
1. Introduction
Multiple myeloma (MM) is characterized by the accumulation of malignant plasma cells into the bone marrow (BM). Despite the recent introduction of new drugs, MM remains an incurable disease with high tendency to relapse and drug resistance [Citation1]. The microenvironment exerts a critical role in MM cell survival and proliferation and recent studies, focused on the relationship between the BM niche and MM cells, highlight the role of hypoxia in MM cell growth and progression [Citation2], identifying the hypoxia-inducible factor-1α as a potential therapeutic target [Citation2,Citation3]. This evidence suggests that MM cells acquire a more aggressive phenotype and modify their metabolism to adapt to the hypoxic microenvironment in which they grow. In several tumors, the metabolic switch induced by hypoxic conditions render cancer cells addicted to nutrients, such as glucose or glutamine (Gln) [Citation4].
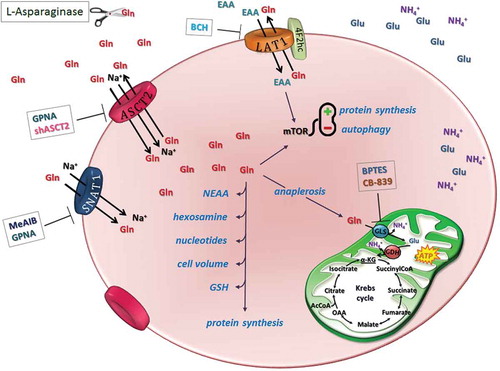
The interest on the role of Gln metabolism in cancer has steadily increased in the last few years. Gln has multiple roles in cell physiology (), since it is not only used for the synthesis of proteins, nucleotides, and hexosamine but also provides glutamate for glutathione and determines the overall amino acid pool, thus contributing to cell volume regulation. Moreover, Gln activates mTOR, a master regulator of protein synthesis and cell proliferation. Importantly, Gln also serves for anaplerosis, a pivotal role in rapidly proliferating cells that must balance energy supply with high biosynthetic rates. Gln-dependent anaplerosis involves glutaminase-dependent Gln hydrolysis to glutamate and ammonium. Glutamate is then deaminated through glutamate dehydrogenase or transaminases to obtain the Krebs cycle intermediate α-ketoglutarate (2-oxoglutarate).
Figure 1. Glutamine transport and metabolism as therapeutic targets in MM cells.
A distinctive Gln metabolism characterizes MM cells: they express GLS but lack a sizable expression of GS. MM cells express three major transporters for Gln: ASCT2, LAT1, and SNAT1. After Gln uptake, MM cells may use Gln for multiple metabolic roles. One of these roles is anaplerosis. Gln transport and metabolism can be targeted with different approaches exploitable for therapeutic use: extracellular Gln can be depleted by L-asparaginase, while the activity of Gln transporters can be hindered with inhibitors or gene silencing; finally, CB-839 and BPTES can be used to block GLS activity.
AcCoA: Acetyl coenzyme A; α-KG: alpha-ketoglutarate; BCH: 2-Amino-2-norbornanecarboxylic acid; BPTES: Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl) ethyl sulfide; EAA: essential amino acid; GDH: glutamate dehydrogenase; Gln: glutamine; GLS: glutaminase; Glu: glutamate; GPNA: l-γ-glutamyl-p-nitroanilide; GSH: glutathione; MeAIB: methylaminoisobutyrate; mTOR: mammalian target of rapamycin; NEAA: non-essential amino acid; OAA: oxaloacetate.
2. How cells get Gln
Several transport systems are devoted to Gln transport in mammalian cells [Citation5]. These transporters widely differ for dependence on ion gradients, affinity, and nature of the transport process. While some transporters are ubiquitous (e.g. LAT1 and ASCT2), others are strictly tissue specific. Gln can be also synthesized by the enzyme glutamine synthetase (GS) from glutamate and ammonium. However, GS expression is not ubiquitous, although several cell types, such as astroglia and skeletal muscle, are endowed with very high levels of the enzyme.
3. Gln metabolism in MM cells: membrane transport as an attractive target
Gln metabolism exhibits distinctive features in MM [Citation6]. Indeed, MM cells perform Gln-dependent ammoniagenesis, use the amino acid for anaplerosis, which is a marker of Gln addiction, and, for the most part, lack a sizable expression of GS. This unique combination justifies the strict dependence of MM cells from extracellular Gln and clearly points to the inhibition of Gln transport as an attractive therapeutic target. Consistently, MM cells in vitro undergo cell death when incubated in the absence of Gln or when the extracellular amino acid is depleted by l-asparaginase (IC50 < 0.1 U/ml), which, besides asparagine, hydrolyses also Gln [Citation6]. Gln depletion induced by l-asparaginase synergizes the cytotoxic effects of bortezomib on MM cells [Citation6]. These experiments have been performed under normoxic conditions, and it is expected that under the hypoxic conditions typical of BM, dependence on Gln should be further increased, as demonstrated for other tumor models [Citation4].
The expression of enzymes involved in Gln metabolism and Gln transporters is coherent with marked Gln dependence of MM cells. Indeed, both MM cell lines and cells from patients express high levels of kidney-type glutaminase (GLS), while they lack a sizable expression of GLS2. Moreover, MM cells express three major Gln transporters, ASCT2, LAT1, and SNAT1 (), the expression of which gradually increases during the progression of plasma cell dyscrasias from normal plasma cells to plasma cell leukemias [Citation6], pointing to their importance for the affirmation of the neoplastic phenotype. Most of Gln influx in MM cells occurs through the ASCT2 transporter, while the contributions of SNAT1 and LAT1 seem minor. Consistently, l-γ-glutamyl-p-nitroanilide (GPNA), the most commonly employed ASCT2 inhibitor, markedly lowers Gln influx and hinders the proliferation of MM cells. Importantly, also ASCT2 silencing partially hampers MM cell proliferation in vitro and delays the growth of human myeloma xenografts in murine models [Citation6]. Thus, among the various approaches that, in theory, target Gln transport or metabolism in MM cells (), interference with ASCT2 expression and/or function has proven to be effective in vivo.
Table 1. Glutamine transporters found operative in MM cells.
The most important advantage of this approach is likely connected with the multiple metabolic roles of the amino acid (). Although glutaminase-dependent anaplerosis is evident in MM cell lines, the glutaminase inhibitors bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES) or CB-839 were only partially effective if used alone [Citation6], suggesting that Gln plays additive critical roles besides anaplerosis in those cells. Indeed, even MM cell lines resistant to BPTES or CB-839 were very sensitive to Gln starvation [Citation6]. These different effects could be explained considering that lowering Gln transport from the extracellular compartment limits the availability of the amino acid for all the metabolic pathways for which Gln is required.
Interestingly, in MM cells, Gln shortage elicits synthetic lethality to venetoclax, a BH3-mimetic drug [Citation7]. Although the elegant molecular mechanism described in that study may not be specific for MM cells, several contributions have reported that Gln shortage synergizes chemotherapeutic agents in a variety of cancers. Consistently, as far as MM is concerned, a limited synergy between Gln depletion and bortezomib has been indeed described [Citation6].
4. Gln transport as a therapeutic target: problems and limitations
As a therapeutic target for MM, Gln transport presents several possible limitations. First, the efficacy of such an approach requires a strict dependence of MM cells on extracellular Gln. However, MM cells relatively resistant to Gln depletion have been recently reported [Citation7]. Given that GS expression was not assessed in that study, the possibility exists that sensitivity to Gln depletion associates with absent-to-low GS expression.
Among the Gln transporters characterized in MM cells, ASCT2 can be knocked down without affecting mice viability [Citation8], thus indicating that its inhibition for therapeutic purposes would be feasible. At present, two ASCT2 inhibitors are employed to inhibit the transporter in cancer cells, GPNA and benzylserine. Unfortunately, both compounds are less than optimal. Although employed to suppress ASCT2 activity, benzylserine has been recently found ineffective in inhibiting the transporter in Xenopus oocytes [Citation9], while it directly inhibits LAT1 in the same model [Citation10]. Conversely, in the same model, GPNA exhibited a marked inhibitory effect on ASCT2, but the inhibitor also suppressed the activity of several SNAT transporters [Citation9]. In spite of lack of a complete structural characterization of the carrier, other ASCT2 inhibitors have been proposed and used in experimental therapy [Citation11], but their specificity awaits confirmation.
In spite of these problems, ASCT2 inhibition remains an attractive target since it would also suppress mTORC1 kinase activity [Citation12], with triggering of autophagy and additional inhibitory effects on protein synthesis and cell proliferation. ASCT2-dependent mTORC1 activation is usually attributed to the stimulation of leucine influx through LAT1 by Gln accumulated through ASCT2, although Gln depletion hinders mTORC1 independently from cell leucine changes [Citation13], thus implying that different mechanisms may underlie the stimulatory effects of ASCT2 on the kinase. Interestingly, a reciprocal regulation exists between Gln metabolism and mTORC1 activity. mTORC1 activates Gln metabolism through a MYC-dependent induction of glutaminase and glutamate dehydrogenase [Citation14], while, conversely, Gln promotes mTORC1 translocation to lysosomes and its activation [Citation15].
Finally, the contribution of the SNAT transporters to Gln influx in MM cells does not appear to be substantial, at least in Gln-rich media [Citation6]. However, these transporters are highly regulated mechanisms that are induced under a variety of stress conditions, which, importantly, include Gln depletion. This would imply that their inhibition may have significant effects on MM cell survival under conditions of Gln shortage in vivo.
5. Conclusion
Recent data indicate that Gln transport inhibition, as well as Gln deprivation, could be suitable targets in MM. However, the best approach to inhibit Gln transport in MM, as well as possible synergistic effects with anti-MM drugs, is yet to be defined.
6. Expert opinion
Absent-to-low expression of GS together with Gln-dependent anaplerosis renders MM an ideal model to test the efficacy of therapeutic strategies based on the inhibition of Gln transport. However, two recent, separate studies have reported GS-positive MM cells [Citation16] and MM cells relatively resistant to Gln depletion [Citation7]. A correlation between GS expression and lack of Gln dependence is therefore likely, although not demonstrated yet. If this hypothesis were confirmed, expression of GS would become an important parameter to predict Gln dependence of MM cells and, hence, sensitivity to Gln-dependent approaches.
Also high levels of expression of glutaminase may be an indicator of dependence on Gln, although, in this case, the situation is more complicated given the existence of several isoforms of the enzyme. Kidney-type glutaminase isoforms, encoded by GLS, have been so far the isoforms more commonly associated with enhanced Gln metabolism in tumors and the usual target of glutaminase inhibitors synthesized to date [Citation11]. Even if the other glutaminase gene (GLS2) has been found expressed in several cancers [Citation17] and both genes are expressed in human leukemia cells [Citation18], its low expression in MM cells [Citation6] suggest that GLS expression may be a reliable indicator of Gln metabolism in MM.
The strict dependence of GS-negative MM cells on Gln suggests that even a partial inhibition of Gln transport, while not highly toxic for normal cells, may have profound consequences on cancer cell proliferation and tumor growth. This hypothesis is consistent with the significant delay in MM xenograft growth observed upon a partial silencing of ASCT2 [Citation6]. Actually, Gln may be more limiting in the tumor microenvironment in vivo than under in vitro conditions, as suggested for other tumors by the greater efficiency of Gln-depleting strategies observed in xenografts compared to cultured cells [Citation19].
While these considerations have been developed for MM, they may also apply to other tumors strictly dependent on Gln. A particularly attractive example is constituted by triple negative breast cancer (TNBC). This tumor is highly Gln dependent [Citation20], upregulates ASCT2 and SNATs transporters, as well as GLS [Citation12], and is sensitive to ASCT2 inhibition [Citation12]. While there are many other types of cancers where ASCT2 expression is increased, MM and TNBC appear the most Gln-sensitive tumors and, thus, privileged targets for therapeutic strategies based on Gln depletion and/or inhibition of Gln transport.
Unfortunately, specific inhibitors of ASCT2 are not yet available. Until this objective is achieved, the aim to employ this transporter as a specific therapeutic target appears hardy feasible. Although the role of ASCT2 in Gln accumulation has been recently discussed [Citation9], its specific inhibition remains a valuable objective, also considering the relationships between its activity and mTORC1 activation. However, the role of the various transporters involved in Gln transport in MM cells, in particular SNAT1, still awaits a complete characterization, especially upon Gln shortage, when this transporter may be upregulated, or under the hypoxic conditions encountered in BM in vivo.
Thus, characterization of SNAT1 role and development of reliable ASCT2 inhibitors appear two promising routes to better understand how to hamper MM cell metabolism through Gln shortage. As for other tumors, this nutritional approach could exert synergistic anti-MM activity with established therapies. In particular, given the variety of examples on synergistic effects between Gln shortage and chemotherapy, further investigations should focus on the combinatory effects between inhibitors of Gln transport and drugs currently in use for MM treatment, such as the proteasome inhibitors.
Declaration of interest
N. Giuliani is funded by a grant from the Associazione Italiana per la Ricerca sul Cancro (IG2014 n.15531) and M. Bolzoni receives a fellowship Fondazione Italiana per la Ricerca sul Cancro (id. n. 18152). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Additional information
Funding
Related Research Data
References
- Yang WC, Lin SF. Mechanisms of drug resistance in relapse and refractory multiple myeloma. Biomed Res Int. 2015;2015:341430. Epub 2015 12 10.
- Colla S, Storti P, Donofrio G, et al. Low bone marrow oxygen tension and hypoxia-inducible factor-1alpha overexpression characterize patients with multiple myeloma: role on the transcriptional and proangiogenic profiles of CD138(+) cells. Leukemia. 2010;24:1967–1970. Epub 2010 09 03.
- Storti P, Bolzoni M, Donofrio G, et al. Hypoxia-inducible factor (HIF)-1alpha suppression in myeloma cells blocks tumoral growth in vivo inhibiting angiogenesis and bone destruction. Leukemia. 2013;27:1697–1706. Epub 2013 01 25.
- Corbet C, Feron O. Metabolic and mind shifts: from glucose to glutamine and acetate addictions in cancer. Curr Opin Clin Nutr Metab Care. 2015;18:346–353. Epub 2015 05 24.
- Bhutia YD, Babu E, Ramachandran S, et al. Amino acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015;75:1782–1788. Epub 2015 04 10.
- Bolzoni M, Chiu M, Accardi F, et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: a new attractive target. Blood. 2016;128:667–679. Epub 2016 06 09.
- Bajpai R, Matulis SM, Wei C, et al. Targeting glutamine metabolism in multiple myeloma enhances BIM binding to BCL-2 eliciting synthetic lethality to venetoclax. Oncogene. 2016;35:3955–3964. Epub 2015 12 08.
- Nakaya M, Xiao Y, Zhou X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40:692–705. Epub 2014 05 06.
- Broer A, Rahimi F, Broer S. Deletion of amino acid transporter ASCT2 (SLC1A5) reveals an essential role for transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to sustain glutaminolysis in cancer cells. J Biol Chem. 2016;291:13194–13205. Epub 2016 04 30.
- Wang Q, Beaumont KA, Otte NJ, et al. Targeting glutamine transport to suppress melanoma cell growth. Int J Cancer. 2014;135:1060–1071. Epub 2014 02 18.
- Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619–634. Epub 201.
- Jeon YJ, Khelifa S, Ratnikov B, et al. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell. 2015;27:354–369. Epub 2015 03 12.
- Chiu M, Tardito S, Barilli A, et al. Glutamine stimulates mTORC1 independent of the cell content of essential amino acids. Amino Acids. 2012;43:2561–2567. Epub 2012 05 09.
- Csibi A, Lee G, Yoon SO, et al. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol. 2014;24:2274–2280. Epub 2014 09 16.
- Jewell JL, Kim YC, Russell RC, et al. Differential regulation of mTORC1 by leucine and glutamine. Science. 2015;347:194–198. Epub 2015 01 09.
- Nguyen TV, Lee JE, Sweredoski MJ, et al. Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol Cell. 2016;61:809–820. Epub 2016 03 19.
- Mates JM, Segura JA, Martin-Rufian M, et al. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med. 2013;13:514–534. Epub 2012 09 01.
- Perez-Gomez C, Campos-Sandoval JA, Alonso FJ, et al. Co-expression of glutaminase K and L isoenzymes in human tumour cells. Biochem J. 2005;386:535–542. Epub 2004 10 22.
- Chiu M, Tardito S, Pillozzi S, et al. Glutamine depletion by crisantaspase hinders the growth of human hepatocellular carcinoma xenografts. Br J Cancer. 2014;111:1159–1167. Epub 2014 07 30.
- Timmerman LA, Holton T, Yuneva M, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24:450–465. Epub 2013 10 08.