
ABSTRACT
Introduction
Progressive familial intrahepatic cholestasis (PFIC) is a group of disorders characterized by inappropriate bile formation, causing hepatic accumulation of bile acids and, subsequently, liver injury. Until recently, no approved treatments were available for these patients.
Areas covered
Recent clinical trials for PFIC treatment have focused on intestine-restricted ileal bile acid transporter (IBAT) inhibitors. These compounds aim to reduce the pool size of bile acids by interrupting their enterohepatic circulation. Other emerging treatments in the pipeline include systemic IBAT inhibitors, synthetic bile acid derivatives, compounds targeting bile acid synthesis via the FXR/FGF axis, and chaperones/potentiators that aim to enhance the residual activity of the mutated transporters.
Expert opinion
Substantial progress has been made in drug development for PFIC patients during the last couple of years. Although data concerning long-term efficacy are as yet only scarcely available, new therapies have demonstrated robust efficacy in a considerable fraction of patients at least on the shorter term. However, a substantial fraction of PFIC patients do not respond to these novel therapies and thus still requires surgical treatment, including liver transplantation before adulthood. Hence, there is still an unmet medical need for long-term effective medical, preferably non-surgical, treatment for all PFIC patients.
Plain Language Summary
Normally, the liver produces bile which is a route of secretion of waste products from the body and also helps in the intestinal absorption of fats from the diet. The bile goes from the liver, through the bile duct to the intestines and components are taken up again at the end of the intestine and transported back to the liver. However, progressive familial intrahepatic cholestasis (PFIC in short) is a group of diseases where bile stays in the liver and damages it. PFIC often causes symptoms already in very young children, like itch and jaundice (getting a slight yellow color). Patients get more and worse symptoms over time and may eventually need a liver transplantation. This review discusses what drugs have been developed for PFIC recently and what drugs are in development now. Two new drugs for PFIC have been developed and approved in the last few years: odevixibat and maralixibat. These drugs help bile in the intestines leave the body via the stool and prevent bile from going back to the liver instead. Drugs in development aim to either 1) do the same, 2) make the bile less toxic, 3) reduce the production of bile, or 4) help bile go from the liver into the bile ducts. There has been a lot of progress in drug development for PFIC in the last few years. The new drugs have helped a considerable number of patients, but many patients still do not respond to these new drugs, keep having symptoms and may need surgery. Therefore, despite considerable progress, research needs to continue for an effective treatment for all PFIC patients.
1. Background
Progressive familial intrahepatic cholestasis (PFIC) represents a group of autosomal recessive disorders characterized by disrupted bile formation, leading to cholestasis [Citation1]. The key features of cholestasis, i.e. the impaired bile formation and subsequent accumulation of bile acids within the liver and systemic circulation, induce liver injury [Citation2]. The impaired bile formation observed in PFIC patients can be caused by loss-of-function mutations in different genes. To date, more than 10 different genes have been related to PFIC and have been numbered, although sometimes inconsistently for the classical subtypes familial intrahepatic cholestasis 1 deficiency (FIC1; PFIC1), bile salt export pump deficiency (BSEP; PFIC2) and MDR3 deficiency (multidrug resistance protein 3; PFIC3). The classical types are indicated with the names of the mutated genes, and the most established ones are discussed in more detail later in this review. Patients with PFIC often present themselves in infancy or early childhood with symptoms such as jaundice and pruritus. The various types of PFIC can evolve quickly to portal hypertension, cirrhosis, liver failure and/or hepatocellular carcinoma [Citation1]. The exact incidence of the different types of PFIC is unknown but is estimated to be 1 in 50,000–100,000 births for PFIC1-3 [Citation3]. Nine to 15% of children with cholestasis suffer from PFIC [Citation3,Citation4].
Under physiological conditions, bile acids are synthesized in hepatocytes and secreted into the bile by BSEP after their conjugation to glycine or taurine [Citation5]. Bile acids are then transported via bile ducts to the gallbladder for temporary storage. Upon ingestion of a meal, the gallbladder contracts and expels its contents into the small intestine to facilitate digestion and solubilization of dietary fat through the formation of mixed micelles [Citation5]. In the terminal ileum about 95% of bile acids are absorbed from the intestinal lumen via the ileal bile acid transporter, also known as the apical sodium-dependent bile acid transporter (IBAT, ASBT) [Citation1,Citation6]. The reabsorbed bile acids are transported via the portal vein back to the liver for resecretion into the bile, thus constituting the enterohepatic circulation. Bile acids that are not reabsorbed from the small intestine enter the colon where they can be metabolized into secondary bile acids and be either passively reabsorbed or excreted via the feces [Citation6]. The net loss of bile acids, estimated around 5% per enterohepatic cycle, is compensated for by de novo bile acid synthesis to maintain the size of the bile acid pool. As the bile acid pool cycles between 6 and 10 times per day, 30–50% of the bile acid pool is replaced every 24 hours in healthy human individuals [Citation6].
De novo bile acid synthesis is subject to negative feedback regulation via bile acid-induced signaling pathways. Reuptake of bile acids from the intestine leads to activation of the farnesoid X receptor (FXR) in enterocytes, which controls gene expression of fibroblast growth factor 19 (FGF19) [Citation7,Citation8]. The produced FGF19 is then secreted into the blood circulation and transported to the liver via the portal vein. In the liver, FGF19 inhibits bile acid synthesis by stimulating the FGF receptor 4/b-klotho complex, which results in downregulation of bile acid synthesis enzyme via as yet incompletely elucidated signaling pathways [Citation9]. Low ileal bile acid levels are associated with relatively low activation of FXR in enterocytes and, consequently, with low FGF19 production and higher bile acid synthesis rates [Citation7,Citation8].
Depending on the type, PFIC is characterized by hepatocyte and/or cholangiocyte injury due to either accumulated bile acids or impaired protection of these cells against the detergent effect of bile acids [Citation10]. Subtypes of PFIC are classified based on the responsible mutations in different genes (, ) [Citation41]. The most common genetic deficiencies are in ATP8B1 (FIC1), ABCB11 (BSEP) and ABCB4 (MDR3), underlying the PFIC subtypes 1, 2 and 3, respectively [Citation1]. These mutations affect bile production in different ways. ATP8B1 encodes an amino phospholipid flippase (FIC1 protein) that is responsible for maintaining an asymmetric phospholipid distribution in the hepatocyte canalicular membrane bilayer by flipping phosphatidylserine from the outer to the inner leaflet [Citation12,Citation13]. FIC1 protein activity preserves membrane integrity and asymmetry of the canalicular membrane, thereby making it more resistant to the high bile acid concentrations in the canalicular lumen [Citation42–44]. Mutations in ATP8B1 destabilize the membrane and disrupt bile acid transport, causing accumulation of bile acids within hepatocytes [Citation44]. ATP8B1 is also expressed in extrahepatic tissues including the intestine, kidney, and pancreas [Citation45]. Accordingly, ATP8B1 deficiency can present with extrahepatic manifestations. ABCB11 encodes BSEP, which is located in the canalicular membrane of hepatocytes [Citation46]. The impact of ABCB11 mutations on bile acid transport activity can vary between individuals. Whereas some patients exhibit complete loss of function of BSEP, for example due to premature truncation of the protein, others may have residual bile acid transport activity due to missense mutations [Citation14,Citation15]. Mutations leading to complete loss-of-function of BSEP are associated with fast deterioration of liver function and a high risk of hepatocellular carcinoma [Citation47]. The ABCB4 gene codes for MDR3, which flops phosphatidylcholine from the inner to the outer leaflet of the canalicular membrane. From here, luminal bile acids induce the secretion of phosphatidylcholine into the bile [Citation19]. Phosphatidylcholine is an important component of bile acid micelles. It decreases the detergent ability of bile acids and thereby protects the bile canaliculi as well as the cells lining the biliary tree, i.e. the cholangiocytes [Citation48]. MDR3 deficient PFIC may manifest later in life as compared to FIC1 and BSEP deficiency, with an onset ranging from 1 month of age to far in adult life [Citation43].
Figure 1. Schematic overview of affected proteins for all discussed PFIC subtypes on a cellular level. Mutations in FIC1, BSEP, MDR3, TJP2, FXR and MYO5B are well established in literature as a cause of PFIC. Mutations in USP53, KIF12, VPS33B and ZFYVE19 putatively cause PFIC. ABCB11, ATP-binding cassette sub-family B member 11 protein; BSEP, bile salt export pump; ESCRT-III, endosomal-sorting complexes required for transport-III; FIC1, familial intrahepatic cholestasis 1; FXR, farnesoid X receptor; KIF12, kinesin family member 12; MDR3, multidrug resistance protein 3; MYO5B, myosin 5B; TJP2, tight junction protein 2; USP53, ubiquitin specific peptidase 53, VPS33B, vacuolar protein sorting-associated protein 33B; ZFYVE19, zinc finger FYVE-type containing 19. Figure created with BioRender.com.
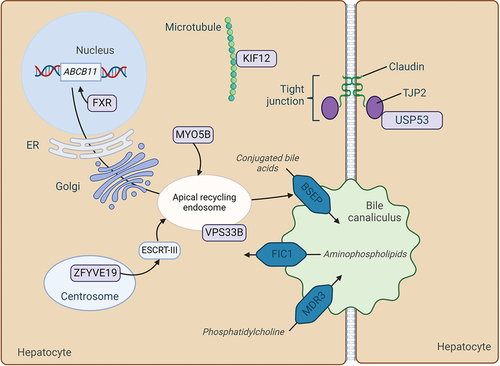
Table 1. Summary of PFIC subtypes [Citation11].
More rare subtypes of PFIC include Tight junction protein-2 (TJP2), NR1H4 (FXR) and Myosin 5B (MYO5B) deficiency [Citation43,Citation49,Citation50]. TJP2 maintains cell polarity and asymmetric distribution of membrane proteins, preserving integrity of tight junctions in hepatocytes and cholangiocytes [Citation22,Citation23]. Loss-of-function mutations result in leakage of bile back into the parenchyma [Citation22,Citation51]. Studies in mice suggest that especially loss of TJP2 in cholangiocytes contributes to liver injury [Citation23]. NR1H4 encodes the bile acid-activated nuclear receptor FXR. NR1H4 mutations are associated with decreased expression of its target genes ABCB11 and to a lesser degree ABCB4, leading to bile acid accumulation in hepatocytes [Citation43,Citation50,Citation24]. MYO5B is involved in trafficking of transporters toward apical domains in enterocytes as well as in hepatocytes [Citation29]. Certain mutations encoding full-length MYO5B with residual function can disrupt intracellular transport of proteins in hepatocytes, including that of BSEP [Citation52]. This leads to decreased protein expression and activity in the membrane [Citation29,Citation30]. Mutations encoding MYOB5 protein without residual function, cause disease in both the liver and the intestine, i.e. cholestasis and microvillus inclusion disease (MVID), respectively. MVID is characterized by intractable diarrhea starting within the first months after birth [Citation29,Citation30].
Children suffering from chronic intrahepatic cholestasis report a poorer quality of life than healthy children [Citation53]. Severe pruritus is one of the major complaints of PFIC and may cause cutaneous mutilation and affect daily life due to irritability, loss of sleep, and poor school performance [Citation54–58]. Patients with pruritus show poor physical health, health-related quality of life and low psychosocial scores compared to healthy individuals [Citation53]. A substantial part of PFIC patients needs a liver transplantation, sometimes even in the absence of end-stage liver disease, due to uncontrollable pruritus [Citation59]. Apart from the debilitating nature of PFIC and the psychological burden of therapy-resistant pruritus, the condition also has important economic consequences [Citation60], because liver transplantation and lifelong post-transplant care are costly. Successful treatment strategies would therefore not only improve the quality of life of the affected individuals but may also decrease financial burdens due to these diseases [Citation60].
Treatments currently in development aim to limit hepatocellular damage by reducing the amount of bile acids in the liver. Approaches aiming to interrupt the enterohepatic circulation of bile acids (e.g. using IBAT inhibitors) and inhibit bile acid synthesis in hepatocytes (e.g. FXR agonists and FGF19 mimetics) are furthest in development [Citation1,Citation51,Citation61]. In addition, efforts are ongoing to target the impaired proteins and underlying mutations using chaperones or gene therapy, although these have not yet reached phase II clinical trials.
2. Medical need
Until recently, there were no approved pharmacological treatments available for PFIC [Citation62]. In the absence of approved drugs, patients were treated by off-label use of medications aiming to decrease pruritus and by supportive care (vitamin supplementation, dietary treatments), each with limited efficacy [Citation49]. Surgical treatments are also applied, including different forms of interruption of the enterohepatic circulation such as partial biliary diversion (PBD) and, ultimately, liver transplantation [Citation51]. Data from the NAtural course and Prognosis of PFIC and Effect of biliary Diversion (NAPPED) study showed that surgical biliary diversion (SBD) significantly prolonged native liver survival (NLS) in BSEP1 and BSEP2 patients (Hazard ratio 0.50) [Citation47]. These patients have at least one missense mutation (p.D482G or p.E297G, which mutations have been associated with residual transport function in vitro, BSEP1), or at least one other missense mutation (and not p.D482G or p.E297G; BSEP2), respectively. The ability of SBD to delay or avert the need for liver transplantation in patients was associated with significantly decreased serum bile acid levels. Both serum bile acid levels <102 µmol/L and ≥75% decrease in serum bile acid levels after SBD were associated with prolonged NLS. However, SBD was not effective for all BSEP-deficient patients. Twenty-nine percent of patients who underwent SBD showed no improvement in pruritus. Thirty percent of patients required a secondary liver transplantation due to end-stage liver disease or refractory pruritus [Citation47]. Effective, preferably non-surgical, therapies are therefore urgently warranted.
3. Existing treatment
3.1. Ursodeoxycholic acid
Ursodeoxycholic acid (UDCA) is a hydrophilic bile acid, used as off-label treatment for PFIC [Citation5,Citation51]. It decreases the detergent activity of the bile acid pool by lowering its overall hydrophobicity. One study reported that liver tests improved in 14 out of 24 patients with MDR3 deficiency upon UDCA treatment, and in 5 patients pruritus resolved. However, UDCA only showed effect in children with a missense mutation, not in children with a truncated protein [Citation63]. These results suggest that at least some extent of residual MDR3 activity is required for the ability of UDCA to ameliorate disease, which has been confirmed by later findings [Citation64]. Patients with FIC1 or BSEP deficiencies may initially experience alleviation of symptoms upon UDCA treatment. However, in most patients, these effects are not sustained [Citation65]. Moreover, no method is available to predict who among these patients may experience some temporary alleviation of symptoms [Citation66]. The efficacy of UDCA in patients with PFIC is thus very limited, and sustained effects are only observed in MDR3 deficient patients with residual MDR3 activity. Norucholic acid (originally referred to as norursodeoxycholic acid [norUDCA]), the C-23 homologue of UDCA which is inefficiently conjugated in the liver, mitigated cholangitis more significantly in Abcb4 knockout mice than UDCA in one study [Citation67]. Norucholic acid also showed a significant reduction in alkaline phosphatase and a favorable safety profile in a phase 2 trial in primary sclerosing cholangitis (PSC) patients [Citation68]. Two other trials for norucholic acid in PBC and PSC are ongoing (EudraCT 2021–001431-56, NCT03872921), but no data from these are available yet.
3.2. Symptom management
Among PFIC patients, >75% experience severe pruritus, scoring ≥3 on a developed scratching scale ranging between 0 (no pruritus) to 4 (most severe pruritus), the Whitington scale [Citation54,Citation69]. Drugs used to manage cholestatic pruritus include cholestyramine, rifampicin, naltrexone, and antihistamines [Citation70]. However, these treatments often show no or limited efficacy, even when used in combination [Citation71]. Cholestyramine sequesters bile acids in the intestinal lumen, forming an insoluble resin complex that prevents bile acid reabsorption and thus increases their excretion via the feces [Citation72]. Rifampicin is a nuclear pregnane X receptor agonist [Citation73,Citation74] and is thought to induce hydroxylation of bile acids [Citation75]. Rifampicin is also proposed to increase metabolism of pruritogenic compounds [Citation76]. Naltrexone is an opioid antagonist, decreasing opioid-mediated neurotransmission associated with cholestatic pruritus [Citation77]. Antihistamines are often tried as initial therapy and may improve sleep through sedative effects in young patients, but ultimately show limited efficacy in cholestatic pruritus [Citation74].
Additionally, patients with PFIC are at risk of chronic malnutrition due to cholestasis-related malabsorption, especially of minerals such as calcium and zinc, and fat-soluble vitamins A, D, E, and K [Citation78,Citation79]. These deficiencies cause decreased bone mineral density and growth retardation, often refractory to supplementation. Nutritional support consists of a high-calorie, medium-chain triglyceride-rich and high protein diet with mineral and vitamin supplementation [Citation78,Citation79].
3.3. Surgical biliary diversion
SBD interventions include partial external biliary diversion (PEBD), partial internal biliary diversion and ileal exclusion (IE) [Citation80–82]. The common goal of the different forms of SBD is to drain the bile acid pool by interrupting their enterohepatic circulation [Citation83]. PEBD has been the most commonly used approach [Citation84]. SBD has been indicated for non-cirrhotic patients with insufficient response to antipruritic medication and aims to improve pruritus and to delay the need for liver transplantation [Citation45,Citation85,Citation86]. Only non-cirrhotic patients are considered for SBD, since retrospective studies indicated that secondary liver transplantation was required in the majority of cirrhotic patients who received SBD [Citation55,Citation59]. However, this concept has been challenged more recently [Citation87]. Biliary diversion is associated with improved health-related quality of life, specifically as a result of decreased pruritus and improved sleep [Citation57]. SBD can alleviate pruritus for sustained periods of time and may inhibit progression of liver fibrosis, but there is large variation in individual responses [Citation85,Citation88]. Favorable outcomes were particularly obtained in patients whose serum bile acid concentration strongly decreased after SBD [Citation47]. Comparing the surgical approaches, one study reported IE to be less effective than PEBD in the long-term, with symptoms recurring in half of patients after 12 months [Citation89]. This was attributed to gradual adaptation of the remaining ileum, leading to increased re-absorption of bile acids over time [Citation89]. Drawbacks of external diversion include stoma-related issues, such as dehydration and leakage [Citation90]. Internal diversion can be associated with malabsorption and choleretic diarrhea, due to supraphysiological quantities of bile acids entering the colon [Citation84]. Also, post-surgery complications may occur and require reoperations [Citation90].
3.4. Liver transplantation
Liver transplantation is indicated for PFIC patients who develop end-stage liver disease with severe complications, like liver cirrhosis or hepatocellular carcinoma, and for patients who have developed severe therapy-resistant pruritus in the absence of cirrhosis or end-stage liver disease [Citation51]. Liver transplantation has proven to be an effective treatment for PFIC. However, liver transplantation does not always ameliorate all symptoms among the various subtypes. Due to its extrahepatic expression, FIC1-deficient patients can still have or even develop extrahepatic symptoms after liver transplantation [Citation50]. Moreover, FIC1-deficient patients often develop inflammatory graft injury and marked graft steatosis post-transplantation [Citation12,Citation88,Citation91]. Furthermore, liver transplantation is associated with exacerbation of diarrhea in FIC1-deficient patients, which may hinder catch-up growth and reduce quality of life [Citation92]. Total external or internal biliary diversion can significantly mitigate graft injury in transplanted FIC1-deficient patients and can be performed concurrently with the transplantation [Citation83,Citation93,Citation94]. BSEP-deficient patients with two protein-truncating mutations typically require liver transplantation during their very first years of life, because of rapid deterioration of liver function and/or development of hepatocellular carcinoma [Citation93]. After liver transplantation, BSEP-deficient patients can develop antibodies that inhibit BSEP function in the transplanted liver, causing the original phenotype to reoccur [Citation61,Citation96–99]. Although limited availability of data hampers accurate assessment, it is estimated that about 10% of BSEP-deficient patients develop anti-BSEP antibodies after liver transplantation [Citation100]. Most patients can be effectively treated with increased immunosuppression, although some require several years of B-cell depletion [Citation100,Citation101]. Retransplantation may be required in selected cases. Other limitations of liver transplantation include the need for life-long immunosuppression, graft rejection and risk of infections [Citation102]. However, liver transplantation remains the ultimate treatment for PFIC patients, especially for those with treatment-resistant pruritus, hepatocellular carcinoma or end-stage liver disease [Citation55].
3.5. Odevixibat
The IBAT inhibitor odevixibat (BylvayTM) strongly reduces reabsorption of bile acids in the terminal ileum, thereby interrupting the enterohepatic circulation by non-surgical means [Citation103]. Odevixibat was approved by the EMA and FDA in 2021 [Citation62,Citation104,Citation105]. The EMA approved it for treating patients with any type of PFIC, aged 6 months and older [Citation104]. The FDA approved odevixibat for treatment of pruritus in PFIC patients of at least 3 months old [Citation105]. The recommended dose is 40 µg/kg body weight once a day in the morning, administered orally [Citation105]. If the response is inadequate after 3 months of continuous use, the dose may be increased up to 120 µg/kg daily, with a maximum daily dose of 7.2 mg in the EU and 6 mg in the USA [Citation105,Citation106]. In a double-blind, randomized, placebo-controlled phase 3 trial (NCT03566238) 2 dosing regiments, 40 µg/kg daily and 120 µg/kg daily, were evaluated [Citation107]. Odevixibat effectively reduced pruritus and serum bile acids in children with FIC1 deficiency (PFIC1) or in BSEP deficiency (PFIC2) without predicted protein truncating mutations. Children diagnosed with FIC1 or BSEP deficiency, with confirmed biallelic mutations and a history of severe pruritus due to PFIC as determined by the investigator, were included in this study. Patients with a predicted complete absence of BSEP activity or who had not responded with decreased pruritus to an IBAT inhibitor previously, were excluded [Citation107]. Based on its mode of action, odevixibat is thought to require at least some transport of bile acids into the bile and thus into the intestine [Citation108]. Accordingly, patients do not respond to odevixibat treatment if BSEP is fully absent or not functional at all [Citation106]. Non-responsiveness was also observed in FIC1 and in BSEP-deficient patients without predicted protein-truncating mutations. Estimated overall responsiveness in BSEP-deficient patients is therefore less than 50% [Citation47]. In the phase 3 trial, the percentage of patients achieving the predefined serum bile acid response was significantly higher in the treatment groups than in the placebo group (43% for 40 μg/kg/day, 21% for 120 μg/kg/day vs 0% for placebo). The serum bile acid response was defined as a reduction in serum bile acids by at least 70% from baseline or serum bile acids concentrations <70 μmol/L after 24 weeks of treatment [Citation107]. The proportion of patients who experienced a significant decrease in pruritus was also significantly higher in both treatment groups compared to the placebo group (58%, 52% vs 30%, respectively). The most common adverse events were transient diarrhea and frequent bowel movements: 31% of all patients treated with odevixibat vs 10% of the placebo group [Citation107]. An open-label extension of this trial is ongoing (NCT03659916) to evaluate the long-term safety and efficacy of odevixibat in PFIC patients after treatment for at least 72 weeks (about 1 and a half years). Interim results show that none of the patients classified as responders after 6 months of treatment required a liver transplantation for 3 years, while only 70% of non-responders survived with their native liver for 3 years [Citation109]. Patients experienced a decrease in pruritus within 4 weeks after initiating odevixibat, with a continued mean improvement over time. Although not significant in all cohorts, improvements in weight, height, and sleep parameters were observed in patients who were treated with odevixibat for up to 48 weeks. Out of 69 patients, 45 had a mild or moderate treatment-emerging adverse event. Eight patients (12%) experienced mild or moderate diarrhea, 2 of which were possibly or definitely related to the odevixibat treatment. No serious drug-related adverse events were observed [Citation109]. Gastrointestinal problems like diarrhea and abdominal pain are hypothesized to be secondary to non-reabsorbed bile acids spilling into the colon [Citation109,Citation110]. IBAT inhibitors increase the amount of bile acids entering the colon by inhibiting their ileal absorption. The latter, however, also interferes with ileal FXR signaling, thereby relieving the FGF19-mediated suppression of bile acid synthesis and thus in augmented bile acid production by the liver [Citation111].
4. Market review
Odevixibat and maralixibat are currently the only approved drugs for PFIC and therefore encompass the entire market size. No market data are available for maralixibat, because it has only been approved by the FDA as recently as March 2024. Off-label use of other drugs, including UDCA, is not accounted for. The FDA and EMA designated odevixibat as an orphan drug in 2012 [Citation112,Citation113]. The patent life of odevixibat extends to November 2031 in both the EU and USA [Citation114]. The EMA estimates a total number of 4000–5000 PFIC patients in the EU [Citation115]. In line, Albireo estimates the prevalence of all types of PFIC in Europe and the US to be 8,000–10,000 patients, taking into account the estimated incidence of PFIC and population statistics [Citation116]. Including only patients who have not received a liver transplantation, are under 17 years old and experience significant pruritus, Ipsen, the current license holder of odevixibat, estimated 500 PFIC patients in the USA to be eligible for odevixibat at its moment of launch [Citation114].
A significant proportion of PFIC patients are not eligible for treatment with IBAT inhibitors, since they have no residual BSEP function due to the specific genetic mutation they harbor [Citation47,Citation106,Citation117]. The genotype–phenotype relationship in PFIC has only been established for a subset of the known mutations [Citation12,Citation47]. However, 22% of patients registered in the NAPPED database have two predicted protein-truncating mutations associated with fully absent BSEP activity. Additionally, approximately 8% of BSEP-deficient patients registered in the NAPPED database have a genotype with a similarly severe phenotype. Based on this, 30% of patients with BSEP deficiency have a BSEP transport activity below the presumed threshold to clinically mitigate the disease with IBAT inhibitors [Citation117]. The aforementioned phase 3 trial excluded these patients in advance, and found that 58% of remaining patients showed a ≥1-point decrease in ObsRO (observation-reported outcome on a scale of 0 to 4) pruritus score, an ObsRO pruritus score of ≤1, a serum bile acid level ≤70 μmol/L, or a ≥70% reduction from baseline in fasting serum bile acid level [Citation107]. Reports on the prevalence of BSEP deficiency among all PFIC patients vary from 38% to 91% [Citation60]. Assuming a relative prevalence for BSEP deficiency of 50%, excluding 30% of whom based on predicted residual BSEP activity, and assuming a response rate of 58% among all remaining PFIC patients, we estimate that 4,000–5,000 patients in the EU and USA are likely to respond to IBAT treatment. However, the majority of these patients have likely already received a liver transplantation and are thus expected to not benefit from odevixibat treatment.
At the end of the third quarter in 2022, a total of 270 PFIC patients used odevixibat globally, yielding $7.5 million in net revenue [Citation118]. Ipsen reported €45.7 million in total global sales for odevixibat from March to October 2023, after finalizing the acquisition of Albireo in March of 2023 [Citation119]. Notably, odevixibat was also approved by the FDA for treatment of cholestatic pruritus in Alagille syndrome in the USA in June 2023 [Citation120]. Both indications are included by Ipsen in the reported sales and future estimated maximum net annual revenue, approximately $400 million assuming successful marketing going forward [Citation114,Citation119]. So far, odevixibat has been launched in nine European countries and is publicly reimbursed in Germany, France, Italy, the United Kingdom, Belgium, and the Netherlands, but has been rejected for reimbursement in Sweden [Citation114,Citation120–122].
5. Current research goals
The main research goal is the development of a treatment for PFIC that effectively achieves at least one of the following, with limited adverse effects: 1) decrease accumulation of bile acids within the liver, 2) reduce pruritus, 3) preserve liver function and 4) increase long-term quality of life for patients [Citation1]. Pharmacological treatments are of specific interest to delay or even prevent the need for surgery and accordingly, the risk on surgical complications [Citation84,Citation90,Citation92]. A major line of research involves pharmacological interruption of the enterohepatic circulation of bile acids. The aim of this approach is to reduce the size of the bile acid pool and, consequently, intrahepatic bile acid concentrations in PFIC patients. Its efficacy can be indirectly estimated by effects on serum bile acid concentrations as well as by improvement of symptoms like pruritus and overall quality of life [Citation123]. Other approaches aim to inhibit bile acid synthesis or increase activity of bile acid and phospholipid transport proteins, like BSEP, ATP8B1, and MDR3, in the canalicular membrane to reduce the hepatic accumulation of bile acids and thereby mitigate tissue damage and disease progression [Citation6,Citation124].
6. Scientific rationale
IBAT inhibitors aim to interrupt the enterohepatic circulation of bile acids by inhibiting reabsorption from the terminal ileum. This decreases the bile acid pool size, lowers serum bile acid concentrations, and reduces pruritus in a subset of patients [Citation107]. Other approaches include inhibition of bile acid synthesis (FXR agonists, FGF19 analogues) and chaperone treatment (4-phenylbutyrate [4-PB]). FXR inhibits bile acid synthesis through intestinal FGF19 secretion and activation of the FGFR4 receptor in the liver [Citation125]. A range of FXR agonists and FGF agonists have been developed as inhibitors of bile acid synthesis [Citation126].
As stated above, the wide variety of genetic mutations correspond with different pathophysiological mechanisms of disease. Depending on the underlying mutation, expression of the transporter protein, transport to and retention in the canalicular membrane may be decreased, or the inherent transport activity of the protein may be inhibited despite being present at the cell surface [Citation127]. Endoplasmic reticulum associated degradation inhibitors such as MG132 inhibit proteasomal degradation of misfolded proteins [Citation127,Citation128]. Chaperones like 4-phenylbutyrate aim to enhance the expression of functional transporter proteins at the cell membrane through the correction of protein misfolding and trafficking of proteins to the membrane [Citation129]. Once the protein is expressed at the cell membrane, potentiators such as ivacaftor may be able to improve its transporter function. Ivacaftor has been shown to significantly increase residual transport activity of BSEP in vitro [Citation131]. The strategies aiming to improve protein folding, localization and transport activity mentioned above may prove beneficial for PFIC patients with certain specific mutations, but will not be effective in all patients. Personalized approaches, in which therapy is tailored to the underlying genetic defect, will be required in order to further preserve liver function and improve the quality of life of more patients with PFIC.
7. Competitive environment
7.1. Search strategy
Phase 2 and phase 3 clinical trials of drugs for the treatment of PFIC were sought using ClinicalTrials.gov, GlobalData, and Orphanet (January 2015 – December 2023). Additional literature was explored using PubMed. Drug structures and chemical formulas were sourced from PubChem. A summary of these emerging drugs for the treatment of PFIC is shown in . The corresponding, completed trials are shown in Supplemental Table S1. Compounds in earlier stages of development are summarized in .
Table 2. Summary of emerging drugs for the treatment of PFIC that have been approved and are currently in phase 2 or 3 clinical trials.
Table 3. Summary of emerging drugs in development for the treatment of patients with cholestatic disease, or used in experimental treatment of PFIC patients.
7.2. Maralixibat
The intestine-restricted IBAT inhibitor maralixibat (LivmarliTM) was first approved for the treatment of cholestatic pruritus in patients with Alagille syndrome [Citation131]. The FDA approved its use in 2021 for patients of at least 3 months old [Citation132]; the EMA gave approval for use in patients aged 2 months and older in 2022 [Citation131,Citation133]. The recommended starting dose for this indication is 190 µg/kg body weight once daily, increased to 380 µg/kg once daily after a week [Citation131,Citation132]. Whereas the content of odevixibat capsules may be mixed with food, maralixibat is available as an oral solution and needs not to be mixed into food [Citation106,Citation131]. In March 2024, maralixibat was also approved by the FDA for the treatment of cholestatic pruritus in PFIC patients aged at least 5 years in the United States [Citation132]. The recommended starting dose for PFIC patients is 285 µg/kg body weight once daily in the morning and may be increased up to 570 µg/kg twice daily with a maximum daily dose of 38 mg [Citation132]. Maralixibat has been submitted for approval in PFIC patients of 2 months and older in Europe [Citation133]. Maralixibat has also been in clinical development for treatment of biliary atresia [Citation134], but recently negative results on the primary outcomes have been reported of a randomized controlled trial [Citation135].
The effects of maralixibat in children with PFIC were studied in a number of trials (NCT02057718, NCT03905330, NCT05543187). The INDIGO trial (NCT02057718), a long-term phase 2 trial, studied the efficacy and safety of maralixibat in 33 patients with FIC1 or BSEP deficiencies [Citation136]. Seven out of 19 patients (37%) with non-truncated BSEP deficiency met the responder criteria: a reduction in serum bile acids of >75% from baseline or concentrations <102.0 µmol/L. Six out of these seven patients responded within 2–4 weeks while using 266 µg/kg maralixibat once daily and mostly maintained this throughout the study. Improved liver biomarkers, pruritus, growth, and quality of life were also observed in these children. None of the FIC1-deficient or truncated-BSEP deficient patients met these responder criteria at any point in the study. All responding patients survived without liver transplantation after 312 weeks, compared to none of the non-responding patients. Responders also had a gradual increase in mean weight and height from baseline to week 72 and maintained this throughout the study. In contrast, non-responders experienced a decrease in weight and height in week 72 compared to baseline. All patients experienced at least one adverse event, most commonly pyrexia (61%), diarrhea (58%) and cough (55%). Fifteen patients reported serious adverse events, of which diarrhea, gastroenteritis, and abdominal pain were reported by more than 1 patient [Citation136].
Recently, the 26-week-long randomized phase 3 trial MARCH-PFIC was completed (NCT03905330) [Citation137,Citation138]. This trial included 93 PFIC patients harboring a larger range of deficiencies than in previous trials, including FIC1, non-truncated BSEP, MDR3, TJP2, and MYO5B-deficiencies. Patients received 570 μg/kg body weight maralixibat twice a day or placebo [Citation137]. The primary outcome was the mean change in the average morning itch (ObsRO) severity score in the primary cohort, consisting of 31 non-truncated BSEP-deficient patients. The secondary measures for this primary cohort were the mean change in total serum bile acid level, the proportion of ObsRO responders and of serum bile acid responders [Citation137,Citation138]. In addition to the primary cohort, 13 FIC1-, 9 MDR3-, 7 TJP2-, and 4 MYO5B-deficient patients were included in the study. Combined with the primary cohort, they formed the ‘all PFIC’ cohort consisting of 64 patients [Citation137]. Secondary outcome measures in the ‘all PFIC’ cohort were the mean change in average morning itch (ObsRO) severity score between baseline and the average of the last 12 weeks, the mean change in total serum bile acid levels, the proportion of ObsRO responders and the proportion of serum bile acid responders [Citation138]. The full-study population of 93 patients included an exploratory cohort of 29 patients besides the all-PFIC cohort. They did not meet inclusion criteria for the all-PFIC cohort due to heterozygosis (n = 2), truncated BSEP (n = 9), unidentified variants (n = 8), fluctuating serum bile acid levels (n = 2) or surgery (n = 8). Outcome measures for the full-study population were mean change in total and direct bilirubin, height, and weight from baseline [Citation137].
Preliminary results show that the most frequent adverse effects in the full-study cohort were transient, such as diarrhea (57.4% vs 19.6%) and abdominal pain (25.5% vs 13%) in the treatment vs. placebo group [Citation137,Citation139]. In the BSEP cohort, maralixibat treatment resulted in a significant mean reduction in pruritus and serum bile acids compared to placebo. The proportion of responders was significantly higher in maralixibat treated patients than in controls for reduced serum bile acids, but it was not significantly different for reduced pruritus [Citation138]. In the all-PFIC cohort, a significant reduction in pruritus and a significant improvement in sleep were observed after 26-weeks maralixibat treatment [Citation140]. In the last 10 weeks of the trial, the median proportion of days with the lowest pruritus scores was 95% in the treatment group vs. 9% in the placebo group [Citation140]. In the all-PFIC cohort, data of 60 patients was assessed for bilirubin normalization. Among patients with abnormal bilirubin levels, 10 out of 25 (40%) patients showed normalization of total bilirubin after maralixibat treatment, compared to 0 out of 18 (0%) patients who received placebo. Among patients with already normal total bilirubin, 0 out of 7 patients treated with maralixibat developed abnormal levels, compared to 3 out of 10 patients in the placebo group. Normalization of total bilirubin in all monitored patients was related to significantly larger reductions of serum bile acid concentrations; 94.9% reduction with normalized total bilirubin vs. 13.3% reduction without normalized total bilirubin [Citation141]. Remarkably, baseline data showed a trend of higher bilirubin levels in the treatment group vs. the placebo group [Citation137].
Notably, the administered dose of maralixibat has been increased throughout the trials. A dose of 280 μg/kg body weight once a day was used in the INDIGO trial (NCT02057718), whereas this was increased to 570 μg/kg body weight twice a day in the MARCH-PFIC trial (NCT03905330). Multiple phase 2 and 3 trials are ongoing (NCT04168385, NCT04729751, NCT04185363) to further study the efficacy of maralixibat in infants and children with cholestatic liver diseases, including PFIC. In these trials, maximum daily doses of 1140 μg/kg/day are used for patients 12 months and older, 570 μg/kg for patients up to 12 months, 600 μg/kg twice daily for patients 1–18 years old, respectively. The ongoing open-label extension trial MARCH-ON (NCT04185363) enrolled 88 patients who had completed the MARCH-PFIC trial, studying efficacy and safety of maralixibat up to 2 years [Citation142]. Despite the higher dose of maralixibat and longer treatment compared to the INDIGO trial, no novel adverse effects were reported in the MARCH-ON trial [Citation142].
7.3. Other drugs in development
Experimental compounds currently in development, but not yet in phase 2 trials (), include 4-PB, protein potentiators, systemic IBAT inhibitors and compounds targeting the FXR-FGF19 signaling axis. 4-PB is a drug approved for the treatment of urea cycle disorders [Citation143]. Yet, in vitro studies indicate that 4-PB can also increase the presence of non-mutated human BSEP as well as the mutants p.D482G, p.E297G and p.T1210P in the canalicular membrane by acting as a chaperone [Citation129,Citation144–146]. It has been suggested that 4-PB inhibits the adaptor complex that mediates BSEP internalization from the canalicular membrane [Citation145]. The effects of sodium phenylbutyrate have been assessed in BSEP-deficient patients with missense mutations associated with decreased canalicular expression of BSEP, while retaining its transport activity when present at the canaliculus. Results showed partial correction of BSEP expression, improvements in liver function biomarkers, liver histology, and bile secretion, while no severe adverse effects were observed during or after treatment [Citation146–148]. The condition of the patients remained stable and treatment tolerance was good with a median follow-up of 40 months [Citation147].
Cystic fibrosis transmembrane conductance regulator (CFTR) potentiators have also been implicated in PFIC treatment, rescuing dysfunctional BSEP that is properly targeted to the canalicular membrane but has reduced transport activity [Citation124,Citation130,Citation149]. The potentiators ivacaftor, GLP1837, SBC040 and SBC219 significantly improved bile acid transport activity of the BSEP A257V, T463I, and G562D missense mutants in HepG2 cells. Moreover, addition of SBC040 or SBC219 to ivacaftor resulted in a significant synergistic effect for T463I BSEP [Citation124]. In another study, ivacaftor fully rescued A257V BSEP function in HepG2 cells [Citation149].
Systemic IBAT inhibitors like A3907, are possibly more potent than ileal IBAT inhibitors by targeting IBAT throughout the body, e.g. enhancing urinary bile acid excretion by preventing their reabsorption by proximal tubular epithelial cells in the kidney [Citation111]. A3907 was found to be potent and act selectively in vitro, and to be distributed to the liver, ileum, and kidneys in Abcb4 knockout mice. A3907 administered by oral gavage reduced serum bile acid concentrations, improved markers of liver injury and increased fecal and urinary excretion of bile acids in male adult Abcb4 knockout mice as well as in male adult C57BL/6 J mice with bile duct ligation. In a phase 1 study in healthy volunteers, oral administration of A3907 resulted in plasma concentrations similar to the therapeutic concentrations in mice [Citation150].
FGF19 mimetics (e.g. aldafermin) and FXR agonists (e.g. obeticholic acid and cilofexor) inhibit bile acid synthesis [Citation151–153] and may thereby limit liver pathology in cholestatic conditions. FXR agonists like obeticholic acid and cilofexor are experimentally used as off-label treatment for primary sclerosing cholangitis (PSC) [Citation151,Citation152]. Obeticholic acid (OCA, 6-α ethyl CDCA, OcalivaTM) is a synthetic steroidal FXR agonist. OCA was approved in the EU and USA in 2016 for the treatment of primary biliary cholangitis (PBC) in combination with UDCA for adult patients with an inadequate response to UDCA monotherapy [Citation154,Citation155]. In male Wistar rats with induced cholestasis, OCA has been shown to induce bile flow and protect hepatocytes against necrosis due to intrahepatic accumulation of hydrophobic bile acids [Citation156]. In PBC patients, OCA monotherapy significantly decreased alkaline phosphatase from baseline and resulted in total bilirubin levels significantly lower than in the placebo group [Citation157]. Cilofexor is a non-steroidal FXR agonist that significantly decreased liver transaminases in a phase 2 trial in PSC patients [Citation151]. However, two other trials for cilofexor in PBC and PSC patients have been terminated, failing to meet the primary end point (NCT02943447, NCT03890120). In fact, both OCA and cilofexor were found to induce pruritus in a dose-dependent manner, making it less suitable for PFIC monotherapy. Nonetheless, inclusion in combination therapy with antipruritic agents remains a line of research [Citation157,Citation158]. For example, a recent study found that combination therapy of the IBAT inhibitor linerixibat and the FXR agonist cilofexor effectively reduced liver injury and lowered the bile salt load in the colon of cholestatic mice [Citation159].
8. Potential development issues
8.1. IBAT inhibitors
The most frequently observed adverse effects of odevixibat and maralixibat are gastrointestinal issues, like diarrhea, abdominal pain, and infrequent bowel movements [Citation107,Citation136]. These are due to the supraphysiological concentration of bile acids in the colon, which affect motility and absorption of water and electrolytes, causing choleretic diarrhea [Citation5]. Since this is a direct consequence of the mechanism of action of IBAT inhibitors, these adverse effects are also anticipated for other IBAT inhibitors still in development. Gastrointestinal issues are therefore expected to limit the tolerability of this entire drug class. Yet, a supraphysiological concentration of bile acids is mainly expected in responders and may be less apparent if the bile acid pool size has been decreased after the initial phase of the IBAT inhibitor treatment.
8.2. Lacking efficacy of current treatment
The major drawback of current and experimental therapies is that they are only efficacious in a part of the patients. In the PEDFIC1 phase 3 trial (NCT03566238), odevixibat was found to significantly decrease pruritus and serum bile acid concentrations [Citation107]. However, only 10 out of 23 patients (low dose) and 4 out of 20 patients (high dose) met the primary endpoints after 24 weeks of treatment. The two primary endpoints were the proportion of a patient’s scratching score ≤1 or a ≥1-point reduction from baseline over 24 weeks, and the proportion of patients with a ≥70% reduction from baseline in fasting serum bile acid level or a serum bile acid level ≤70 μmol/L at week 24. Importantly, BSEP deficiency patients with predicted protein-truncating mutations were excluded from this study [Citation107]. Therefore, the overall response rate of BSEP-deficient patients to odevixibat most conceivably is considerably lower when taking those a priori excluded, suppositionally non-responding, patients into account. Indeed, maralixibat only showed improvements in a fraction of the treated patients with non-truncated BSEP mutations, while no improvements were observed in patients with truncated BSEP [Citation136]. At present, no effective pharmacological treatments are available for patients with complete loss of BSEP activity. Furthermore, patients with residual BSEP function also do not always respond to available off-label treatment. Although widely used, UDCA is only effective in a limited number of patients and even then, frequently not in sustained fashion [Citation57]. A recent case report describes a patient with ZFYVE deficiency who only responded when odevixibat was added to the combination therapy of rifampicin and UDCA [Citation38]. In short, currently used therapies are often not effective in a substantial part of PFIC patients. Although the degree of residual function plays a clear role for therapies interrupting the enterohepatic circulation of bile acids (e.g. IBAT inhibitors), the mechanisms underlying the (un)responsiveness to other therapeutic approaches are still largely enigmatic.
8.3. Drug development in context of approved IBAT inhibitors
Since both odevixibat and maralixibat have already been approved, the development and marketing of new therapies have become more challenging. A case series indicates that odevixibat may also be effective in TJP2 deficient patients, supporting its position as an effective drug, also for the more uncommon PFIC subtypes [Citation160]. Patients will primarily be treated with established, approved treatment options before entering experimental treatments or clinical trials. Consequently, future research will mostly be conducted in patients who do not respond to these currently available treatments. Patients only become available for clinical trials once other approved or off-label drugs, like odevixibat and UDCA, have been proven ineffective. Likely, this will result in fewer patients eligible for inclusion in future clinical trials. Furthermore, the patients that are eligible for inclusion have been selected for a more challenging phenotype and can thus be expected to be less responsive to treatments, creating a bias in the study population. As a result, the beneficial effects of an experimental drug may be underestimated if such a study were undertaken. Classically, a high number of patients are needed for a randomized controlled study to obtain regulatory approval of a new drug. However, the (ultra)rarity of the different subtypes of PFIC, in combination with the large genetic variability within the different subtypes, does not accord with this classic concept. Particularly, very few patients can be included in the development of treatments tailored to specific mutations. Alternatively, new designs of clinical trials may be required, including n = 1 trials. Here, patients act as their own control, requiring fewer patients for sufficiently powered results. The investigational drug and the control are administered in separate time periods with intermittent wash-out periods. Regardless, future clinical trials will conceivably take a longer time to complete which will increase the time needed for a novel drug to be approved by the responsible agencies and reach the market. This will shorten the time span between market authorization and patent expiration, thus limiting market protection under its patent. Market protection may however also be upheld by orphan designation. Thus, paradoxically, new therapies for hitherto IBAT inhibitor unresponsive patients will encounter hurdles from the recent successes obtained by odevixibat and maralixibat.
9. Conclusion
UDCA is a regularly used off-label treatment but is only efficacious in a subset of patients and shows limited, and often only temporal, benefit. Currently, odevixibat and maralixibat are the only approved drugs for treatment of PFIC with efficacy toward reducing both serum bile acids and pruritus. Not all patients, however, are responsive to these IBAT inhibitors. SBD does not offer guaranteed success (prolonged relief of pruritus and prolonged survival with native liver), may cause complications and require extensive post-operative care. It is still unclear to what extent the recent availability of odevixibat will decrease the indication for SBD. The surgical approach of liver transplantation successfully alleviates pruritus, but obviously is an invasive procedure with perioperative morbidity and mortality and with the need for long-term immunosuppressive therapy. Other experimental compounds in earlier stages of development include chaperones (4-PB), potentiators (e.g. ivacaftor), systemic IBAT inhibitors, FGF mimetics and FXR agonists. Further research into these promising compounds is needed to develop a treatment that effectively prevents liver injury, relieves pruritus, and improves patients’ prognoses and quality of life.
10. Expert opinion
The development of the IBAT inhibitors gave rise to the first two drugs being approved for treatment of PFIC: odevixibat and maralixibat. Moreover, compounds such as systemic IBAT inhibitors [Citation150], protein potentiators [Citation130], chaperones [Citation161,Citation162,Citation163], FXR agonists [Citation151], FGF19 mimetics [Citation164] and norucholic acid [Citation165] have shown promising results in vitro or as experimental treatments. However, years of further studies regarding their safety and efficacy in PFIC patients are required before clinical use, not only in patients that are not responsive to IBAT inhibition, but in patients with newly identified subtypes of PFIC.
Most strategies that are currently envisioned to have potential for the treatment of cholestatic liver diseases such as PFIC face inherent disadvantages, as they do not fully restore bile acid homeostasis. This applies especially to approaches other than restoring biliary excretion of bile acids from the hepatocytes into the bile canaliculi. For example, interruption of the enterohepatic circulation may reduce the accumulation of bile acids in the liver but may also decrease bile acid concentrations in the small intestine. Low intraluminal bile acid concentrations can decrease solubilization of dietary fat and fat-soluble vitamins, resulting in the need for nutritional supplements. Additionally, IBAT inhibition may lead to a supraphysiological bile acid concentration in the lumen of the colon, particularly in the initial phase of treatment, causing gastrointestinal adverse effects in many patients as illustrated in multiple clinical studies for IBAT inhibitors [Citation107,Citation136,Citation142]. The long-term effects of IBAT inhibition on colonic mucosa have not yet been identified. Inhibition of bile acid synthesis may reduce bile acid accumulation in the liver more directly but could require relatively long-term treatment of PFIC patients before obtaining clinically meaningful results due to the enterohepatic circulation.
The ideal pharmacological treatment for PFIC patients would restore the hepatobiliary secretion of bile acids, thereby preventing cholestasis and its harmful consequences. Additionally, sufficient biliary secretion of bile acids allows effective solubilization of fat-soluble nutrients, which may prevent the need for medium-chain triglyceride-rich diets and supplementation of fat-soluble vitamins. Restoration of biliary bile acid secretion would result in (partial) restoration of bile acid homeostasis and therefore can be expected to facilitate, at least to some extent, the physiological modulation of glucose and energy metabolism by bile acids [Citation166]. Also, the first-pass extraction from the portal vein would increase, decreasing the spillover to the peripheral blood. This may be associated with alleviation of symptoms like pruritus, and prolonged native liver survival.
Systemic IBAT inhibitors aim to divert bile acids toward both the kidneys and the colon. This may prove more effective than intestinal IBAT inhibitors for patients with fully absent biliary bile acid secretion, e.g. patients with truncated BSEP. Afterall, its mechanism of action does not only rely on the enterohepatic circulation of bile acids but also facilitates urinary excretion of bile acids from the systemic blood circulation. Although systemic IBAT inhibitors show promising preliminary results, the systemic exposure may also raise new potential issues compared to the ileal IBAT inhibitors, mostly related to pharmacokinetics. For example, there could be a higher risk of additional adverse effects, since a major part of the body will be exposed to the systemic drug, instead of only the lumen of the gastrointestinal tract. Another point of interest is the alternative route of excretion for bile acids. Serum bile acids will more readily be excreted via the kidneys to avoid gastrointestinal adverse effects [Citation111]. Theoretically, this raises the possibility of urinary supraphysiological bile acid concentrations causing kidney– or urinary tract-related adverse effects, although the measured bile acid concentrations remain still relatively low. On the contrary, recent data have shown that no increased urinary bile acid levels or adverse effects were observed after 13 weeks in a phase 1 study with healthy adults receiving 9 mg, 27 mg, or 67.5 mg A3907 once daily [Citation150]. Moreover, data indicate that the systemic IBAT inhibitor protects against cholestatic nephropathy. In bile duct ligated mice, inhibiting renal IBAT increased renal excretion of bile acids, but also decreased bile acid uptake into tubular epithelial cells, preventing kidney damage [Citation167]. Nonetheless, this may yield different results in pediatric PFIC patients. Altogether, further research is required to fully explore the benefits and possible adverse effects of systemic IBAT inhibitors.
Other than the systemic IBAT inhibitors, an interesting emerging field is combination therapy of a potentiator, like ivacaftor, and a chaperone, like 4-PB or its derivative glycerol phenylbutyrate [Citation124,Citation168]. This may be applied to increase the fraction of BSEP successfully targeted at the canalicular membrane and to optimize its activity once located there. This approach would specifically target the underlying pathology of PFIC. Research into this area can build upon available data of these drugs that have already been approved for other indications, and in some cases used experimentally to treat PFIC patients [Citation146,Citation164]. However, efficacy of the chaperone/potentiator combination would likely depend on the specific mutation and may only benefit a small number of patients.
Gene therapy is another approach to address the root cause of PFIC, the genetic mutations. Its affinity for the liver is an advantage for adeno-associated virus (AAV) mediated gene transfer targeting the liver [Citation169]. Pre-clinical research in Abcb4 knockout mice has indicated the feasibility of gene therapy mediated by AAV for ABCB4 deficiency [Citation171]. Previous data had shown that targeting a proportion of hepatocytes resulted in partial normalization of the phosphatidylcholine level in bile, achieving a therapeutic effect [Citation3,Citation172]. However, there are a number of challenges for clinical application of gene therapy for all types of PFIC. First, AAV vectors remain in the nucleus of the host tissue but are not integrated into the genome and thus are not copied into daughter cells during cell division [Citation173]. Consequently, hepatocyte proliferation in infancy and proliferation induced by liver damage limit the long-term efficacy of the AAV vector [Citation170]. Additionally, other types of PFIC require targeting of most or all hepatocytes to effectively prevent accumulation of bile acids within hepatocytes, for example by targeting BSEP in all hepatocytes. Therefore, genome-integrating gene therapy may be more suitable, but this has not yet reached the stage of clinical trials. Ex vivo gene therapy is limited by the need for a large number of preferably patient-derived, healthy hepatocytes to repopulate the liver. Thus, the development of gene therapy for PFIC is still in its infancy and will take a substantial amount of time to completely develop. Furthermore, a wide array of treatments will be required for the different mutations in personalized gene therapy for each patient. Although likely possible in the future, this would be a very expensive procedure. Lastly, PFIC presents itself in infancy or early childhood in many patients. This involves practical challenges, since little if any experience exists regarding gene therapy for such young patients.
The main challenges in the development of a treatment for PFIC patients are to alleviate the consequences of the mutations on bile formation, ideally by restoring as much as possible the normal bile acid homeostasis. To prevent a decline in the further development of novel therapeutic strategies for PFIC, a reorientation on regulatory requirements for drug approval may be necessary to allow personalized approaches in patients with these ultrarare diseases, frequently with unique underlying genetic mutations.
Abbreviations
4-PB, 4-phenylbutyrate; AAV, adeno-associated virus; ABCB11, ATP-binding cassette sub-family B member 11 protein; ALT, alanine aminotransferase; ASBT, apical sodium-dependent bile acid transporter; AST, aspartate aminotransferase; ATP8B1, ATPase phospholipid transporting 8B1; BSEP, bile salt export pump; CFTR, cystic fibrosis transmembrane conductance regulator; FIC1, familial intrahepatic cholestasis 1; FGF19, fibroblast growth factor 19; FXR, farnesoid X receptor; GGT, gamma-glutamyl transferase; IBAT, ileal bile acid transporter; IE, ileal exclusion; KIF12, kinesin family member 12; MDR3, multidrug resistance protein 3; MVID, microvillus inclusion disease; MYO5B, myosin 5B; MASLD, metabolic-dysfunction associated steatotic liver disease; NAPPED, NAtural course and Prognosis of PFIC and Effect of biliary Diversion; MASH, metabolic dysfunction-associated steatohepatitis; NLS, native liver survival; norUDCA, norursodeoxycholic acid; OCA, obeticholic acid; PBC, primary biliary cholangitis; PBD, partial biliary diversion; PEBD, partial external biliary diversion; PFIC, progressive familial intrahepatic cholestasis; PSC, primary sclerosis cholangitis; sBA, serum bile acids; SBD, surgical biliary diversion; TJP2, tight junction protein 2; UDCA, ursodeoxycholic acid; USP53, ubiquitin-specific peptidase 53.
Declaration of interest
HJ Verkade has been a consultant for Ausnutria, Albireo AB/Ipsen, Mirum, Vivet, Intercept and Orphalan (each on ad interim basis) and has received unrestricted research grants from Albireo AB/Ipsen and Mirum. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
A reviewer on this manuscript has disclosed that they are a Consultant for IPSEN, CAMP4 therapeutics and SIGNANTHEALTH. They have also received speaker’s fee from ADVANZ. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Supplemental Material
Download MS Word (410.9 KB)Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14728214.2024.2336986.
Additional information
Funding
References
- Kamath BM, Stein P, Houwen RHJ, et al. Potential of ileal bile acid transporter inhibition as a therapeutic target in Alagille syndrome and progressive familial intrahepatic cholestasis. Liver Int. 2020;40(8):1812–1822.
- Li T, Udayan A. Bile acid metabolism and signaling in cholestasis, inflammation and cancer. Adv Pharmacol. 2015;74(1):263–302.
- Davit-Spraul A, Gonzales E, Baussan C, et al. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4(1):1–12.
- Ruth ND, Gray Z, McKay K, et al. P1248 identifying incidence of inherited metabolic disorders in patients with infantile liver disease. J Hepatol. 2014;60(1).
- Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159(22):2647–2658.
- Stellaard F, Lütjohann D. Dynamics of the enterohepatic circulation of bile acids in healthy humans. Am J Physiol Gastrointest Liver Physiol. 2021;321(1):G55–66.
- Keitel V, Dröge C, Häussinger D. Targeting FXR in Cholestasis. In: Fiorucci S, Distrutti E, editors. Handbook of Experimental Pharmacology. Cham, Switzerland: Springer Nature Switzerland; 2019. p. 299–324.
- Cariello M, Piccinin E, Garcia-Irigoyen O, et al. Nuclear receptor FXR, bile acids and liver damage: introducing the progressive familial intrahepatic cholestasis with FXR mutations. Biochim Biophys Acta - Mol Basis Dis. 2018;1864(4):1308–1318.
- Kurosu H, Ogawa Y, Miyoshi M, et al. Regulation of fibroblast growth factor-23 signaling by Klotho. J Biol Chem. 2006;281(10):6120–6123.
- Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2012;36(SUPPL.1):S26–35.
- Johns Hopkins University. Online Mendelian inheritance in man PFIC [Internet]. 2023. Available from: https://www.omim.org/search?index=entry&start=1&limit=10&sort=score+desc%2C+prefix_sort+desc&search=pfic
- Davit-Spraul A, Fabre M, Branchereau S, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase Progressive Familial Intrahepatic Cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. 2010;51(5):1645–1655.
- Groen A, Romero MR, Kunne C, et al. Complementary functions of the flippase ATP8B1 and the floppase ABCB4 in maintaining canalicular membrane integrity. Gastroenterology. 2011;141(5):1927–1937.e4.
- Strautnieks SS, Byrne JA, Pawlikowska L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134(4):1203–1214.
- Evason K, Bove KE, Finegold MJ, et al. Morphologic findings in progressive familial intrahepatic cholestasis 2 (PFIC2): correlation with genetic and immunohistochemical studies. Am J Surg Pathol. 2011;35(5):687–696.
- Gudbjartsson DF, Helgason H, Gudjonsson SA, et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet. 2015;47(5):435–444.
- Tougeron D, Fotsing G, Barbu V, et al. ABCB4/MDR3 gene mutations and cholangiocarcinomas. J Hepatol. 2012;57(2):467–468.
- Jacquemin E, De Vree JML, Cresteil D, et al. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. 2001;120(6):1448–1458.
- Crawford AR, Smith AJ, Hatch VC, et al. Hepatic secretion of phospholipid vesicles in the mouse critically depends on mdr2 or MDR3 P-glycoprotein expression: visualization by electron microscopy. J Clin Invest. 1997;100(10):2562–2567.
- Kamarajah S, Patten D, Wadkin J, et al. Reduced expression of TJP-2 is associated with chronic liver disease and hepatic malignancy. Gut. 2016;65(1):A159.
- Sambrotta M, Thompson RJ. Mutations in TJP2, encoding zona occludens 2, and liver disease. Tissue Barriers. 2015;3(3):1–5.
- Sambrotta M, Strautnieks S, Papouli E, et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet. 2014;46(4):326–328.
- Xu J, Kausalya PJ, Van Hul N, et al. Protective functions of ZO-2/Tjp2 expressed in hepatocytes and cholangiocytes against liver injury and cholestasis. Gastroenterology. 2021;160(6):2103–2118.
- Gomez-Ospina N, Potter CJ, Xiao R, et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat Commun. 2016;7:1–8.
- Qiu YL, Gong JY, Feng JY, et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low γ-glutamyltransferase cholestasis. Hepatology. 2017;65(5):1655–1669.
- Amirneni S, Haep N, Gad MA, et al. Molecular overview of progressive familial intrahepatic cholestasis. World J Gastroenterol. 2020;26(47):7470–7484.
- Gissen P, Arias IM. Structural and functional hepatocyte polarity and liver disease. J Hepatol. 2015;63(4):1023–1037.
- Gonzales E, Taylor SA, Davit-Spraul A, et al. MYO5B mutations cause cholestasis with normal serum gamma-glutamyl transferase activity in children without microvillous inclusion disease. Hepatology. 2017;65(1):164–173.
- Aldrian D, Vogel GF, Frey TK, et al. Congenital diarrhea and cholestatic liver disease: phenotypic spectrum associated with myo5b mutations. J Clin Med. 2021;10(3):1–15.
- Uyar Aksu N, Görükmez O, Ö Görükmez, et al. A novel homozygous mutation in the MYO5B gene associated with normal-gamma-glutamyl transferase progressive familial intrahepatic cholestasis. Cureus. 2021;13(11):1–5.
- Bull LN, Ellmers R, Foskett P, et al. Cholestasis due to USP53 deficiency. J Pediatr Gastroenterol Nutr. 2021;72(5):667–673.
- Samanta A, Parveen N, Sen Sarma M, et al. Cholestatic liver disease due to novel USP53 mutations: a case series of three Indian children. J Clin Exp Hepatol. 2024;14(2):101290.
- Nakagawa T, Tanaka Y, Matsuoka E, et al. Identification and classification of 16 new kinesin superfamily (KIF) proteins in mouse genome. Proc Natl Acad Sci U S A. 1997;94(18):9654–9659.
- Stalke A, Sgodda M, Cantz T, et al. KIF12 variants and disturbed hepatocyte polarity in children with a phenotypic spectrum of cholestatic liver disease. J Pediatr. 2022;240:284–291.
- Luan W, Hao CZ, Li JQ, et al. Biallelic loss-of-function ZFYVE19 mutations are associated with congenital hepatic fibrosis, sclerosing cholangiopathy and high-GGT cholestasis. J Med Genet. 2021;58(8):514–525.
- Mandato C, Siano MA, Nazzaro L, et al. A ZFYVE19 gene mutation associated with neonatal cholestasis and cilia dysfunction: case report with a novel pathogenic variant. Orphanet J Rare Dis. 2021;16(1):1–9.
- Wu SH, Chang MH, Chen YH, et al. The ESCRT-III molecules regulate the apical targeting of bile salt export pump. J Biomed Sci. 2021;28(1):1–18.
- Pepe A, Colucci A, Carucci M, et al. Case Report: add-on treatment with odevixibat in a new subtype of progressive familial intrahepatic cholestasis broadens the therapeutic horizon of genetic cholestasis. Front Pediatr. 2023;11(February):1–5.
- Satomura Y, Bessho K, Nawa N, et al. Novel gene mutations in three Japanese patients with ARC syndrome associated mild phenotypes: a case series. J Med Case Rep. 2022;16(1):1–6.
- Fu KL FK, Chen P, Zhou YY, et al. Hepatic Vps33b deficiency aggravates cholic acid-induced cholestatic liver injury in male mice. Acta Pharmacol Sin. 2022;43(4):933–940.
- Van Mil SWC, Houwen RHJ, Klomp LWJ. Genetics of familial intrahepatic cholestasis syndromes. J Med Genet. 2005;42(6):449–463.
- Srivastava A. Progressive Familial Intrahepatic Cholestasis. J Clin Exp Hepatol. 2014;4(1):25–36.
- Vitale G, Gitto S, Vukotic R, et al. Familial intrahepatic cholestasis: new and wide perspectives. Dig Liver Dis. 2019;51(7):922–933.
- Paulusma CC, De Waart DR, Kunne C, et al. Activity of the bile salt export pump (ABCB11) is critically dependent on canalicular membrane cholesterol content. J Biol Chem. 2009;284(15):9947–9954.
- Lemoine C, Superina R. Surgical diversion of enterohepatic circulation in pediatric cholestasis. Semin Pediatr Surg. 2020;29(4):150946.
- Jansen PLM, Strautnieks SS, Jacquemin E, et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117(6):1370–1379.
- Van Wessel DBE, Thompson RJ, Gonzales E, et al. Genotype correlates with the natural history of severe bile salt export pump deficiency. J Hepatol. 2020;73(1):84–93.
- Alissa FT, Jaffe R, Shneider BL. Update on progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2008;46(3):241–252.
- Bosma PJ, Wits M, Oude-Elferink RPJ. Gene therapy for progressive familial intrahepatic cholestasis: current progress and future prospects. Int J Mol Sci. 2021;22(1):1–13.
- Bull LN, Thompson RJ. Progressive Familial Intrahepatic Cholestasis. Clin Liver Dis. 2018;22(4):657–669.
- Martínez-García J, Molina A, González-Aseguinolaza G, et al. Gene therapy for acquired and genetic cholestasis. Biomedicines. 2022;10(6):1–20.
- Girard M, Lacaille F, Verkarre V, et al. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology. 2014;60(1):301–310.
- Kamath BM, Chen Z, Romero R, et al. Quality of life and its determinants in a multicenter cohort of children with alagille syndrome. J Pediatr. 2015;167(2):390–396.e3.
- Lee WS, Chai PF, Looi LM. Progressive familial intrahepatic cholestasis in Malaysian patients - A report of five cases. Med J Malaysia. 2009;64(3):216–219.
- Schukfeh N, Metzelder ML, Petersen C, et al. Normalization of serum bile acids after partial external biliary diversion indicates an excellent long-term outcome in children with progressive familial intrahepatic cholestasis. J Pediatr Surg. 2012;47(3):501–505.
- Yang H, Porte RJ, Verkade HJ, et al. Partial external biliary diversion in children with progressive familial intrahepatic cholestasis and alagille disease. J Pediatr Gastroenterol Nutr. 2009;49(2):216–221.
- Jones-Hughes T, Campbell J, Crathorne L. Epidemiology and burden of progressive familial intrahepatic cholestasis: a systematic review. Orphanet J Rare Dis. 2021;16(1):1–14.
- Mehl A, Bohorquez H, Serrano MS, et al. Liver transplantation and the management of progressive familial intrahepatic cholestasis in children. World J Transplant. 2016;6(2):278.
- Englert C, Grabhorn E, Richter A, et al. Liver transplantation in children with progressive familial intrahepatic cholestasis. Transplantation. 2007;84(10):1361–1363.
- Baker A, Kerkar N, Todorova L, et al. Systematic review of progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2019;43(1):20–36.
- Kubitz R, Dröge C, Kluge S, et al. Autoimmune BSEP disease: disease recurrence after liver transplantation for progressive familial intrahepatic cholestasis. Clin Rev Allergy Immunol. 2015;48:273–284.
- Deeks ED. Odevixibat: first approval. Drugs. 2021;81(15):1781–1786.
- Jacquemin E. Role of multidrug resistance 3 deficiency in pediatric and adult liver disease: one gene for three diseases. Semin Liver Dis. 2001;21(4):551–562.
- Gordo-Gilart R, Andueza S, Hierro L, et al. Functional analysis of ABCB4 mutations relates clinical outcomes of progressive familial intrahepatic cholestasis type 3 to the degree of MDR3 floppase activity. Gut. 2015;64(1):147–155.
- European Association for the Study of the Liver. EASL clinical practice guidelines: management of cholestatic liver diseases. J Hepatol [Internet]. 2009;51(2):237–267. doi: 10.1016/j.jhep.2009.04.009
- Stapelbroek JM, van Erpecum KJ, Klomp LWJ, et al. Liver disease associated with canalicular transport defects: current and future therapies. J Hepatol. 2010;52(2):258–271.
- Fickert P, Wagner M, Marschall HU, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130(2):465–481.
- Fickert P, Hirschfield GM, Denk G, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol. 2017;67(3):549–558.
- Whitington PF, Freese DK, Alonso EM, et al. Clinical and biochemical findings in progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 1994;18:134–141.
- Bhalerao A, Mannu GS. Management of pruritus in chronic liver disease. Dermatol Res Pract. 2015;2015.
- Hegade VS, Kendrick SFW, Jones DEJ. Drug treatment of pruritus in liver diseases. Clin Med J R Coll Physicians London. 2015;15(4):351–357.
- Scaldaferri F, Pizzoferrato M, Ponziani FR, et al. Use and indications of cholestyramine and bile acid sequestrants. Intern Emerg Med. 2013;8(3):205–210.
- Li T, Chiang JYL. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7α-hydroxylase gene transcription. Am J Physiol Gastrointest Liver Physiol. 2005;288(1 51–1):74–84.
- Beuers U, Wolters F, Oude Elferink RPJ. Mechanisms of pruritus in cholestasis: understanding and treating the itch. Nat Rev Gastroenterol Hepatol. 2023;20(1):26–36.
- Wietholtz H, Marschall HU, Jan S, et al. Stimulation of bile acid 6α-hydroxylation by rifampin. J Hepatol. 1996;24(6):713–718.
- Marschall H, Wagner M, Zollner G, et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology. 2005;129(2):476–485.
- Murray-Brown FL. Naltrexone for cholestatic itch: a systematic review. BMJ Support Palliat Care. 2021;11(2):217–225.
- Yang CH, Perumpail BJ, Yoo ER, et al. Nutritional needs and support for children with chronic liver disease. Nutrients. 2017;9(10):1–16.
- Agarwal S, Lal BB, Rawat D, et al. Progressive familial intrahepatic cholestasis (PFIC) in Indian children: clinical spectrum and outcome. J Clin Exp Hepatol. 2016;6(3):203–208.
- Whitington PF, Whitington GL. Partial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasis. Gastroenterology. 1988;95(1):130–136.
- Gunaydin M, Tander B, Demirel D, et al. Different techniques for biliary diversion in progressive familial intrahepatic cholestasis. J Pediatr Surg. 2016;51(3):386–389.
- Van Vaisberg V, Tannuri ACA, Lima FR, et al. Ileal exclusion for pruritus treatment in children with progressive familial intrahepatic cholestasis and other cholestatic diseases. J Pediatr Surg. 2020;55(7):1385–1391.
- Usui M, Isaji S, Das BC, et al. Liver retransplantation with external biliary diversion for progressive familial intrahepatic cholestasis type 1: a case report. Pediatr Transplant. 2009;13(5):611–614.
- Wang KS, Tiao G, Bass LM, et al. Analysis of surgical interruption of the enterohepatic circulation as a treatment for pediatric cholestasis. Hepatology. 2017;65(5):1645–1654.
- Hüpper MN, Pichler J, Huber WD, et al. Surgical versus medical management of progressive familial intrahepatic cholestasis—case compilation and review of the literature. Children. 2023;10(6).
- Van Der Woerd WL, Houwen RHJ, Van De Graaf SFJ. Current and future therapies for inherited cholestatic liver diseases. World J Gastroenterol. 2017;23(5):763–775.
- Pfister ED, Jaeger VK, Karch A, et al. Native liver survival in bile salt export pump deficiency: results of a retrospective cohort study. Hepatol Commun. 2023;7(4).
- Bull LN, Pawlikowska L, Strautnieks S, et al. Outcomes of surgical management of familial intrahepatic cholestasis 1 and bile salt export protein deficiencies. Hepatol Commun. 2018;2(5):515–528.
- Kaliciński PJ, Ismail H, Jankowska I, et al. Surgical treatment of progressive familial intrahepatic cholestasis: comparison of partial external biliary diversion and ileal bypass. Eur J Pediatr Surg. 2003;13(5):307–311.
- Bjornland K, Hukkinen M, Gatzinsky V, et al. Partial biliary diversion may promote long-term relief of pruritus and native liver survival in children with cholestatic liver diseases. Eur J Pediatr Surg. 2021;31(4):341–346.
- Miyagawa-Hayashino A, Egawa H, Yorifuji T, et al. Allograft steatohepatitis in progressive familial intrahepatic cholestasis type 1 after living donor liver transplantation. Liver Transplant. 2007;13(5):767–768.
- Siebold L, Dick AAS, Thompson R, et al. Recurrent low gamma-glutamyl transpeptidase cholestasis following liver transplantation for bile salt export Pump (BSEP) disease (Posttransplant Recurrent BSEP Disease). Liver Transplant. 2010;16:856–863.
- Aydogdu S, Cakir M, Arikan C, et al. Liver transplantation for progressive familial intrahepatic cholestasis: clinical and histopathological findings, outcome and impact on growth. Pediatr Transplant. 2007;11(6):634–640.
- Nicastro E, Stephenne X, Smets F, et al. Recovery of graft steatosis and protein-losing enteropathy after biliary diversion in a PFIC 1 liver transplanted child. Pediatr Transplant. 2012;16(5):177–182.
- Mali VP, Fukuda A, Shigeta T, et al. Total internal biliary diversion during liver transplantation for type 1 progressive familial intrahepatic cholestasis: a novel approach. Pediatr Transplant. 2016;20(7):981–986.
- Knisely AS, Strautnieks SS, Meier Y, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology. 2006;44(2):478–486.
- Dröge C, Bonus M, Baumann U, et al. Sequencing of FIC1, BSEP and MDR3 in a large cohort of patients with cholestasis revealed a high number of different genetic variants. J Hepatol. 2017;67(6):1253–1264.
- Jara P, Hierro L, Martínez-Fernández P, et al. Recurrence of bile salt export pump deficiency after liver transplantation. N Engl J Med. 2009;361(14):1359–1367.
- Keitel V, Burdelski M, Vojnisek Z, et al. De novo bile salt transporter antibodies as a possible cause of recurrent graft failure after liver transplantation: a novel mechanism of cholestasis. Hepatology. 2009;50(2):510–517.
- Stindt J, Kluge S, Dröge C, et al. Bile salt export pump-reactive antibodies form a polyclonal, multi-inhibitory response in antibody-induced bile salt export pump deficiency. Hepatology. 2016;63(2):524–537.
- Grammatikopoulos T, Knisely AS, Dhawan A, et al. Anti-CD20 monoclonal antibody therapy in functional bile salt export pump deficiency after liver transplantation. J Pediatr Gastroenterol Nutr. 2015;60(6):e50–3.
- Jadlowiec CC, Taner T. Liver transplantation: current status and challenges. World J Gastroenterol. 2016;22(18):4438–4445.
- Graffner H, Gillberg PG, Rikner L, et al. The ileal bile acid transporter inhibitor A4250 decreases serum bile acids by interrupting the enterohepatic circulation. Aliment Pharmacol Ther. 2016;43(2):303–310.
- European Medicines Agency. Bylvay (odevixibat) medicine overview [Internet]. 2021 cited 2023 Oct 24. Available from: https://www.ema.europa.eu/en/documents/overview/mylotarg-epar-summary-public_en.pdf
- Albireo Pharma. Bylvay Prescribing Information [Internet]. 2023 cited 2023 Oct 24. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215498s000lbl.pdf
- European Medicines Agency. Bylvay Summary of Product Characteristics [Internet]. 2021 cited 2023 Oct 24. Available from: https://www.ema.europa.eu/en/documents/product-information/bylvay-epar-product-information_en.pdf
- Thompson RJ, Arnell H, Artan R, et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: a randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. 2022;7(9):830–842.
- Ellinger P, Stindt J, Dröge C, et al. Partial external biliary diversion in bile salt export pump deficiency: association between outcome and mutation. World J Gastroenterol. 2017;23(29):5295–5303.
- Thompson RJ, Artan R, Baumann U, et al. Interim results from an ongoing, open-label, single-arm trial of odevixibat in progressive familial intrahepatic cholestasis. JHEP Rep. 2023;5(8):100782.
- Gonzales E, Hardikar W, Stormon M, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Lancet. 2021;398:1581–1592.
- Kunst RF, de Waart DR, Wolters F, et al. Systemic ASBT inactivation protects against liver damage in obstructive cholestasis in mice. JHEP Rep. 2022;4(11):100573.
- Food & Drug Administration. Orphan drug designations and approvals [Internet]. 2021. Available from: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=370912
- European Medicines Agency. Orphan maintenance assessment report bylvay (Odevixibat) [Internet]. 2021. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/evrysdi
- Ipsen. The acquisition of Albireo [Internet]. 2023. Available from: https://www.ipsen.com/websites/ipsen_com_v2/wp-content/uploads/2023/01/23204433/The-acquistion-of-Albireo.pdf
- Committee for medicinal products for human use. Assessment report - Bylvay (Odevixibat). EMA/319560/2021. 2021.
- Albireo Pharma. Current report exhibit 99.2 [Internet]. 2020. Available from: https://www.sec.gov/Archives/edgar/data/1322505/000110465920008111/tm205897d2_ex99-2.htm
- Felzen A, van Wessel DBE, Gonzales E, et al. Genotype-phenotype relationships of truncating mutations, p.E297G and p.D482G in bile salt export pump deficiency. JHEP Rep. 2023;5(2).
- Albireo Pharma. Albireo reports Q3 2022 financial results and business update. 2022.
- Ipsen. Ipsen delivers solid sales growth in the first nine months of 2023 and confirms its full-year guidance. YTD 2023 - sales announcement. 2023.
- Ipsen. U.S. FDA approves Bylvay® for patients living with cholestatic pruritus due to Alagille syndrome. 2023.
- Tandvårds- och läkemedelsförmånsverket (TLV). TLV decision bylvay. 2023.
- Zorginstituut Nederland. GVS advice odevixibat (Bylvay®). 2023.
- Van de Peppel IP, Verkade HJ, Jonker JW. Metabolic consequences of ileal interruption of the enterohepatic circulation of bile acids. Am J Physiol Gastrointest Liver Physiol. 2020;319(5).
- Mareux E, Lapalus M, Ben Saad A, et al. In vitro rescue of the bile acid transport function of ABCB11 variants by CFTR potentiators. Int J Mol Sci. 2022;23(18):10758.
- Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Mol Cell Endocrinol. 2013;368(1–2):17–29.
- Floreani A, Gabbia D, De Martin S. Update on the pharmacological treatment of primary biliary cholangitis. Biomedicines. 2022;10(8):1–17.
- Gonzales E, Jacquemin E. Mutation specific drug therapy for progressive familial or benign recurrent intrahepatic cholestasis: a new tool in a near future? J Hepatol. 2010;53(2):385–387.
- Garrison P, Bangs JD. p97 Inhibitor CB-5083 Blocks ERAD in Trypanosoma brucei. Mol Biochem Parasitol. 2020;239(Sep):111313.
- Hayashi H, Sugiyama Y. 4-Phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type and mutated bile salt export pumps. Hepatology. 2007;45(6):1506–1516.
- Mareux E, Lapalus M, Amzal R, et al. Functional rescue of an ABCB11 mutant by ivacaftor: a new targeted pharmacotherapy approach in bile salt export pump deficiency. Liver Int. 2020;40(8):1917–1925.
- European Medicines Agency. Livmarli summary of product characteristics [Internet]. 2022 cited 2023 Oct 26. Available from 2023 Oct 26: https://www.ema.europa.eu/en/documents/product-information/livmarli-epar-product-information_en.pdf
- Food & Drug Administration. Livmarli Prescribing Information [Internet]. 2024 cited 2024 May 7. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/214662s005s008lbl.pdf
- Mirum Pharma. Mirum submits for European approval of LIVMARLI in progressive familial intrahepatic cholestasis [Internet]. Available from: https://ir.mirumpharma.com/news-events/News/news-details/2023/Mirum-Submits-for-European-Approval-of-LIVMARLI-in-Progressive-Familial-Intrahepatic-Cholestasis/default.aspx
- Shirley M. Maralixibat: First Approval. Drugs. 2022;82(1):71–6. doi: 10.1007/s40265-021-01649-0
- Mirum Pharma. Mirum Pharmaceuticals announces data from EMBARK phase 2b study for biliary atresia. Available from: https://ir.mirumpharma.com/news-events/News/news-details/2023/Mirum-Pharmaceuticals-Announces-Data-from-EMBARK-Phase-2b-Study-for-Biliary-Atresia/default.aspx
- Loomes KM, Squires RH, Kelly D, et al. Maralixibat for the treatment of PFIC: long-term, IBAT inhibition in an open-label, Phase 2 study. Hepatol Commun. 2022;6(9):2379–2390.
- Miethke A, Moukarzel A, Porta G, et al. Efficacy and safety of maralixibat in patients with progressive familial intrahepatic cholestasis (march-pfic): a randomized placebo-controlled phase 3 study. In: Journal of pediatric gastroenterology and nutrition. 2023. p. 709‐710.
- A study to evaluate the efficacy and safety of maralixibat in subjects with progressive familial intrahepatic cholestasis (MARCH-PFIC) NCT03905330 [Internet]. 2023 cited 2023 Dec 8]. Available from 2023 Dec 8: https://clinicaltrials.gov/study/NCT03905330
- Miethke A, Moukarzel A, Porta G, et al. Analysis of safety in maralixibat-treated participants with progressive familial intrahepatic cholestasis: data from MARCH-PFIC. J Hepatol. 2023;78:S387–8.
- Thompson R, Mogul D, Nunes T, et al. Maralixibat leads to significant reductions in pruritus and improvements in sleep for children with progressive familial intrahepatic cholestasis: data from MARCH-PFIC. J Hepatol. 2023;78:S370.
- D’Antiga L, Miethke A, Mogul D, et al. Maralixibat leads to significant reductions in bilirubin for patients with progressive familial intrahepatic cholestasis: data from MARCH-PFIC. J Hepatol. 2023;78:S61.
- Miethke AG, Moukarzel A, Porta G, et al. Long-term maintenance of response and improved liver health with maralixibat in patients with progressive Familial intrahepatic cholestasis (PFIC): 2-year data from the MARCH-ON Study. In: American Association for the Study of Liver Diseases (AASLD) The Liver Meeting [Internet]. 2023. p. 5048C. Available from: https://mirumpharma.com/wp-content/uploads/2023/11/Thompson-RJ-AASLD-2023-Longterm-maintenance-of-response-and-improved-liver-health-with-MRX-in-PFIC.pdf
- Nakano S, Osaka S, Sabu Y, et al. Effect of food on the pharmacokinetics and therapeutic efficacy of 4-phenylbutyrate in progressive familial intrahepatic cholestasis. Sci Rep. 2019;9(1):1–12.
- Lam P, Pearson CL, Soroka CJ, et al. Levels of plasma membrane expression in progressive and benign mutations of the bile salt export pump (Bsep/Abcb11) correlate with severity of cholestatic diseases. Am J Physiol Cell Physiol. 2007;293(5):1709–1716.
- Hayashi H, Inamura K, Aida K, et al. AP2 adaptor complex mediates bile salt export pump internalization and modulates its hepatocanalicular expression and transport function. Hepatology. 2012;55(6):1889–1900.
- Gonzales E, Grosse B, Cassio D, et al. Successful mutation-specific chaperone therapy with 4-phenylbutyrate in a child with progressive familial intrahepatic cholestasis type 2. J Hepatol. 2012;57(3):695–698.
- Gonzales E, Grosse B, Schuller B, et al. Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: evidence for improvement of cholestasis with 4-phenylbutyrate. Hepatology. 2015;62(2):558–566.
- Naoi S, Hayashi H, Inoue T, et al. Improved liver function and relieved pruritus after 4-phenylbutyrate therapy in a patient with progressive familial intrahepatic cholestasis type 2. J Pediatr. 2014;164(5):1219–1227.e3.
- Mareux E, Lapalus M, Ben-Saad A, et al. In vitro functional rescue by ivacaftor of an ABCB11 variant involved in PFIC2 and intrahepatic cholestasis of pregnancy. Orphanet J Rare Dis. 2021;16(1):1–4.
- Caballero-Camino FJ, Rodrigues PM, Wångsell F, et al. A3907, a systemic ASBT inhibitor, improves cholestasis in mice by multiorgan activity and shows translational relevance to humans. Hepatology. 2023;78(3):709–726.
- Trauner M, Gulamhusein A, Hameed B, et al. The nonsteroidal farnesoid x receptor agonist cilofexor (GS-9674) improves markers of cholestasis and liver injury in patients with primary sclerosing cholangitis. Hepatology. 2019;70(3):788–801.
- Kowdley KV, Vuppalanchi R, Levy C, et al. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J Hepatol. 2020;73(1):94–101.
- Mayo MJ, Wigg AJ, Leggett BA, et al. NGM282 for treatment of patients with primary biliary cholangitis: a multicenter, randomized, double-blind, placebo-controlled trial. Hepatol Commun. 2018;2(9):1037–1050.
- Food & Drug Administration. OCALIVA® (obeticholic acid) Prescribing Information [Internet]. 2016. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/207999s008lbl.pdf
- European Medicines Agency. Ocaliva summary of product characteristics [Internet]. 2022. Available from: https://www.ema.europa.eu/en/documents/product-information/ocaliva-epar-product-information_en.pdf
- Pellicciari R, Fiorucci S, Camaioni E, et al. 6r-Ethyl-Chenodeoxycholic Acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. Society. 2002;45(17):15–18.
- Nevens F, Andreone P, Mazzella G, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631–643.
- Kunst RF, Bolt I, van Dasselaar RDJ, et al. Combined inhibition of bile salt synthesis and intestinal uptake reduces cholestatic liver damage and colonic bile salts in mice. JHEP Rep. 2023;6(1):100917.
- Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965.
- Di Giorgio A, Sciveres M, Fuoti M, et al. Treatment with an ileal bile acid transporter inhibitor in patients with TJP2 deficiency. Clin Res Hepatol Gastroenterol. 2023;47(8):102185.
- Soroka CJ, Boyer JL. Biosynthesis and trafficking of the bile salt export pump, BSEP: therapeutic implications of BSEP mutations. Mol Aspects Med. 2014;37:3–14.
- Jacquemin E, Hermans D, Myara A, et al. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology. 1997;25(3):519–523.
- Misawa T, Hayashi H, Sugiyama Y, et al. Discovery and structural development of small molecules that enhance transport activity of bile salt export pump mutant associated with progressive familial intrahepatic cholestasis type 2. Bioorg Med Chem. 2012;20(9):2940–2949.
- Luo J, Ko B, Elliott M, et al. Liver disease: a nontumorigenic variant of FGF19 treats cholestatic liver diseases. Sci Transl Med. 2014;6(247):1–12.
- Halilbasic E, Steinacher D, Trauner M. Nor-ursodeoxycholic acid as a novel therapeutic approach for cholestatic and metabolic liver diseases. Dig Dis. 2017;35(3):288–292.
- Kuipers F, Bloks VW, Groen AK. Beyond intestinal soap - Bile acids in metabolic control. Nat Rev Endocrinol. 2014;10(8):488–498.
- Ghallab A, González D, Strängberg E, et al. Inhibition of the renal apical sodium dependent bile acid transporter prevents cholemic nephropathy in mice with obstructive cholestasis. J Hepatol. 2023.
- Almes M, Jobert A, Lapalus M, et al. Glycerol phenylbutyrate therapy in progressive familial intrahepatic cholestasis type 2. J Pediatr Gastroenterol Nutr. 2020;70(6):E139.
- Hinderer C, Katz N, Buza EL, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. 2018;29(3):285–298.
- Cunningham SC, Spinoulas A, Carpenter KH, et al. AAV2/8-mediated correction of OTC deficiency is robust in adult but not neonatal Spfash mice. Mol Ther. 2009;17(8):1340–1346.
- Aronson SJ, Bakker RS, Shi X, et al. Liver-directed gene therapy results in long-term correction of progressive familial intrahepatic cholestasis type 3 in mice. J Hepatol. 2019;71(1):153–162.
- De Vree JML, Ottenhoff R, Bosma PJ, et al. Correction of liver disease by hepatocyte transplantation in a mouse model of progressive familial intrahepatic cholestasis. Gastroenterology. 2000;119(6):1720–1730.
- Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clin Microbiol Rev. 2008;21(4):583–593.