalimab), in patients (Pts) with untreated metastatic pancreatic adenocarcinoma (mPDAC): phase (Ph) 2 final results. J Clin Oncol. 2021;39(15):2497–2505.
- Le DT, Wang-Gillam A, Picozzi V, et al. Safety and Survival With GVAX pancreas prime and listeria Monocytogenes –expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. 2015;33(12):1325–1333.
- Singhi AD, Ali SM, Lacy J, et al. Identification of targetable ALK rearrangements in pancreatic ductal adenocarcinoma. J Natl Compr Canc Netw. 2017;15(5):555–562.
- Subbiah V, Bauer T KB, Bauer T. Abstract CT011: efficacy and safety of selpercatinib in RET fusion-positive cancers other than lung or thyroid cancers. Cancer Res. 2021;81(13):CT011–CT011.