
ABSTRACT
Introduction
Mutations in the RPGR gene are responsible for one of the most prevalent and severe types of retinitis pigmentosa. Gene therapy has shown great promise to treat inherited retinal diseases, and currently, four RPGR gene therapy vectors are being evaluated in clinical trials.
Areas covered
This manuscript reviews the gene therapy products that are in development for X-linked retinitis pigmentosa caused by mutations in RPGR, and the challenges that scientists and clinicians have faced.
Expert opinion
The development of a gene therapy product for RPGR-associated retinal degeneration has been a great challenge due to the incomplete understanding of the underlying genetics and mechanism of action of RPGR, and on the other hand, due to the instability of the RPGR gene. Three of the four gene therapy vectors currently in clinical trials include a codon-optimized version of the human RPGR sequence, and the other vector contains a shortened version of the human RPGR. To date, the only Phase I/II results published in a peer-reviewed journal demonstrate a good safety profile and an improvement in the visual field using a codon optimized version of RPGRORF15.
1. Background
The development of new tools for gene therapy and the better understanding of the pathophysiology of different rare genetic diseases have boosted the gene therapy market over the last years. In 2019, 20 gene therapy products (gene and cell-based) had been approved and over two thousand human gene therapy clinical trials were ongoing worldwide [Citation1]. Inherited retinal diseases (IRDs) are a genetically and clinically heterogeneous group of disorders characterized by a progressive retinal degeneration that leads to severe visual impairment and complete blindness. Gene therapy has shown great promise in proof-of-concept studies and human clinical trial to treat IRDs. The best example to date is Luxturna® (voretigene neparvovec), a gene therapy drug to treat Leber congenital amaurosis type 2 (LCA2) and retinitis pigmentosa type 20, both caused my mutations in the RPE65 gene. Luxturna®, developed by Spark Therapeutics, was approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) in 2017 and 2018, respectively, and constitutes the first and only treatment for an IRD [Citation2–4]. This success that led to the development of an AAV-based gene therapy has brought hope to patients affected by IRDs for which there is no treatment to date.
In the present review, we summarize the development of a gene therapy product for X-linked retinitis pigmentosa (X-linked RP) caused by mutations in the RPGR gene, one of the most prevalent and severe forms of RP. We will review the pathophysiology associated with the disease, the particular characteristics of the RPGR gene, the obstacles to develop a gene therapy vector and the current clinical development.
2. Medical need and existing treatment
The IRDs are the leading cause of vision loss in persons between 15 and 45 years of age, with an estimated incidence of 1:3,000. The loss of sight causes enormous human suffering for the affected individuals and their families. It also represents a public health, social, and economic problem for countries. A study report commissioned by the IRD COUNTS consortium analyzed the socioeconomic burden of IRDs in a base period (2019) in the United Kingdom (UK). 22.1% (£114.1 million) of the cost burden due to IRDs is related to productivity costs (reduce workforce participation, absenteeism, and presentism), whilst wellbeing costs (years of quality of life lost due to IRDs) amount to 38.4% (£196.1 million) [Citation5]. The substantial disease burden and the lack of an effective treatment for these IRDs highlight the need for searching innovative therapies that can stop, delay, or even revert the progression of the disease.
Retinitis Pigmentosa (RP) which is the clinical phenotype of photoreceptor degeneration is the most frequent form of IRD with a prevalence of 1:4,000 cases worldwide [Citation6]. The genetic basis and mutations that cause RP are complex and highly heterogeneous. More than 70 genes have been identified to be linked to RP, with usually different disease-causing mutations reported in each gene [Citation7,Citation8]. Mutations in different genes may lead to the same disease, and different mutations in the same gene may be responsible for several diseases. RP can also occur as part of syndromes, in which other organs are affected. The most common syndromic form of RP is Usher syndrome, characterized by congenital or early-onset hearing impairment followed by vision loss [Citation9]. The second most common syndromic form is Bardet-Biedl syndrome, which besides RP includes obesity, intellectual disability, polydactyly, and kidney abnormalities [Citation10]. RP also presents clinical heterogeneity. The same mutation in different individuals may result in different progression of the disease and clinical manifestations. Generally, the initial degeneration of the rods, which are concentrated in the peripheral retina, causes night blindness and visual field constriction (tunnel vision). In the advanced stage of RP, cones are also affected, causing the loss of visual acuity [Citation11]. Although the rod-cone phenotype is the most common clinical presentation, when the causative gene is expressed in rods and cones, patients can present with a cone-rod dystrophy phenotype. In this case, the cone-rich central region is primarily affected, and the initial clinical manifestations are poor visual acuity, photophobia, or abnormal color vision [Citation12].
Significant advances for the treatment of IRDs have been made in recent years and the future looks more optimistic than in the 20th Century. Although there is no approved effective treatment, there are several options available to manage the progression of the RP condition, regardless the mutation or the gene affected [Citation11]. Some of these options are vitamins and supplements such as vitamin A or DHA, an omega-3 polyunsaturated fatty acid and an antioxidant, which are being studied with the purpose of delaying the progression of the disease by tackling the cellular responses triggered by the genetic mutation, e.g. oxidative stress [Citation13–18]. The results obtained with this type of treatment are not consistent among groups, and more research would be needed to determine their safety and efficacy. On the other hand, the clinical investigation on advanced therapies that encompass gene therapy, cell therapy, and tissue engineering, is very active at the moment [Citation19,Citation20]. These therapies have the potential to prevent and halt the progression of the disease, or even revert the pathological phenotype, constituting a long-lasting cure.
IRDs are excellent candidates for gene and cell therapies – the accessibility of the retina allows relatively noninvasive procedures, the effect of the treatment can be easily monitored and the blood-retinal barrier limits the immunological response to the treatment by reducing systemic spread. Cell therapy aims to inject or implant viable cells (usually progenitor stem cells) that can replace the affected cells in the eye. Photoreceptor replacement therapy involves the differentiation of stem cells or progenitor cells into photoreceptors, the integration, and survival of these cells in the retina and the establishment of synaptic connectivity to bipolar cells. Over the last years, numerous preclinical studies have been developed to establish protocols to overcome the obstacles and advance toward the generation and characterization of transplantable photoreceptors [Citation21,Citation22]. Several cell therapy clinical trials to treat RP are ongoing ().
Table 1. Cell therapy clinical trials.
Unlike cell therapy, gene therapy is a therapeutic strategy directed against the primary genetic defect. Depending on the mutated gene, the cellular function affected, and the type of the genetic mutation there are three main approaches: (1) gene replacement or augmentation strategy to target genetic mutations resulting in loss of protein function, for example in autosomal recessive disorders – the introduction of a healthy copy of the gene would provide the cell with the lost function; (2) gene silencing to disable the expression of a mutated gene that results in a gain of function, characteristic of autosomal dominant disorders; and (3) genome editing strategies aimed at correcting the altered genetic sequence directly. The field of genome editing, with new and improved techniques being developed constantly, has risen tremendously since it has the potential to treat most of the human diseases [Citation23,Citation24]. There are several gene therapy clinical trials ongoing to treat RP caused by mutations in different genes ().
Table 2. Gene therapy clinical trials.
One of the major contributions to RP is made by mutations in the retinitis pigmentosa GTPase regulator (RPGR) gene, that account for 10–20% of all RP cases and for over 70% of X-lined RP-affected families [Citation25]. Unlike others IRDs whereby the symptoms appear during the adulthood and progress slowly without vision being impaired significantly, RPGR-associated dystrophies are severe with the symptom onset starting during childhood and patients becoming legally blind within the 4th decade of life [Citation26,Citation27]. Mutations in the RPGR gene can be associated with a rod-cone or cone-rod dystrophy phenotype [Citation28,Citation29]. The harshness and the relative high prevalence of RPGR-associated dystrophies make the finding of a treatment highly necessary.
For RPGR-related RP, as for other recessive disorders, the potential benefits of gene replacement therapy are indisputable. The delivery of a functioning copy of RPGR to supplement the mutant gene in surviving photoreceptors would provide these cells with normal RPGR protein and restore function. A potential regeneration mechanism by which the outer segments of the degenerated photoreceptors are rehabilitated after gene therapy has also been suggested [Citation30,Citation31]. Being an X-linked disease, one could assume that only hemizygous males are affected whilst heterozygous females can only carry the mutation and present a mild, subclinical phenotype. However, differences in the X chromosome inactivation ratio result in a phenotypic variability in female carriers. In cases where the normal X chromosome is inactivated, randomly or because some mutations may skew the inactivation process and select the mutant allele, females can present a severe male-pattern phenotype, with clinically significant visual impairment [Citation32–34]. These severely affected females could also benefit from the gene replacement therapy.
Currently, there are four gene therapy clinical trials to treat X-linked RP caused by mutations in RPGR. These studies evaluate the safety and efficacy of the subretinal and intravitreal administration of AAV vectors containing the human RPGR gene in subjects with X-linked RP caused by mutations in RPGRORF15. The main and most important difference between the four studies, besides the route of administration, is the therapeutic vector (viral capsid and transgene) used, which will be discussed in greater detail in the sections below.
3. Current research goals
The design of an optimal gene therapy vector that will deliver a healthy copy of the gene or a repair system to the target cells in the retina involves the selection of a vector (viral, non-viral) that will act as a vehicle, the coding sequence that will be expressed in the patient’s retinal cells and the basic elements required for this expression to happen, i.e. promoter. The delivery of the therapeutic molecule into the cells and the consequent release into the nucleus in a stable manner entails the first challenge. Adeno-associated virus (AAV) is currently the vector of choice for retinal gene therapy – it is the best characterized vector in ophthalmology and has been widely used in gene therapy studies to target photoreceptors, the retinal pigment epithelium (RPE) and ganglion cells [Citation35,Citation36]. AAV vectors present several advantages such as the high transduction efficiency, the ability to transduce non-dividing cells with long-lasting expression, and the target selectivity since different serotypes with tropism for different retinal cell types have been identified. In addition, AAV are versatile vectors and modifications to derive variants of known AAVs with different attributes have been analyzed in several proof-of-concept studies in animal models [Citation37–39]. Although AAV vectors have shown low immunogenicity compared to the other viral vectors, they have the potential to trigger an immune/inflammatory response. This, together with the capacity constraints (packaging limit of 4.7 kilobases), has fostered the search for alternatives to AAV vectors. Non-viral vectors (naked DNA, liposome-DNA complexes, etc) have a better safety profile and a larger cargo capacity; however, the transfection efficiency is lower compared to viral vectors. There is an increasing number of research studies intended for a better understanding of the factors that limit the transfection and to improve the vector engineering (antisense oligonucleotides, minicircle DNA, nanoformulations, etc.) to achieve long-term expression [Citation40,Citation41].
The selection of the optimal promoter is important to limit transgene expression to the target cells and minimize off-target expression. To this effect, ideally, the promoter has to be selectively expressed in the target cells, where it regulates the expression of the gene of interest. The promoter sequence used in the gene therapy vector can also include enhancers such as transcription factor-binding sites, nuclear localization signals or other elements to boost transcription and to increase the level of expression of the therapeutic transgene in the target cells [Citation42,Citation43].
The coding sequence determines the sequence of amino acids of the protein that will be synthetized upon transduction and activation of the promoter. In some cases, altering the human wild-type sequence to be delivered with the gene therapy vector might be appropriate to increase the fidelity and stability of the sequence and reduce the possibility of deleterious events, such as the generation of mutated proteins. In the case of RPGR, given the inherent instability of its sequence, this approach whereby the wild type coding sequence is modified without altering the protein sequence, can be used for the generation of a RPGR gene therapy vector to treat X-linked RP – this will be discussed in more detail in the sections below.
The RPGR gene, located in the short arm of the X chromosome, is composed of 19 exons that, through the mechanism of alternative splicing, can result in up to 19 splice variants. Most of these splice variants are part of the large non-coding transcriptome, and only nine of these transcripts are predicted to encode for proteins [Citation44,Citation45]. The canonical sequence with 19 exons, encodes the 815 amino acid protein RPGR1-19, known as constitutive variant, since it is expressed in a wide range of ciliated cells. Exons 1 to 10 encode for a domain homologous to the Regulator of Chromosome Condensation 1 (RCC1), known as RCC1-like domain (RLD). This domain is highly conserved across species, and it is present in all predicted isoforms. It is thought that this domain acts as a guanine nucleotide exchange factor (GEF) for small GTPases, such as RAB8A [Citation46]. The second major transcript encodes the RPGRORF15 isoform, a 1152 amino acid protein that is only expressed in rod and cone photoreceptors of the retina. This isoform, RPGRORF15, shares exon 1–14 with the constitutive isoform and includes the alternatively spliced exon 15 and intron 15, ORF15. The ORF15 domain is unique to vertebrates, and it is believed to enable the ciliary-based transport of cargoes between inner and outer segments, contributing to uphold the high metabolic rate characteristic of photoreceptors. An in-depth analysis of the alternative splicing of human RPGR performed by Vervoort and colleagues (2000) in 47 unrelated XLRP patients concluded that, not only the exon ORF15 is the variant preferentially expressed in retinal tissues but that this isoform is essential for the normal function of RPGR in the human retina. Interestingly, RPGRORF15 is the only known isoform that contains the highly conserved basic domain in the carboxy-terminal [Citation47], which has been identified to interact with several proteins that have been associated with retinal degeneration as well, such as whirlin or TTLL5 [Citation48,Citation49]. To date, the presence of other protein-coding transcripts expressed at higher or similar level to the retinal isoform ORF15 in the human eye, with biological activity and function, has not been demonstrated.
RPGR-related phenotype has been mainly associated to retinal degeneration. Mutations identified in exon 1–14 account for approximately 25% of cases, whilst the ORF15 region, which comprises mostly adenine (A) and guanine (G) nucleotides, harbors the disease-causing variants responsible for up to 80% of RPGR-associated cases and is regarded as a mutation hot spot [Citation44]. The ORF15 region is prone to mutations due to the purine-rich repetitive sequence that results in a reduced fidelity during DNA replication and transcription, and in atypical DNA conformations. A recent analysis of the reported variants in the literature showed that 84% of the identified variants are truncations, 14.4% are missense changes and 1.7% are in-frame variants [Citation50]. Within ORF15, small deletions that cause frameshifts producing truncated forms of the protein are the most prevalent. Small in-frame deletions and missense changes have also been identified in the ORF15 region, although these variants seem to be well tolerated [Citation51]. Mutations in the RPGR gene can result in two different clinical phenotypes: the more prevalent mainly affecting rods (classic RP) and the other phenotype primarily affecting cones (cone/cone-rod dystrophy). Some studies have presented a genotype-phenotype correlation in RPGR patients, in which the location of the mutation determines the phenotype observed: those who have mutations in exons 1–14 present a classic RP phenotype, whereas those affected by a cone phenotype have mutations in the distal end of the ORF15 region [Citation52,Citation53–56]. To date, there is no clear phenotype difference between patients with null mutations, those with missense changes or those with ORF15 frameshifts. All these mutations appear to result in a similarly severe rod-cone dystrophy. The only exception being a splice site mutation which allows processing of some wild-type mRNA transcripts and leads to a mild phenotype with preservation of cones and visual acuity [Citation57].
The unusual genetic code of RPGRORF15 renders the cDNA unstable and this may also present challenges for manufacturing gene therapy vectors able to express the full-length protein with biological activity [Citation58–60]. Overcoming this challenge is the first step to succeed in a gene therapy for X-linked RP caused by mutations in RPGR.
4. Scientific rationale
RPGR-associated RP is a good candidate for treatment with gene therapy for several reasons: (1) the considerable disease burden, given by the relatively high prevalence and its severity; (2) the affected gene has been identified and its size (3,459 base pairs) is small enough to fit within the capacity of an AAV vector; (3) the target tissue, the retina, is accessible using relatively noninvasive procedures; (4) the delivery of the normal gene can revert the pathological phenotype caused by loss-of-function mutations in RPGR, and (5) the severe and rapid progression of the disease eases the clinical trial endpoints assessment within a relatively short period of time [Citation61]. However, the development of a recombinant viral vector to treat RPGR-associated RP is not straightforward. As mentioned above, the ORF15 region is a purine-rich repetitive region – this feature not only makes it a mutational hotspot but also constitutes a challenge for therapeutic vector production. The poor sequence stability of this region makes difficult to control the introduction of random, spontaneous mutations in the transgene and maintain the complete sequence of the therapeutic gene.
A further difficulty relates to the limited knowledge on the underlying genetics and the mechanism of action of RPGRORF15, which is subject to a considerable debate. It is known that the RPGR1-19 isoform is involved in the development of photoreceptors, whilst the RPGRORF15 isoform is responsible for maintaining mature photoreceptors [Citation62]. Evidence in animal models and patient autopsy specimens has shown that the absence of RPGR or the synthesis of abnormal RPGR protein led to opsin mislocalization [Citation63–66], suggesting that it plays an important role in the protein trafficking along the photoreceptor connecting cilia () [Citation67–70]. The connecting cilia, with a structure similar to the transition zone of primary cilia, connect the inner segment of photoreceptors, where the proteins are synthesized, with the outer segment, where those proteins are required for phototransduction. Besides the traffic of proteins required for phototransduction, 10% of the outer segments of rods and cones is daily replaced to prevent the accumulation of toxic metabolites that could compromise the photoreceptor survival – discs are phagocytized in the distal end by the retinal pigment epithelium (RPE) and renewed at the proximal end (disc morphogenesis). Photoreceptors are neurons with an extremely high metabolic rate and for this neuroprotective mechanism to succeed a highly reliable transport of proteins is essential [Citation71]. Hence, a well-regulated targeting and transport of molecules across this connecting cilium is crucial for the homeostasis of the photoreceptor cells. The regulation of cilia genesis, maintenance, and function is accomplished by a protein interaction network whereby RPGR plays an important part [Citation72]. Several proteins have been identified to interact with the RLD of RPGR. The phosphodiesterase 6 delta subunit (PDE6δ) was the first RPGR interacting protein identified in a yeast two-hybrid screen using the RLD (amino acid 51–370) [Citation73]. RPGR interacting protein 1 (RPGRIP1) C-terminus interacts with the RLD of RPGR and it is thought that it tethers RPGR to the ciliary membrane [Citation74]. Other interactions with the RLD of RPGR include the proteins SMC1, SMC3, and RAB8A. The two chromosome-associated proteins, SMC1 and SMC3, are involved in microtubule-based movement of chromosomes and also associated with ciliary microtubules in the retina [Citation64]. RAB8A is a critical GTPase involved in photoreceptor protein trafficking and is proposed to be activated by the GEF activity of RPGR [Citation46]. Whirlin and nucleophosmin have been also identified as binding partners for RPGRORF15, interacting specifically with the highly conserved basic domain (amino acid 1079–1152). Whirlin is expressed in photoreceptors and cochlear hair cells – mutations in this gene may cause Usher syndrome and non-syndromic congenital deafness [Citation48]. Nucleophosmin is a multifunctional protein chaperone associated with centrosomal division and with the assembling of RPGRORF15 containing protein complexes [Citation63,Citation75]. Other proteins in the cilia have been identified as binding partners for RPGRORF15, interacting outside the functional domains (amino acid 370–585). Some of these are IFT88, CEP290, KIF3A, or IQCB1, which have been found to be implicated in retinal degeneration [Citation64,Citation68,Citation76]. Mutations that result in the absence, truncation, or alteration of RPGRORF15 protein can cause the disruption of these interactions, compromising the cilia homeostasis, and eventually the photoreceptor cell survival [Citation46,Citation69,Citation77].
Figure 1. RPGRORF15 is located in the connecting cilia of rods and cones. Its proposed function is to enable the transport of different molecules along the connecting cilia, for which its glutamylation and its interaction with other proteins seem to be essential.
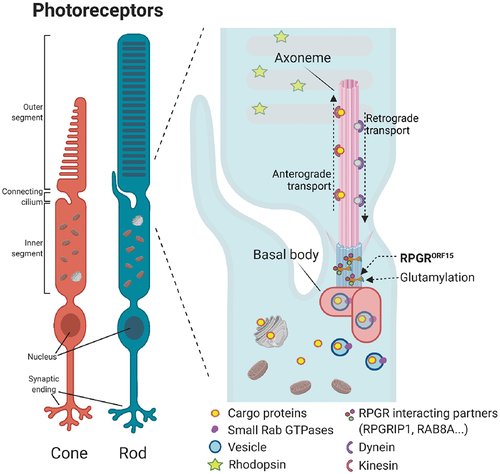
The activation of the proteins that constitute the regulatory network in the connecting cilia, and the large diversity of functions that these develop are mechanistically controlled by post-translational modifications. Specialized functions of microtubule-associated proteins have been related to glutamylation, glycylation, and tyrosination/detyrosination [Citation78]. Glutamylation, the addition of glutamate side chains on glutamate residues catalyzed by Tubulin Tyrosine Ligase-like (TTLL) enzymes, contributes to the stabilization of microtubules and regulates the recruitment and activity of microtubule-interacting proteins [Citation79]. RPGRORF15 is a glutamylation substrate [Citation69,Citation80]. Exon ORF15 is translated into a glutamic acid-glycine rich domain comprising G and E residues as repeat-like sequence such as ‘EEEGEGEGE'. This region can be considered as an intrinsically disordered region since it contains a high proportion of glutamic acid residues (60.1%) which could prevent a protein domain to mediate cooperative folding. The computation of various physical and chemical parameters of the ORF15 region determines that this unique sequence has an instability index of 87.52, which classifies this domain as unstable [Citation81]. But what appears to be a disordered region with unstable structure, could be providing specialized functional advantages [Citation82]. TTLL5 enzyme, expressed in rod and cone photoreceptors, interacts with the basic domain of RPGRORF15 and catalyzes the addition of glutamates to the 11 consensus motifs within the ORF15. The addition of negative charges to the ORF15 region might affect the stabilization of the RPGRORF15 protein, and the integrity of the RPGRORF15 containing protein complexes in the connecting cilia [Citation69,Citation83]. To date, there is no evidence that supports that RPGRORF15 can preserve fully its function with a reduced level of glutamylation and it has been suggested that a complete or partial lack of glutamylation may compromise RPGRORF15 function [Citation80]. Indeed, loss-of-function variants in TTLL5, that lead to a lack of glutamylation, have been identified as a cause of recessive retinal dystrophy, similar to RPGR deficiency [Citation49].
To ensure the success of the treatment in patients with retinal degeneration caused by mutations in the RPGR gene, and avoid unpredictable effects, the goal of the gene replacement strategy must be to provide the patients’ eye with a healthy copy of the RPGRORF15 isoform, this is, the 1152 amino acid protein. Considering the instability of the human RPGRORF15 nucleotide sequence, the generation of mutated forms of the gene during the standard cloning procedures is highly likely, thus alternative strategies need to be applied. Besides an optimal molecular approach to design and synthesize the vector to avoid the generation of mutated proteins, careful consideration is required to translate the findings in preclinical studies to clinical trials. The large repetitive glutamic acid-rich region of RPGRORF15 is present in all vertebrates, but it is poorly conserved. The comparison of the amino acid sequence between orthologues suggests that different lengths of this region can be well tolerated as long as the reading frame and the acidic charge are maintained [Citation25]. The fact that the length of the amino acid sequence of the ORF15 region varies between species, highlights the need of a precise interpretation of the preclinical data to facilitate translational studies. A proof-of-concept study published in 2015, showed that the delivery of an AAV vector containing a mutated form of the human RPGRORF15 gene into the subretinal space of the Rpgr null mice, reversed some of the pathological features associated to the absence of RPGR, i.e. opsin mislocalization, outer segment shortening and photoreceptor function [Citation84]. This shortened form contained an in-frame deletion of 126 codons (residues 862–987) in the ORF15 region similar to that currently being tested in a clinical trial sponsored by MeiraGTx (NCT03252847). The therapeutic effect achieved with this construct containing a deletion in the mouse model can be explained by the differences between the mouse and the human RPGRORF15 protein sequence. The orthologous mouse isoform (NCBI reference sequence NP_001171421 and the partial coding sequence AF286473) does not contain the amino acid residues that are absent in this mutated construct (residues 862–987 of the human sequence), suggesting that this region might not be essential to maintain the integrity and functionality of mature photoreceptors in the mouse retina. Hence, the addition of a shorter version of the human RPGRORF15, containing the conserved domains RLD and the basic domain, would provide the mouse retina with the required biological activity to revert the pathological phenotype in the Rpgr null mouse, which is characterized by a very mild phenotype compared to the severe degeneration observed in humans. The deletion of these 126 amino acids in the glutamic acid-glycine rich region (one third of the ORF15) means that 6 of the 11 consensus motifs for glutamylation are absent, and this truncation results indeed, in a reduction of the 70% of the level of glutamylation [Citation69]. The delivery of a shortened RPGRORF15 sequence to the human retina that generates a truncated protein unable to be completely glutamylated would be suboptimal since it has been suggested that this modification is essential to preserve RPGRORF15 function, as discussed above. It is still not possible to predict the long-term effects of a gene replacement therapy using an RPGRORF15 truncated sequence in the human retina. Indeed, Hong et al. suggested that truncated RPGR may cause a severe phenotype due to a dominant gain-of-function mechanism [Citation85].
In order to produce the human full length RPGRORF15 protein (1152 amino acids), investigators have designed AAV.RPGR vectors introducing silent substitutions – this is, changing the nucleotide sequence without altering the translated amino acid sequence of the transgene. This strategy, known as codon optimization, reduces the GA (purines) repeats and also, reduces the risk of anomalous splicing or premature polyA signals. This generates more stable RPGR cDNA with increased sequence fidelity. In 2017, two preclinical studies were published using codon optimized versions of the human RPGRORF15 sequence (patent applications WO2017042584A1 and US20150353938A1) [Citation59,Citation60]. In these codon-optimized vectors, the splice donor site at the exon 15 boundary was disabled in order to prevent aberrant splicing, which might occur with the addition of pyrimidine bases in the ORF15 region where there are numerous potential ‘AG’ splice acceptor sites. This modification prevents the synthesis of truncated RPGR proteins, or RPGR proteins with large in-frame deletions. The codon optimized RPGR vector developed by the University of Oxford (now in clinical trial sponsored by Biogen Inc.), showed higher stability and sequence integrity than the wild-type sequence in in vitro studies and demonstrated safety and therapeutic effect in two mouse models of the disease (Rpgr knock out and Rd9 mice) [Citation60,Citation86,Citation87] and in humans [Citation30]. The codon optimized RPGR vector in trial by AGTC was tested in a canine model with a naturally occurring deletion in ORF15, showing a good safety profile and efficacy, with evidence of preservation of photoreceptor structure and function [Citation59,86]. Both optimized vectors are currently being tested in Phase I/II/III clinical trials (NCT03116113 and NCT03316560).
5. Competitive environment
Four gene therapy vectors are currently being developed for X-linked RP caused by mutations in RPGR (). There are three main differences between these vectors: the route of administration, the viral capsid and the human RPGR sequence (). Nightstar Therapeutics Ltd. (now Biogen Inc.) and the University of Oxford developed a recombinant AAV8 vector with a stable transgene containing the photoreceptor-specific promoter rhodopsin kinase promoter (GRK1), which drives the transgene expression in rods and cones, and a codon optimized version of the human RPGRORF15 (AAV2/8.GRK1.coRPGRORF15) [Citation60]. Recombinant AAV2/8 vectors have been shown to be very efficient and have a fast onset of gene expression in photoreceptors and RPE cells following subretinal injection into mice and monkeys’ eyes [Citation88,Citation89]. The first gene therapy clinical trial for an RPGR-associated dystrophy was initiated in March 2017 using this vector (NCT03116113). This Phase I/II study, a dose-escalation interventional study, has recruited over 50 patients in the UK and USA, and the initial results were published in 2020 in Nature Medicine [Citation30]. Applied Genetic Technologies Corp (AGTC) also opted to use the codon optimization strategy to ensure the stability of the sequence during the production process, hence avoiding the introduction of mutations into the therapeutic gene that could alter or eliminate the RPGR protein function. The AGTC codon optimized human RPGRORF15, 99% identical to the sequence used by Biogen, is expressed under the control of a similar GRK1 promoter and packaged into an AAV2 capsid variant with three tyrosine to phenylalanine mutations (AAV2tYF), described to enhance the efficiency of transduction [Citation90–92]. The safety, tolerability, and efficacy of this vector, AAV2tYF.GRK1.coRPGRORF15, is being evaluated in a Phase I/II human clinical trial (NCT03116113) initiated in April 2018, and has recently entered Phase III (NCT04850118).
Figure 2. Recombinant AAV.RPGR vectors currently being tested in human clinical trials. The three AAV vectors administered in the subretinal space use the human GRK1 promoter to drive the expression of RPGRORF15. The vectors developed by Biogen Inc., AGTC and 4D Molecular Therapeutics contain a codon-optimized (co) version of the human RPGRORF15 gene whilst the vector developed by MeiraGTx contains a shortened version of the human RPGRORF15. The AAV serotype capsids used to develop these vectors are AAV8 (Biogen), AAV5 (MeiraGTx), an AAV2 variant, with three tyrosine to phenylalanine mutations (AAV2tYF) (AGTC), and an AAV capsid variant named 4D-R100 suitable for intravitreal administration (4D Molecular Therapeutics).
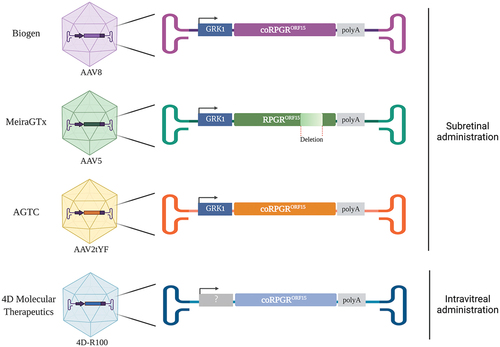
As mentioned in the previous section, another vector containing a shortened version of the human wild type RPGRORF15 in which 10% of the ORF15 region is missing, is currently being tested in a Phase III human clinical trial (NCT04671433) sponsored by MeiraGTx. In this vector, whose scientific name is Botaretigene Sparoparvovec, the shortened version of the human RPGRORF15 is also under the control of the GRK1 promoter, and the transgene is packaged into AAV5 capsid proteins (AAV2/5.GRK1.RPGRORF15). AAV5 has very good efficiency at transducing all subclasses of cones (foveal and parafoveal), as demonstrated in non-human primate retinas where vector was delivered subretinally [Citation93]. Botaretigene Sparoparvovec was granted Fast Track and orphan designation from the US FDA and orphan designation from the EMA in 2018. In 2020, MeiraGTx announced six, nine and 12-month results from the dose-escalation phase of the Phase I/II trial, however a peer-reviewed publication is not yet available at the time of the preparation of this manuscript. Lastly, 4D Molecular Therapeutics initiated in 2020 a Phase 1/2 clinical trial to assess the safety and tolerability of an intravitreal injection of their proprietary vector, which comprises an AAV capsid variant (4D-R100) carrying a codon-optimized version of the human RPGRORF15 (NCT04517149). This gene therapy product candidate, named 4D-125, has been granted Fast Track Designation by the FDA.
6. Potential development issues
Clinical and scientific advancements in next-generation gene and cell therapies are beginning to fulfil the promise of personalized medicine since the realization of the first human genome project in 2003 with attempts to cure patients suffering from devastating conditions including blindness. Multiple approved genetic products have been launched in global gene therapy markets and the number of clinical trials continues to grow. The global gene therapy market, valued at $1 billion in 2018, is projected to grow to ~$12 billion by 2025 [Citation94]. However, the gene therapy market is distinctly different from traditional biopharma products and despite considerable efforts in gene therapy segment, only six of the twenty approved products have been translated into the clinic.
Development of gene therapy medicinal products is still a young field with many challenges that stand in the way of realizing their full potential. These include a dynamic regulatory environment, market access for these costly therapies, clinical long-term safety and efficacy which have yet to be established, provider and health-care economic disruption, manufacturing costs with variable yields and limited capacity and even patient access and recruitment for gene therapies targeting rare diseases. There are over 300 different genes that can cause retinal degeneration. The plan for gene therapy ophthalmology sector to develop treatments for each one of these diseases, presents unique challenges with huge health care and economic implications. Even when pathogenic variants are known, the fact that they affect very small populations, makes the clinical development pathway economically non-viable in many instances.
Life sciences companies and academic institutions are at present key players that drive advancements in gene therapy market of rare genetic diseases. They need to address challenges along all stages of product development. Clinical trials need to be designed with patient-centric approaches to reach small populations and meet complex endpoints. The advantages that orphan diseases tend to have over common conditions are clear genomic targets as well as high unmet need in small patient populations, often underserved by more traditional therapies, if any exist at all. In addition, pathways to approval of therapies for orphan diseases by regulatory authorities are often accelerated, shifting the paradigm of clinical trials by consolidating the Phase I, II, III process into Phase I/II/III and confirmatory Phase IV studies post approval. Companies are also allowed to ‘experiment’ with innovative trials designs and new or surrogate endpoints. As the gene therapy technology matures, large pharma companies are becoming more excited about owning the technology and driving the clinical research forward. For example, acquisition of Nightstar Therapeutics in 2019 by Biogen, one of the leading gene therapy companies in the global market, was a huge step for developing clinical pipelines of gene therapy treatments in ophthalmology, including gene therapy for RPGR-related RP.
In addition, Luxturna®, the first approved gene therapy for another inherited retinal degeneration, has set a precedent for other trials. The success was due to a better clinical and scientific understanding of safety profiles of the gene therapy vector. The regulatory approval also included recognition of the AAV vector manufacturing process that met the quality standards required for clinical scale production. The trial endpoint was an improvement in visual function with no obligation to demonstrate a slowing of retinal degeneration, which would have taken many years to validate and which would not be practical for a company to undertake. Spark Therapeutics, which ran the trial, was able to demonstrate to the authorities that another new test (multi luminance mobility test) could provide a meaningful Phase III trial endpoint, in place of traditional outcomes, such as visual acuity. Similarly, following successes of Phase I/II RPGR clinical trials [Citation30], Biogen was able to show the authorities that retinal sensitivity as measured by microperimetry can be used as the primary endpoint for Phase III efficacy trial. It must be emphasized that potential participants must be familiar with these new investigative techniques and able to perform tests accurately to ensure reliable detection of potential treatment effects.
The size of the coding sequence of RPGRORF15 (3.5 kb) is within the AAV carrying capacity and the relatively high prevalence and disease severity have justified development of this gene therapy treatment. However, the repetitive sequence of ORF15 not only makes it challenging for therapeutic vector production, as discussed above, but also for diagnostic and thereby recruitment purposes. Despite significant progress in genetic diagnosis, genetic confirmation is still missing in many patients with clear X-linked family history. The ORF15 region is a mutational hotspot in RPGR associated with majority of patients with X-linked RP. Yet, sequencing of ORF15 region remains notoriously difficult and error-prone and any potential small deletions that do not actually exist, would lead to an incorrect genetic diagnosis. Since the phenotype of RPGR-related RP is indistinguishable from any other RP, the diagnosis can easily remain unchallenged and potentially compromise patient’s safety if the patient is recruited in the trial. Clinicians treating patients with gene therapy must therefore be prepared to scrutinize genetic testing reports until they are satisfied of the correct diagnosis. A simple way to confirm X-linked inheritance is to establish the presence of a female carrier phenotype in the patient’s family [Citation51]. In addition, developing molecular assays to confirm genetic diagnosis in uncertain cases is critical for increasing patient access to clinical trials and indeed treatments once they become available [Citation57].
7. Conclusion
A huge progress has been made in gene therapy over the last years to translate the applied research advances to the clinic and the market. These advances include the optimization of the vectors to increase the transduction efficiencies, improve the transgene expression and increase the packaging capacity, among others. The development of a gene therapy vector to deliver a healthy copy of the RPGR gene into the rod and cone photoreceptors cells has not been a direct path. Its unusual sequence has challenged the scientific community to produce a vector able to express the protein in its full length and hence, with biological activity. Currently, four different gene therapy vectors are being evaluated in human clinical trials. Three of these vectors, with an optimized version of the human RPGR gene that increases its stability and produces the full-length protein. The other vector uses a shortened version of the wild type RPGRORF15 sequence. The wild type RPGRORF15 sequence is known to be less stable and prone to mutations. The long-term effects of replacing the endogenous wild-type protein by a RPGR protein containing a large in-frame deletion in the human eye are currently unknown. The results of these clinical trials are emerging and although they look positive and encouraging, a deep and careful interpretation of the data is still needed as these trials move forward.
8. Expert opinion
Gene therapy for X-linked retinitis pigmentosa caused by mutations in RPGR is being developed in four clinical trials that use adeno-associated viral (AAV) vectors to deliver the transgene to rod and cone photoreceptor cells. The normal RPGR mRNA is, however, very unstable and to overcome this, three of the clinical trials use a codon-optimized DNA sequence which encodes the full-length RPGR protein. The other trial, in contrast, uses an RPGR transgene that harbors an in-frame deletion in a region shown to be critical for post-translational modification. Unlike the codon-optimized RPGR which has also been tested extensively in the large animal (dog) model, the deleted RPGR has only been tested in mice, which have a shorter RPGR and much milder disease compared to humans.
The progress made over the last years to find the cure for different IRDs, including X-linked RP, is self-evident. However, scientists and clinicians are still facing significant challenges and trying to overcome the limitations to make different investigational products a reality for patients. Complications and unreliable results can arise from surgical intervention, inadequate clinical endpoints, safety of a bilateral administration, varied patient response to the treatment, stage of the disease, short follow-up, or low number of patients. In the case of RPGR-related RP, an additional challenge is the development of a gene therapy vector with the ability to alter or reverse the pathological phenotype whose underlying mechanism remains unclear. To date, only the results of a six-month follow-up of one of the Phase I/II clinical trials (NCT03116113) have been made available in a peer-reviewed journal. While the Phase I/II/III gene therapy trials are active, patients and professionals are keenly awaiting their outcomes whose ultimate goal is to provide a long-lasting treatment for patients affected by RPGR disease.
Article highlights
Retinitis pigmentosa caused by mutations in the RPGR gene is a severe and relatively high prevalent type of IRD. This disease has proven to be a good candidate for gene replacement therapy using AAVs, the most common vector used in ophthalmology.
The molecular and biological complexity of RPGR represents the biggest challenge to develop a gene therapy vector and to predict the likelihood of success and long-term effect of a gene therapy treatment.
The instability of the human RPGRORF15 nucleotide sequence makes difficult to produce the full-length RPGR gene without introducing spontaneous mutations that could alter or eliminate the RPGR protein function. Codon optimization can be used as a strategy to improve the stability of the sequence during the production process.
The understanding of the pathophysiology of RPGR-associated RP is incomplete. The underlying genetics, mechanism of action and pathogenesis of RPGR require additional investigations that will contribute to the generation of more effective and targeted gene therapies.
Different RPGR gene therapy vectors and delivery routes are currently being investigated in clinical trials to identify a safe and effective treatment for RPGR-associated X-linked RP. To date, the initial results of one of the clinical trials have been made available in a per-reviewed journal, demonstrating a good safety profile and an improvement in the visual field using a codon optimized version of RPGRORF15.
This box summarizes key points contained in the article.
Declaration of interest
RE MacLaren has previously received grant funding from Biogen and has previously provided independent consultancy advice on X-linked retinitis pigmentosa to Biogen Inc. and Janssen Pharmaceuticals. RE MacLaren is also listed as an inventor on a patent for X-linked retinitis pigmentosa gene therapy owned by the University of Oxford. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
A reviewer on this manuscript is an investigator for MeiraGTx-sponsored trials of gene therapy for RPGR. Another reviewer on this manuscript is on the Scientific Advisory Board of AGTC, one of the companies developing RPGR gene therapy. MeiraGTx reviewed and provided comments on the manuscript. The views expressed in the manuscript are those of the authors and do not reflect the views of MeiraGTx. MeiraGTx has provided a Letter to the Editor (https://doi.org/10.1080/14728214.2022.2152202). Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Shahryari A, Saghaeian Jazi M, Mohammadi S, et al. Development and clinical translation of approved gene therapy products for genetic disorders. Front Genet. 2019;10:868.
- Ciulla TA, Hussain RM, Berrocal AM, et al. Voretigene neparvovec-rzyl for treatment of RPE65 -mediated inherited retinal diseases: a model for ocular gene therapy development. Expert Opin Biol Ther. 2020;20(6):565–578.
- Voretigene neparvovec-rzyl (Luxturna) for inherited retinal dystrophy. Med Lett Drugs Ther. 2018;60(1543):53–55.
- Darrow JJ. Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov Today. 2019;24(4):949–954.
- Economics DA. The socioeconomic impact of inherited retinal dystrophies (IRDs) in the United Kingdom. 2019.
- Ferrari S, S. Sorrentino F, Ponzin D, et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12(4):238–249.
- Daiger SP. Retinal Information Network. 1996-2020. [cited 13 Jan 2021]; Available from: https://sph.uth.edu/retnet/home.htm.
- Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84(2):132–141.
- Mathur P, Yang J. Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochim Biophys Acta. 2015;1852(3):406–420.
- Suspitsin EN, Imyanitov EN. Bardet-biedl syndrome. Mol Syndromol. 2016;7(2):62–71.
- Dias MF, Joo K, Kemp JA, et al. Molecular genetics and emerging therapies for retinitis pigmentosa: basic research and clinical perspectives. Prog Retin Eye Res. 2018;63:107–131.
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–1809.
- Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761–772.
- Berson EL, Rosner B, Sandberg MA, et al. Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: subgroup analyses. Arch Ophthalmol. 2004;122(9):1306–1314.
- Hughbanks-Wheaton DK, Birch DG, Fish GE, et al. Safety assessment of docosahexaenoic acid in X-linked retinitis pigmentosa: the 4-year DHAX trial. Invest Ophthalmol Vis Sci. 2014;55(8):4958–4966.
- Berson EL, Rosner B, Sandberg MA, et al. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch Ophthalmol. 2010;128(4):403–411.
- Schwartz SG, Wang X, Chavis P, et al. Vitamin A and fish oils for preventing the progression of retinitis pigmentosa. Cochrane Database Syst Rev. 2020;6(6):CD008428.
- Zhao Y, Feng K, Liu R, et al. Vitamins and mineral supplements for Retinitis pigmentosa. J Ophthalmol. 2019;2019:8524607.
- Vazquez-Dominguez I, Garanto A, Collin RWJ. Molecular therapies for inherited retinal diseases-current standing. Opportunities and Challenges. Genes (Basel). 2019. 10(9)
- MacLaren RE, Bennett J, Schwartz SD. Gene therapy and stem cell transplantation in retinal disease: the new frontier. Ophthalmology. 2016;123(10S):S98–S106.
- Stern JH, Tian Y, Funderburgh J, et al. Regenerating eye tissues to preserve and restore vision. Cell Stem Cell. 2018;23(3):453.
- Gagliardi G, Ben M’Barek K, Goureau O. Photoreceptor cell replacement in macular degeneration and retinitis pigmentosa: a pluripotent stem cell-based approach. Prog Retin Eye Res. 2019;71:1–25.
- Fry LE, Peddle CF, Barnard AR, et al. RNA editing as a therapeutic approach for retinal gene therapy requiring long coding sequences. Int J Mol Sci. 2020;21(3):777.
- Kantor A, McClements ME, MacLaren RE. CRISPR-cas9 DNA base-editing and prime-editing. Int J Mol Sci. 2020;21(17):6240.
- Raghupathy RK, Gautier P, Soares DC, et al. Evolutionary characterization of the Retinitis pigmentosa GTPASE regulator gene. Invest Ophthalmol Vis Sci. 2015;56(11):6255–6264.
- Sandberg MA, Rosner B, Weigel-DiFranco C, et al. Disease course of patients with X-linked retinitis pigmentosa due to RPGR gene mutations. Invest Ophthalmol Vis Sci. 2007;48(3):1298–1304.
- Talib M, van Schooneveld MJ, Thiadens AA, et al. Clinical and genetic characteristics of male patients with RPGR-associated Retinal dystrophies: a long-term follow-up study. Retina. 2019;39(6):1186–1199.
- Campochiaro PA, Mir TA. The mechanism of cone cell death in retinitis pigmentosa. Prog Retin Eye Res. 2017;62:24–37 .
- Demirci FY, Rigatti BW, Wen G, et al. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet. 2002;70(4):1049–1053.
- Cehajic-Kapetanovic J, Xue K, Martinez-Fernandez de la Camara C, et al., Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med. 2020. 26(3): 354–359.
- Menghini M, Jolly JK, Nanda A, et al. Early cone photoreceptor outer segment length shortening in RPGR X-linked retinitis pigmentosa. Ophthalmologica. 2020.
- Nanda A, Salvetti AP, Clouston P, et al. Exploring the variable phenotypes of RPGR carrier females in assessing their potential for retinal gene therapy. Genes (Basel). 2018;9(12):643.
- Salvetti AP, Nanda A, MacLaren RE. RPGR-related X-linked Retinitis pigmentosa carriers with a severe “male pattern.” Ophthalmologica. 2020;1:1–8.
- Fahim AT, Sullivan LS, Bowne SJ, et al. X-chromosome inactivation is a biomarker of clinical severity in female carriers of RPGR-associated x-linked retinitis pigmentosa. Ophthalmol Retina. 2020;4(5):510–520.
- MacLaren RE. Gene therapy for Retinal disease: what lies ahead. Ophthalmologica. 2015;234(1):1–5.
- DiCarlo JE, Mahajan VB, Tsang SH. Gene therapy and genome surgery in the retina. J Clin Invest. 2018;128(6):2177–2188.
- Surace EM, Auricchio A. Versatility of AAV vectors for retinal gene transfer. Vision Res. 2008;48(3):353–359.
- Hickey DG, Edwards TL, Barnard AR, et al. Tropism of engineered and evolved recombinant AAV serotypes in the rd1 mouse and ex vivo primate retina. Gene Ther. 2017;24(12):787–800.
- Naso MF, Tomkowicz B, Perry WL, et al. Adeno-Associated Virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31(4):317–334.
- Bigot K, Gondouin P, Benard R, et al. Transferrin Non-Viral Gene Therapy for Treatment of Retinal Degeneration. Pharmaceutics. 2020;12(9).
- Bordet T, Behar-Cohen F. Ocular gene therapies in clinical practice: viral vectors and nonviral alternatives. Drug Discov Today. 2019;24(8):1685–1693.
- Buck TM, Wijnholds J. Recombinant Adeno-Associated Viral Vectors (rAAV)-Vector elements in ocular gene therapy clinical trials and transgene expression and bioactivity assays. Int J Mol Sci. 2020;21(12):4197.
- Hulliger EC, Hostettler SM, Kleinlogel S. Empowering retinal gene therapy with a specific promoter for human rod and cone ON-Bipolar cells. Mol Ther Methods Clin Dev. 2020;17:505–519.
- Vervoort R, Lennon A, Bird AC, et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25(4):462–466.
- Howe KL, Achuthan P, Allen J, et al. Ensembl 2021. Nucleic Acids Res. 2021;49(D1):D884–D891.
- Murga-Zamalloa CA, Atkins SJ, Peranen J, et al. Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: implications for cilia dysfunction and photoreceptor degeneration. Hum Mol Genet. 2010;19(18):3591–3598.
- UniProt C, Martin M-J, Orchard S. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49(D1):D480–D489.
- Wright RN, Hong DH, Perkins B. RpgrORF15 connects to the usher protein network through direct interactions with multiple whirlin isoforms. Invest Ophthalmol Vis Sci. 2012;53(3):1519–1529.
- Sergouniotis Panagiotis I, Chakarova C, Murphy C, et al. Biallelic variants in TTLL5, encoding a Tubulin glutamylase, cause retinal dystrophy. Am J Hum Genet. 2014;94(5):760–769.
- Yang J, Zhou L, Ouyang J, et al. Genotype–phenotype analysis of RPGR variations: reporting of 62 Chinese families and a literature review. Front Genet. 2021;12(827):600210.
- Cehajic Kapetanovic J, McClements ME, Martinez-Fernandez de la Camara C, et al. Molecular strategies for RPGR gene therapy. Genes (Basel). 2019;10(9):674.
- Cehajic-Kapetanovic J, Martinez-Fernandez de la Camara C, Birtel J, et al. Impaired glutamylation of RPGR ORF15 underlies the cone-dominated phenotype associated with truncating distal ORF15 variants. Proc. Natl. Acad. Sci. U.S.A. 2022;119(49). DOI: 10.1073/pnas.2208707119.
- Hadalin V, Sustar M, Volk M, et al. Cone dystrophy associated with a novel variant in the terminal codon of the RPGR-ORF15. Genes (Basel). 2021;12(4):499.
- Mawatari G, Fujinami K, Liu X, et al. Clinical and genetic characteristics of 14 patients from 13 Japanese families with RPGR-associated retinal disorder: report of eight novel variants. Hum Genome Var. 2019;6:34.
- Nguyen XT, Talib M, van Schooneveld MJ, et al. RPGR-Associated dystrophies: clinical, genetic, and histopathological features. Int J Mol Sci. 2020;21(3):835.
- Yang L, Yin X, Feng L, et al. Novel mutations of RPGR in Chinese retinitis pigmentosa patients and the genotype-phenotype correlation. PLoS One. 2014;9(1):e85752.
- Cehajic-Kapetanovic J, McClements ME, Whitfield J, et al. Association of a novel intronic variant in RPGR with hypomorphic phenotype of X-Linked Retinitis Pigmentosa. JAMA Ophthalmol. 2020;138(11): 1151–1158.
- Deng WT, Dyka FM, Dinculescu A, et al. Stability and safety of an AAV vector for treating RPGR-ORF15 X-linked retinitis pigmentosa. Hum Gene Ther. 2015;26(9):593–602.
- Beltran WA, Cideciyan AV, Boye SE, et al. Optimization of retinal gene therapy for X-linked retinitis pigmentosa due to RPGR Mutations. Mol Ther. 2017;25(8):1866–1880.
- Fischer MD, McClements ME, Martinez-Fernandez de la Camara C, et al., Codon-optimized RPGR improves stability and efficacy of AAV8 gene therapy in two mouse models of x-linked retinitis pigmentosa. Mol Ther. 2017;25(8):1854–1865.
- Martinez-Fernandez De La Camara C, Cehajic-Kapetanovic J, MacLaren RE. RPGR gene therapy presents challenges in cloning the coding sequence. Expert Opin Biol Ther. 2019;20(1):1–9.
- Wright RN, Hong DH, Perkins B. Misexpression of the constitutive Rpgr(ex1-19) variant leads to severe photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2011;52(8):5189–5201.
- Shu X, Fry AM, Tulloch B, et al. RPGR ORF15 isoform co-localizes with RPGRIP1 at centrioles and basal bodies and interacts with nucleophosmin. Hum Mol Genet. 2005;14(9):1183–1197.
- Khanna H, Hurd TW, Lillo C, et al. RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J Biol Chem. 2005;280(39):33580–33587.
- Adamian M, Pawlyk BS, Hong D-H, et al. Rod and cone opsin mislocalization in an autopsy eye from a carrier of X-linked retinitis pigmentosa with a Gly436Asp mutation in the RPGR gene. Am J Ophthalmol. 2006;142(3):515–518.
- Hong DH, Pawlyk BS, Shang J, et al. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc Natl Acad Sci U S A. 2000;97(7):3649–3654.
- Wright AF, Shu X. Focus on molecules: RPGR. Exp Eye Res. 2007;85(1):1–2.
- Patnaik SR, Raghupathy RK, Zhang X, et al. The role of RPGR and its interacting proteins in ciliopathies. J Ophthalmol. 2015;2015:414781.
- Sun X, Park JH, Gumerson J, et al. Loss of RPGR glutamylation underlies the pathogenic mechanism of retinal dystrophy caused by TTLL5 mutations. Proc Natl Acad Sci U S A. 2016;113(21):E2925–34.
- Natarajan K, Gadadhar S, Souphron J, et al. Molecular interactions between tubulin tails and glutamylases reveal determinants of glutamylation patterns. EMBO Rep. 2017;18(6):1013–1026.
- Frederick JM, Hanke-Gogokhia C, Ying G, et al. Diffuse or hitch a ride: how photoreceptor lipidated proteins get from here to there. Biol Chem. 2020;401(5):573–584.
- Megaw RD, Soares DC, Wright AF. RPGR: its role in photoreceptor physiology, human disease, and future therapies. Exp Eye Res. 2015;138:32–41.
- Linari M, Ueffing M, Manson F, et al. The retinitis pigmentosa GTPase regulator, RPGR, interacts with the delta subunit of rod cyclic GMP phosphodiesterase. Proc Natl Acad Sci U S A. 1999;96(4):1315–1320.
- Won J, Gifford E, Smith RS, et al. RPGRIP1 is essential for normal rod photoreceptor outer segment elaboration and morphogenesis. Hum Mol Genet. 2009;18(22):4329–4339.
- Hosch J, Lorenz B, Stieger K. RPGR: role in the photoreceptor cilium, human retinal disease, and gene therapy. Ophthalmic Genet. 2011;32(1):1–11.
- Anand M, Khanna H. Ciliary transition zone (TZ) proteins RPGR and CEP290: role in photoreceptor cilia and degenerative diseases. Expert Opin Ther Targets. 2012;16(6):541–551.
- Zhang Q, Giacalone JC, Searby C, et al. Disruption of RPGR protein interaction network is the common feature of RPGR missense variations that cause XLRP. Proc Natl Acad Sci U S A. 2019;116(4):1353–1360.
- Kai He KL, Jinghua H. The emerging role of tubulin posttranslational modifications in cilia and ciliopathies. Biophysics Reports. 2020;6(4):89–104.
- Garnham CP, Vemu A, Wilson-Kubalek E, et al. Multivalent microtubule recognition by tubulin tyrosine ligase-like family glutamylases. Cell. 2015;161(5):1112–1123.
- Rao KN, Anand M, Khanna H. The carboxyl terminal mutational hotspot of the ciliary disease protein RPGRORF15 (retinitis pigmentosa GTPase regulator) is glutamylated in vivo. Biol Open. 2016;5(4):424–428.
- Gasteiger E, Gattiker A, Hoogland C, et al. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31(13):3784–3788.
- Latysheva NS, Flock T, Weatheritt RJ, et al. How do disordered regions achieve comparable functions to structured domains? Protein Sci. 2015;24(6):909–922.
- Mitchell DR. Polyglutamylation: the GLU that makes microtubules sticky. Curr Biol. 2010;20(5):R234–6.
- Pawlyk BS, Bulgakov OV, Sun X, et al. Photoreceptor rescue by an abbreviated human RPGR gene in a murine model of X-linked retinitis pigmentosa. Gene Ther. 2015;23(2):196–204.
- Hong DH, Pawlyk BS, Adamian M, et al. Dominant, gain-of-function mutant produced by truncation of RPGR. Invest Ophthalmol Vis Sci. 2004;45(1):36–41.
- Song C, Conlon TJ, Deng W-T, et al. Toxicology and pharmacology of an AAV vector expressing codon-optimized RPGR in RPGR-deficient rd9 mice. Hum Gene Ther Clin Dev. 2018;29(4):188–197.
- Dufour VL, Cideciyan AV, Ye G-J, et al. Toxicity and efficacy evaluation of an adeno-associated virus vector expressing codon-optimized RPGR delivered by subretinal injection in a canine model of X-linked retinitis pigmentosa. Hum Gene Ther. 2020;31(3–4):253–267.
- Natkunarajah M, Trittibach P, McIntosh J, et al. Assessment of ocular transduction using single-stranded and self-complementary recombinant adeno-associated virus serotype 2/8. Gene Ther. 2008;15(6):463–467.
- Vandenberghe LH, Bell P, Maguire AM, et al. Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci Transl Med. 2011;3(88):88ra54.
- Ye GJ, Budzynski E, Sonnentag P, et al. Safety and biodistribution evaluation in CNGB3-deficient mice of rAAV2tYF-PR1.7-hCNGB3, a recombinant AAV vector for treatment of achromatopsia. Hum Gene Ther Clin Dev. 2016;27(1):27–36.
- Ye GJ, Budzynski E, Sonnentag P, et al. Safety and biodistribution evaluation in cynomolgus macaques of rAAV2tYF-PR1.7-hCNGB3, a recombinant AAV vector for treatment of achromatopsia. Hum Gene Ther Clin Dev. 2016;27(1):37–48.
- Song C, Dufour VL, Cideciyan AV, et al. Dose range finding studies with two RPGR transgenes in a canine model of X-linked retinitis pigmentosa treated with subretinal gene therapy. Hum Gene Ther. 2020;31(13–14):743–755.
- Boye SE, Alexander JJ, Boye SL, et al. The human rhodopsin kinase promoter in an AAV5 vector confers rod- and cone-specific expression in the primate retina. Hum Gene Ther. 2012;23(10):1101–1115.
- Global cell and gene therapy market to reach $11.96 billion by 2025. Bloomberg, UK: Markets Insider. 2019