
1. Introduction
Epithelial plasticity is the pathological state of tumor cells that renders them to undergo series of events characterized by metastatic cascade and an advantage to survive multiple anticancer therapies across diverse cancer lineages. These tumor cells undergo bidirectional conversions among epithelial (E), mesenchymal (M), and partial/hybrid E/M phenotypes via conserved process of epithelial-to-mesenchymal transition (EMT) and its reverse process of mesenchymal-to-epithelial transition (MET). EMT endows tumor cells to transiently adapt altered morphological traits with reduced requirements of cell–cell contact, immune evasion, gain migratory potential, and infiltrate the surrounding basement membrane. Tumor cells enter the peripheral circulatory/lymphatic system and become circulating tumor cells (CTCs). These heterogeneous populations of tumor cells are composed of (1) subset of cancer stem cells (CSCs)/tumor initiating cells (TICs) with self-renewal and aberrant differentiation property and (2) metastatic precursors with tumorigenic and invasive potential. CTCs extravasate, colonize to distant organ sites, establish pre-metastatic niches through conserved regulatory mechanisms, accelerate tumor progression, and pose clinical challenges in the treatment of solid tumors. Understanding the molecular regulatory determinants that influence CSCs to acquire enhanced stemness properties and epithelial plasticity, adapt to the sites of metastasis and survive in an adverse environment through constant evolution, leads to the development of effective therapeutics to combat metastasis.
2. Epithelial plasticity and its core regulators
At molecular level, EMT is characterized by the transcriptional and translational repression of epithelial junction proteins including α-catenin, γ-catenin/plakoglobin, E-cadherin, desmosomes, and cytokeratins; enhanced expression of mesenchymal proteins including N-cadherin, vimentin, fibronectin, cell surface proteins, CD44 (cluster of differentiation 44), integrin β6, and matrix metalloproteinases (MMPs), and degradation of extracellular matrix. Molecular reprogramming during EMT is triggered and orchestrated by EMT core regulators, also known as drivers of cellular plasticity and are classified as (a) extracellular inducers, (b) EMT-activating transcription factors (EMT-ATFs), and (c) downstream effectors. Wnt, TGF-β, Hedgehog (Hh), Notch, RAS/Receptor tyrosine kinases, and NF-kB signal transduction pathways triggered by extracellular inducers/growth factors in a tumor microenvironment interact through cross talk, mobilize EMT-ATFs [(i) two-handed zinc-finger factors of d-crystallin/E2 box factor (dEF1) family proteins/zinc-finger E-box binding homeobox (ZEB)1; (ii) Smad-interacting protein (SIP)1/ZEB2; (iii) Snail family of zinc-finger transcription factors: Snail1 (Snail), Snail2 (Slug), and Snail3 (Smuc); (iv) basic helix-loop-helix factors (Twist1 and Twist2); and (v) E12/E47 and Tbx3, and orchestrate the EMT transcriptome by inducing the downstream effectors/target genes [Citation1].
MicroRNAs, conserved small noncoding RNA molecules, influence the phenotype of tumor cells by interacting and forming mutually inhibitory feedback loops with EMT-ATFs/inducers. This leads to bistable switching between distinct E (high miR-200 and miR-34; low Zeb and Snail expression) and M phenotypes (low miR-200 and miR-34; high Zeb and Snail) of tumor cells. Fold change differences in the quantitative manifestation of partial EMT state in different types of cancer cells are due to the variation in (a) number of binding sites of miR-200 and miR-34 on Zeb mRNA and on Snail mRNA, respectively; (b) regulation at translational or transcriptional level; and (c) self-inhibition of Snail versus self-activation of Zeb [Citation2,Citation3].
2.1. Epithelial plasticity, cancer stemness, and metastasis
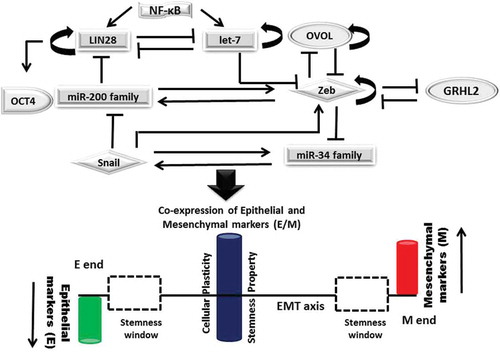
Partial/hybrid E/M phenotype of tumor buds is characterized by the co-expression of epithelial and mesenchymal markers. It influences CTCs to acquire increased stemness, plasticity, higher metastatic potential, and drug resistance, allows them to migrate as multicellular aggregates and thereby associates them with poor clinical outcomes [Citation4]. Higher expression of phenotypic stability factors (PSFs) including GRHL2 and OVOL coupled with EMT core regulators are known to stabilize hybrid E/M phenotype of tumor cells. Computational model described by Jolly et al. emphasizes on the interplay between EMT core regulators and PSFs in shifting the position of stemness window on EMT axis. LIN28/let-7 regulation controlled by miR-200/Zeb finely tunes the stemness of E/M hybrid tumor cells. Further coupling of OVOL with miR200/ZEB/LIN28/let-7, an EMT core decision-making circuit, allows the tumor cells with E/M phenotype to gain stemness and pulls the stemness window away from the M end of EMT axis [Citation5] (). Its association with aggressive tumor progression strongly correlates it with poor patient outcome.
Figure 1. Interplay betweenEMT decision making circuit, PSFs and stemness: Mutually exclusive inhibitory loops including miR200 family/Zeb; miR-34 family/Snail; LIN28/let-7 bring about bistable switch between epithelial (E) and mesenchymal (M) phenotypes, control EMT/MET and stemness. Phenotypic stability factors (PSFs) like OVOL and GRHL2 couple to core-EMT decision making circuits and stabilize hybrid E/M phenotype. NF-κB controls LIN28/let-7 regulation, elevates the likelihood of hybrid E/M phenotype and shifts the stemness window on EMT axis. Solid arrows represent the activation; solid lines represent the repression and circular loops represent the self-activation.
Cellular plasticity contributes to the dedifferentiation of committed epithelial cells into replicating progenitors/functional multipotent stem cells in vivo, thereby regulate complex remodeling of gene expression programs and drive tumor invasion and metastasis [Citation6]. In vivo xenograft tumor growth experiments report the loss of expression of stem cell markers and tumorigenicity upon trans-differentiation of hybrid E/M ovarian tumor cells into cells with either M or E phenotype and strengthen the contribution of plastic tumor cells to multiple stages of metastatic cascade [Citation7]. Autochthonous murine model of prostate cancer was examined to harbor cancerous cells of hybrid E/M phenotype with enhanced plasticity, stemness, and invasive property [Citation8]. Enhanced proliferative capacity and tumorigenic potential was noted in a cluster of human breast cancer cells colonized and spread to axillary lymph nodes in a severe combined immunodeficiency mouse model [Citation9]. Endometrial CTCs with plastic phenotype expressing ALDH and CD44 are linked with stemness and examined to promote metastatic process in an in vivo mouse model [Citation10].
Extrinsic factors (arise from many complex interactions between tumor cells, host cells, and tumor microenvironment) and intrinsic factors define the functions/behavior of tumor cells, impart plasticity to them and therefore, play central mechanism in cancer progression. Potent androgen receptor (AR) antagonist enzalutamide-induced epithelial plasticity requires concomitant suppression of AR signaling, de-repression of Snail in multiple preclinical models of prostate cancer and patient tissues and therefore, has been linked to metastasis, stemness, and drug resistance [Citation11]. Differences in the intrinsic properties of the cell of origin examined by transcriptional and epigenomic profiling dictate different tumor phenotypes and EMT. Due to distinct chromatin landscapes and gene regulatory networks possessed by interfollicular epidermis (IFE) and hair follicle (HF) tumor-initiating cells, squamous cell carcinomas (SCC) derived from IEF are well differentiated, nevertheless, SCC derived from HF cells frequently exhibit epithelial plasticity and form secondary tumors with increased metastatic potential [Citation12]. Increased resistance conferred by mesenchymal-like prostate tumor cells to PI3K (phosphoinositide 3-kinase)/AKT and RAS/MAPK (mitogen-activated protein kinase) inhibitors suggests the fact that epigenetic mechanisms not only regulate EMT but also dictate heterogeneous therapeutic response of cancer cells [Citation13]. High-risk neuroblastoma metastatic site-derived aggressive cells (HR-NB MSDACs) when maintained in alternated culture conditions (serum-free stem cell medium/growth medium with serum), instigate self-renewal capacity, pluripotency maintenance, stem cell-related signaling events that depict aggressive adaptive plasticity and evade intensive multimodal therapy with poor clinical outcomes [Citation14].
3. Expert opinion
Multiple challenges are associated with the liquid biopsy for the efficient sampling of CTCs and or cell free nucleic acids (cfDNA)/genetic fragments that are shed from either the primary or metastatic tumors into the circulation of cancer patients. Owing to the extremely low levels of CTCs and cfDNA, cost-effective newer technologies are required to capture enough material to enrich and sequence the patients’ DNA. Screening and profiling of global CTCs populations for quantitative identification of expression of informative biomarkers/signature molecules improves clinical utility for monitoring the disease recurrence. Biomarkers/regulators that define the plastic nature of tumor cells may not actually block primary tumor growth but can suppress metastatic spread. Metastatic suppressors are functionally characterized to block various stages of metastatic cascade. Although their mechanism of action is poorly defined, nevertheless they are known to work in tumor microenvironment and possess therapeutic potential by suppressing tumor spread. Knockdown of HMGA2 with the histone deacetylase inhibitor LBH589 inhibits epithelial plasticity and stemness activities, reduces metastatic castration-resistant prostate cancer (mCRPC) development through successful targeting of EMT and mesenchymal-like tumor cells, prolongs survival following castration by enhancing p53 and AR acetylation, and in turn sensitizes mCRPC to androgen deprivation therapy [Citation13]. Greater understanding of the regulation of epithelial plasticity by EMT core inducers encourages the use of miR-based inhibitors as metastatic suppressors. Suppression of tumor mesenchymalization and the concomitant loss of tumor stemness via either overexpression of tumor suppressor miRs or repression of oncogenic miRs both in vivo and in vitro modulate (a) expression of EMT-ATFs; (b) cell cycle-associated proteins; (c) activate DNA damage response; (d) downregulate drug efflux pumps of the family of ATP-biding cassette (ABC) transporters; (e) significantly enhance susceptibility to conventional therapeutics; and (f) block phenotypic transition, or restore epithelial-like differentiation in tumor cells that have already undergone mesenchymalization. Expression of miR-424 regulates Twist1 or Snail1-induced EMT and cancer stemness-associated genes, including TGFβR3 while its downregulation exclusively promotes mesenchymal phenotypes but not tumor-initiating phenotypes; and represses later stages of breast cancer metastasis by regulating an EMT-MET axis [Citation15]. MiR-200c, an illustrious tumor suppressor is another most versatile and potential prognostic/diagnostic biomarker in human cancer that coordinates many signaling cascades such as TGF-β signaling, PI3K/Akt signaling, Notch signaling, VEGF signaling, and NF-κB signaling and plays significant functions in stemness, proliferation, EMT, therapy resistance, and metastasis [Citation16]. Targeted anticancer therapies based on the use of inhibitors of epithelial plasticity/miRs as metastatic suppressors to regulate EMT core inducers may facilitate differentiation of tumor cells with enhanced plasticity into mature and committed cells, and provide unique therapeutic alternatives to treat metastatic spread and tumor relapse.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Jung H-Y, Fattet L, Yang J. Molecular pathways: linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res. 2015;21:962–968.
- Adam L, Zhong M, Choi W, et al. MiR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clin Cancer Res. 2009;15:5060–5072.
- Garg M. Targeting microRNAs in epithelial mesenchymal transition induced cancer stem cells: therapeutic approaches in cancer. Expert Opin Ther Targets. 2015;19(2):285–297.
- Garg M. Epithelial, mesenchymal and hybrid epithelial/mesenchymal phenotypes and their clinical relevance in cancer metastasis. Expert Rev Mol Med. 2017;19:e3.
- Jolly MK, Jia D, Boareto M, et al. Coupling the modules of EMT and stemness: a tunable ‘stemness window’ model. Oncotarget. 2015;6:25161–25174.
- Tata PR, Mou H, Pardo-Saganta A, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503(7475):218–223.
- Strauss R, Li ZY, Liu Y, et al. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS One. 2011;6:e16186.
- Ruscetti M, Quach B, Dadashian EL, et al. Tracking and functional characterization of epithelial-mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. 2015;75:2749–2759.
- Hendrix MJ, Seftor EA, Seftor RE, et al. Experimental co-expression of vimentin and keratin intermediate filaments in human breast cancer cells results in phenotypic interconversion and increased invasive behavior. Am J Pathol. 1997;150:483–495.
- Alonso-Alconada L, Muinelo-Romay L, Madissoo K, et al. Molecular profiling of circulating tumor cells links plasticity to the metastatic process in endometrial cancer. Mol Cancer. 2014;13:223.
- Miao L, Yang L, Li R, et al. Disrupting androgen receptor signaling induces snail-mediated epithelial-mesenchymal plasticity in prostate cancer. Cancer Res. 2017;77(11):3101–3112.
- Latil M, Nassar D, Beck B, et al. Cell-type-specific chromatin states differentially prime squamous cell carcinoma tumor-initiating cells for epithelial to mesenchymal transition. Cell Stem Cell. 2017;20(2):191–204.e5.
- Ruscetti M, Dadashian EL, Guo W, et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene. 2016;35(29):3781–3795.
- Pandian V, Ramraj S, Khan FH, et al. Metastatic neuroblastoma cancer stem cells exhibit flexible plasticity and adaptive stemness signaling. Stem Cell Res Ther. 2015;6:2.
- Drasin DJ, Guarnieri AL, Neelakantan D, et al. TWIST1-induced miR-424 reversibly drives mesenchymal programming while inhibiting tumor initiation. Cancer Res. 2015;75(9):1908–1921.
- Mutlu M, Raza U, Saatci Ö, et al. miR-200c: a versatile watchdog in cancer progression, EMT, and drug resistance. J Mol Med (Berl). 2016;94(6):629–644.