
ABSTRACT
Introduction: The proteasome is a multi-subunit enzyme complex responsible for the turnover of short-lived, abnormal or damaged proteins in eukaryotic cells. As organisms that undergo rapid growth and cell division, protozoan parasites exist on the knife-edge of proteotoxic catastrophe and thus rely heavily on their protein quality control machinery for survival. Because of this, the proteasome has recently emerged as a desirable drug target.
Area covered: This review focuses on efforts to identify protozoan parasite-specific proteasome inhibitors using substrate profiling, library screening, and in vitro evolution of resistance approaches to inform medicinal chemistry. Targeting the parasite’s 20S proteasome chymotrypsin-like (β5) activity and selectively inhibiting protein turnover in parasites compared to human cells are critical properties of potent, selective inhibitors.
Expert opinion: Proteasome inhibitors have the potential for rapid action against all stages, all species and all strains of plasmodium and kinetoplastid parasites. Given the high level of conservation of proteasome active sites in eukaryotes, an important challenge is achieving inhibitors that show sufficient selectivity while maintaining properties consistent with drug development.
1. Introduction
The World Health Organization has ranked malaria and kinetoplastid diseases among the world’s greatest global health problems and has highlighted the urgent need to develop new drugs for their treatment [Citation1].
Half the world’s population lives in sub-tropical regions where malaria parasites are endemic, resulting in more than 200 million cases and over 400,000 deaths each year, mostly caused by Plasmodium falciparum [Citation2]. Infection is initiated when the Anopheles mosquito vector bites a human host, injecting infectious sporozoites that enter the blood stream and invade liver cells. The subsequent blood stage of the infection is associated with fever and debilitating metabolic symptoms, and in some cases, coma and death [Citation3]. Worryingly, after a decade of gains, progress in tackling malaria is stalling [Citation2].
Infections with the kinetoplastid parasites, Trypanosoma cruzi, Trypanosoma brucei spp. and Leishmania spp. cause Chagas disease, sleeping sickness and leishmaniasis. Again the world’s poorest communities are most affected, with over 20 million infections and 50,000 deaths annually [Citation2]. Human leishmaniasis comprises a spectrum of diseases that range from ulcerating and highly disfiguring cutaneous sores to visceral forms of the disease, involving infection of the liver, spleen and bone morrow, which are invariably lethal if untreated. Acute cases of Chagas Disease can result in significant morbidity and mortality [Citation4], while long term chronic infections result in megacolon and heart disease [Citation5,Citation6]. Sustained control efforts are yielding benefits in reducing the number of cases of sleeping sickness, but it remains a problem in some regions of Africa [Citation7]. It is invariably fatal if not treated adequately and is much feared due to its neurological symptoms and the pain associated with treatment [Citation8].
2. Current treatment and the need for new therapeutics
While a recently licensed malaria vaccine (RTS,S/AS01) showed some efficacy in trials in children [Citation9], the efficacy wanes with time and does not meet the target set by the Malaria Vaccine Technology Roadmap [Citation10–Citation12]. Antimalarial control remains highly dependent on drug treatments, particularly artemisinin combination therapies (ACTs). Thus, it is extremely concerning that parasites with decreased sensitivity to artemisinins have emerged in South East Asia [Citation13–Citation15]. In areas with concomitant partner drug resistance, up to ~50% treatment failure is now observed, and the decreased efficacy of artemisinins places further selection pressure on the few available partner drugs [Citation16,Citation17]. In response to this impending crisis, the peak body, Medicines for Malaria Venture (MMV), has declared that new antimalarial therapies need to be developed, not only to mitigate the emergence of resistance, but as tools to drive an eradication agenda (i.e. compounds with chemoprevention and/or transmission blocking activity). That plasmodium proteasome inhibitors have activity across the lifecycle makes the target a high priority.
Implementation of treatments for kinetoplastid diseases lags behind that for malaria [Citation18]. There are no vaccines and current drug therapies are often inadequate and/or suffer from serious side effects [Citation19–Citation21]. For example, the two drugs used for the treatment of Chagas disease (benznidazole and nifurtimox) are not suitable for use during pregnancy and can have serious adverse effects. Similarly, treatments for Leishmania (antimonials, amphotericin B, paromomycin sulfate, and miltefosine) have significant toxicity, variable efficacy and require extended treatment regimens.
MMV has published Target Product Profiles for new antimalarial compounds. They recommend that new compounds should preferably have a novel mechanism of action and show activity against all stages of the parasite and against all currently known resistant parasite strains [Citation22]. They should be orally available, kill the parasite quickly and stay in the blood long enough to ensure complete parasite clearance, ideally with a single dose. It is recognized that these are formidable criteria [Citation23].
The Drugs for Neglected Diseases initiative (DNDi), a collaborative, nonprofit organization that maintains a research and development portfolio of kinetoplastid-targeted drugs, proposed a similar set of Target Product Profiles for new drugs for kinetoplastid parasite diseases [Citation19].
3. The proteasome functions as a shredder that chews up damaged and no longer needed proteins
Proteasomes are found in all eukaryotes and archaea, and in some bacteria. The proteasome plays critical roles in cellular homeostasis in eukaryotes, turning over damaged and unwanted proteins and regulating key processes such as the cell cycle. Its barrel-shaped 20S catalytic core comprises two heptameric β rings sandwiched between two heptameric α rings (). In eukaryotic proteasomes, the catalytic sites are formed on the inner walls of the central chamber by auto-processing of three of the β subunit sequences to expose N-terminal threonine residues (). The alcohol side chain of the catalytic threonine acts as a nucleophile, attacking amide bonds and resulting in peptide bond hydrolysis [Citation24,Citation25]. Natural protein or peptide substrates of the proteasome interact with one or more of the active sites via specificity or substrate binding pockets (termed S1, S2, S3 etc and S1ʹ, S2ʹ, S3ʹ etc), on either side of target amide bond [Citation26] (). The β1 active site provides caspase-like activity (cleaves after acidic P1 residues), the β2 site, trypsin-like activity (cleaves after basic residues), and the β5 site, chymotrypsin-like activity (cleaves after nonpolar residues) [Citation27]. Alternative (immunoproteasome) β forms, denoted β1i, β2i, and β5i, are expressed in hematopoietic tissue and can be induced in most cells in response to exposure to pro-inflammatory signals.
Figure 1. Molecular architecture of the 20S proteasome. (a) Side view of the P. falciparum 20S proteasome (PDB: 6 muw) showing two β-rings sandwiched between two α-rings. (b) The catalytic subunits are highlighted. (c) Cross-section of the P. falciparum 20S core showing the catalytic chamber with β1 and β2 (top β-ring) and β5 (bottom β-ring) active sites indicated. In the absence of activator, the α subunit gate is closed. (d) Residues (P1, P2, P3 and P1ʹ, P2ʹ, P3ʹ) on either side of target amide bond in the protein substrate interact with active sites in the proteasome core via substrate binding pockets (termed S1, S2, S3 and S1ʹ, S2ʹ, S3ʹ). (e) The gate to the 20S core is opened by the binding of regulators, including the ubiquitin-dependent, 19S regulatory particle, and the ubiquitin-independent, PA28 activator. (f) Modeled structures (space-filling representation of resolved residues) for the unbound and PA28-bound α-ring of the P. falciparum 20S proteasome (PDB: 6 muw) showing the blocked (left) and opened (right) central pore.
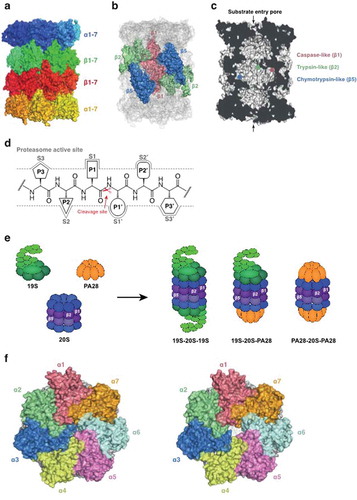
To avoid uncontrolled degradation of cellular proteins, access to the 20S proteasome is controlled by protein complexes that open an α-subunit ‘gate’ (). The most well-studied activator is the 19S regulatory particle, a multi-protein complex that combines with the 20S core to form the 26S complex [Citation28]. Damaged and unfolded proteins are marked for proteasomal degradation by modification with ubiquitin (the so-called ‘molecular kiss of death’) [Citation29]. Polyubiquitinated proteins bind to receptors within the 19S complex and are first passed to deubiquitinases that remove and recycle the ubiquitin and then to AAA-ATPases that unfold the protein and direct it into the catalytic chamber of the 20S core particle for degradation [Citation28,Citation30–Citation32]. Another activator, the PA28 heptamer (also known as 11S or REG) [Citation33], stimulates the 20S peptidase activity in vitro. PA28 may facilitate release of products from 20S and help degrade unstructured proteins [Citation33,Citation34].
4. Proteasome inhibitor drug classes
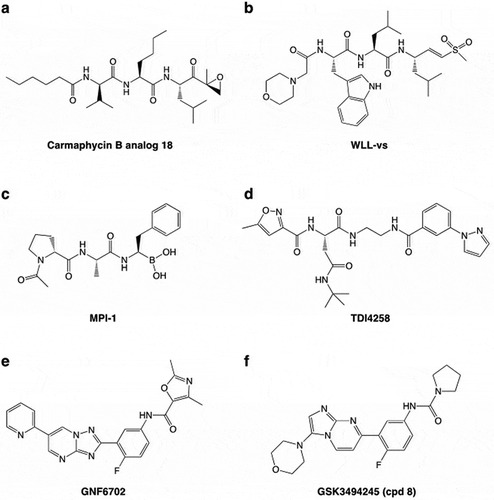
Proteasome inhibitors are structurally diverse and fall into two large groups based on whether they form a covalent bond with the active site threonine or inhibit via non-covalent interactions. The covalent inhibitors are generally peptidyl electrophiles with a warhead that forms a covalent bond with the oxygen of the catalytic threonine. Based on the nature of their reactive head groups, they are classified as irreversible inhibitors, e.g. vinyl sulfones, epoxyketones and β-lactones, or reversible inhibitors, such aldehydes and boronates. Examples of epoxyketone, vinyl sulfone and boronate inhibitors that have been investigated as antimalarial candidates [Citation35–Citation37] are illustrated in .
Figure 2. Some examples of proteasome inhibitors. The epoxyketone, Carmaphycin B analogue 18 (a) and the vinyl sulfone, WLL-vs (b) are irreversible covalent inhibitors. The peptide boronate, MPI-1 (c) is a covalent reversible inhibitor. TDI4258, GNF6701 and GSK3494245 (Compound 8) are non-covalent inhibitors.
Peptide boronates, like peptide aldehydes, are covalent, slowly reversible inhibitors [Citation26,Citation38,Citation39]. The two classes differ in that the boronic acid moiety is more metabolically stable than the aldehyde, and the peptide boronates are selective for serine and N-terminal threonine proteases. In contrast, peptide aldehydes can potently inhibit many cysteine proteases [Citation40]. Peptide epoxyketones are irreversible, covalent inhibitors that react to form a unique morpholino ring system between the epoxyketone functional group and the N-terminal catalytic threonine, underpinning their specificity for the proteasome compared with other proteases [Citation41]. Peptide vinyl sulfones are less reactive than epoxyketones and boronates for N-terminal threonine proteases (e.g. the proteasome). They are capable of a high degree of specificity, which can be exploited to decrease toxicity [Citation42].
As detailed below, non-covalent inhibitors have also been investigated as potential candidates for inhibition of P. falciparum and kinetoplastid proteasomes. Examples of these compounds are presented in .
5. Lessons from the development of proteasome inhibitors as anti-cancer agents
Inactivation of chymotrypsin-like (β5) activity in eukaryotic cells causes a significant retardation of growth, an increase in stress sensitivity, and an accumulation of proteasome substrates [Citation43,Citation44]. Specific inhibitors of the β2 (trypsin-like) proteasome activity are nontoxic to mammalian cell lines when used as a single agent; but synergize the toxicity of β5-targeted inhibitors [Citation45,Citation46].
Malignant plasma cells are particularly sensitive to proteasome inhibition due to the very high levels of proteotoxic stress associated with uncontrolled antibody production [Citation47]. Proteasome inhibition also decreases the degradation of an NF-κB binding protein, thus keeping NF-κB out of the nucleus and permitting apoptosis [Citation48].
The reversible, covalent peptide boronate proteasome inhibitors, bortezomib (Velcade® [Citation49],) and ixazomib (Ninlaro® [Citation50]) and the irreversible peptide epoxyketone, carfilzomib (Kyprolis® [Citation51]), are used to treat multiple myeloma. These inhibitors primarily target the β5 active site of the human proteasome. Partial inhibition of β5 is tolerated in nonmalignant cells, if β2 activity is also maintained, providing a therapeutic window [Citation52]. For example, ixazomib is administered orally and offers a favorable efficacy/safety profile, with weekly dosing [Citation53,Citation54]. Bortezomib has been tested in off-label trials of Systemic Lupus Erythematosus [Citation55], with efficacy shown to be dependent on co-inhibition of the constitutive and immuno-proteasomes [Citation56].
6. Structures of the human 20S proteasome with bound inhibitors
Our understanding of the determinants of proteasome inhibition has been greatly aided by high resolution structures of the 20S core particle determined using X-ray crystallography [Citation26,Citation57–Citation59]. The structure of the yeast 20S proteasome with bound bortezomib [Citation26,Citation60] (PDB: 5lf3) confirmed that the boronate forms a covalent bond with the active site threonine (Thr1). The interaction with the β5 subunit is further stabilized by a hydrogen bonding network between bortezomib and several residues lining the β5 active site pocket, including (in the human structure) Thr21, Gly47, Ala49 and Ala50, and β6 Asp125 [Citation26,Citation61]. Similarly, a crystal structure of the human 20S proteasome in complex with carfilzomib revealed that the S3 and S4 binding pockets are important in carfilzomib binding, with similar interactions with conserved amino acids, Thr21, Gly47, and Ala49 [Citation58]. The binding of carfilzomib or bortezomib causes a shift in the position of Met45 [Citation26,Citation58]. These data underscore the need to obtain high resolution structures of the proteasome with bound inhibitor to fully understand the binding mechanism and to aid further drug design.
7. P. falciparum is highly susceptible to proteotoxic stress
During development in its human host, P. falciparum morphs through different stages, requiring rapid turn-over of proteins via its ubiquitin/proteasome system. Moreover, the intraerythrocytic parasite is under oxidative stress, but is equipped with only limited endogenous anti-oxidant defense mechanisms [Citation62], making it particularly reliant on turnover of oxidatively damaged proteins. For example, a recent saturation mutagenesis study revealed that 54 of the 72 genes in the P. falciparum proteasome degradation pathway are essential [Citation63].
Artemisinin-based antimalarials exert their activity by forming free radical species that cause widespread damage to parasite proteins [Citation64,Citation65], a mechanism of action that is associated with compromised proteasome function – likely by clogging the proteasome with polyubiquitinated proteins [Citation66]. Accordingly, inhibitors of the proteasome strongly synergize artemisinin-induced killing of P. falciparum in culture and P. berghei in vivo [Citation35,Citation67,Citation68]. Similarly, knock-out of the P. falciparum homologue of PA28 renders ring stage P. falciparum more susceptible to dihydroartemisinin [Citation34]. Consistent with this observation, single-insertion mutants of an artemisinin sensitivity cluster of genes show increased sensitivity to the proteasome inhibitor, bortezomib [Citation63]. Similarly, mutations in the deubiquitinase, UBP1, have been observed in artemisinin-pressured parasites [Citation69], and UBP1 mutations are also associated with decreased ACT effectiveness in Africa [Citation70] and South East Asia [Citation71]. Thus, P. falciparum appears to exist on a razor edge of proteotoxic stress, leaving it particularly susceptible to proteasome inhibitors.
8. Targeting the plasmodium proteasome
8.1. The proteasome is a well-validated antimalarial target
Proteasome inhibitors exhibit parasiticidal activity against all stages of development, including early asexual blood stages, late stage gametocytes and liver stage trophozoites [Citation36,Citation42,Citation68,Citation72–Citation74] – life stages that are resistant to most other chemotherapeutic agents. Diverse warheads such as epoxyketone derivatives [Citation37,Citation72], vinyl sulfones [Citation35,Citation73] and peptide boronates [Citation36,Citation75] have been explored. Parasite killing has been shown to be closely correlated with the extent of inhibition of the β5 subunit, as monitored using active site probes [Citation36,Citation72], although synergism is observed if Pf20S β2 activity is also inhibited [Citation68].
A major challenge for the development of proteasome inhibitors as antimalarial agents is achieving selectivity for the plasmodium proteasome over the human proteasome. Efforts to design and synthesize selective inhibitors have relied on substrate profiling, library screening and structural studies, as discussed below.
8.2. Profiling the substrate preferences of the plasmodium proteasome
Several groups have investigated the substrate preferences of the plasmodium proteasome. A diverse set of synthetic natural tetradecapeptides was subjected to degradation by Pf20S and the most favored amino acids in the subsites (P1, P2 and P3) were assessed by mass spectrometry [Citation35]. In an alternative approach, which has the advantage of being able to assign P2 and P3 preferences to a particular P1 residue, cleavage preferences for large libraries of fluorescent 7-amino-4-methyl coumarin (AMC) natural and non-natural peptide substrates were assessed [Citation36,Citation42].
These studies revealed that, in general, the human constitutive and immuno-proteasomes are more active and have a broader specificity profile than P. falciparum 20S; however, residues in some positions do confer some specificity. Pf20S prefers aromatic residues at P1 and P3. Many of the preferred substrates with hydrophobic residues at P1, such as Leu (i.e. likely a substrate for the β5 active site) have P3 tryptophan [Citation36]. Similarly, substrates with tryptophan at both the P2 and P3 positions are preferred by Pf20S [Citation35], particularly for basic residues at P1 [Citation36]. This is potentially due to the more open conformation of the β2-binding pocket compared to the human proteasome (see below). Substrates incorporating the non-natural amino acid, homophenylalanine, at the P3 position provide further selectivity [Citation42], while 2-thienylalanine gives additional specificity compared to leucine at P1.
An inhibitor, WLL-vs ()), designed on the basis of amino acid preferences, proved a potent inhibitor of the P. falciparum β2 and β5 active sites. It also inhibited the human β5 active site but showed little activity against the human β2 active site [Citation35]. This translated into high potency against P. falciparum cultures (50% Effective Concentration (EC50) ~6 nM) and more than 200-fold lower activity against human foreskin fibroblasts (HFF). A single bolus dose (i.v.) of WLL-vs (35 mg/kg administered 3 days after inoculation) provided almost complete reduction of parasite burden in a P. chabaudi model [Citation35].
8.3. Screening libraries for inhibitors of the plasmodium proteasome
Preparation of P. falciparum for screening purposes involves chromatographic purification from parasite lysates [Citation36,Citation76]. Relatively low yields make this a barrier step. Recombinant P. falciparum PA28 or human PA28 can be generated or purchased as an activator. Proteasome activity is assessed using fluorogenic substrates: Ac-nLPnLD-AMC for caspase-like (β1) activity, Ac-WLR-AMC for trypsin-like (β2) activity and Ac-WLA-AMC or Suc-LLVY-AMC for chymotrypsin-like (β5) activity.
A library of 1600 short N,C-capped peptide non-covalent proteasome inhibitors was screened against purified P. falciparum 20S [Citation76]. One cyclic biphenyl ether compound inhibited intraerythrocytic growth of P. falciparum with an EC50 value of 35 nM, with >1000-fold lower activity against human foreskin fibroblasts (HFF).
A recent screen made use of a focussed library of peptide boronates. The screen identified inhibitors of the growth of cultures of P. falciparum with efficacy at low nM concentrations. The most selective compound, MPI-1 ()), which has very low activity against the human β2 active site, exhibited more than 100-fold improvement in selectivity, relative to bortezomib, against the mammalian cell line, HepG2 [Citation36].
Another screen of a focused proteasome inhibitor library identified non-covalent asparagine ethylenediamine (AsnEDA) inhibitors, with the front-runner, TDI4258 ()), exhibiting excellent selectivity (>1000-fold) compared with the mammalian cell line, HepG2 [Citation68]. One difficulty that has been encountered in the biochemical assay of these non-covalent inhibitors is that P. falciparum β2 appears to possess some activity against the β5 substrates, resulting in remnant signal with inhibitors that specifically and reversibly target P. falciparum β5 activity. A similar issue was encountered in a screen of T. cruzi proteasome inhibitors [Citation77]. In the P. falciparum assay, co-inhibition with the β2-specific inhibitor, WLW-vs, was used to assess the β5 activity of AsnEDA inhibitors. Unfortunately, this class of inhibitors is unstable in vivo (half-life ~30 min). The front-runner compound is not active (when used alone) in the P. yoelii model of malaria, but shows efficacy when used at 15 mg/kg in combination with WLW-vs (60 mg/kg; for 4 days beginning 1 day after inoculation) [Citation68].
8.4. Using in vitro evolution of resistance of P. falciparum proteasome inhibitors to validate the target and assess resistance risk
In vitro evolution of resistance has been used successfully to identify, validate and characterize the targets of a number of antiparasitic compounds [Citation78,Citation79]. When applied to P. falciparum blood stages cultures, multiple cloned lines are exposed to the compound of interest over a period of weeks to several months. If cultures are retrieved and exhibit an increase in the EC50 value, it is likely that resistance-conferring mutations have occurred. Whole genome sequencing of cloned resistant lines reveals the causative mutations.
In one study [Citation36], selection of the P. falciparum 3D7 line for resistance to bortezomib, over a period of 4 months, yielded two clones with ~10-fold decreased sensitivity. Sequencing revealed that the clones had acquired a single Met45Ile mutation in the Pf20S β5 subunit, or the same mutation plus a Leu53Phe mutation in the same subunit. The mutated residues are adjacent to the predicted bortezomib-binding pocket [Citation80]. In the yeast and human β5 active sites, binding of bortezomib is driven by a change in the conformation of Met45 in the S1 pocket [Citation26,Citation61]. When resistance to bortezomib was evolved in a human cell line, a related Met45Val mutation was selected [Citation80]. Met45Val was also selected when yeast was rendered resistant to a peptide epoxyketone inhibitor, Carmaphycin B, an analogue of which ()) exhibits good activity against P. falciparum [Citation37]. The equivalent Met120Ile mutant has been observed in a multiple myeloma patient under prolonged treatment with bortezomib [Citation81]. This work confirms that inhibition of Pf20S β5 activity underpins the anti-plasmodial activity of bortezomib and suggests a very similar binding mode in plasmodium and mammalian cells.
Another study selected K13 wildtype and mutant P. falciparum lines for resistance to WLL-vs and WLW-vs, achieving only ~5-fold resistance. A range of mutations was observed in the Pf20S β2, β5 and β6 subunits or in components of the 19S proteasome regulatory particle [Citation82].
In another study, the Dd2 line of P. falciparum was subjected to gradually increasing concentrations of the AsnEDA inhibitor, PKS21004, over a period of 4 months [Citation68]. A stably resistant parasite line with a ∼130-fold decreased sensitivity to PKS21004 was obtained, but with no change in sensitivity to bortezomib. Of interest, the resistant clones exhibited an Ala117Asp mutation in the β6 subunit, with no mutations in the β5 subunit. This work suggests that PKS21004 binds outside the β5 active site, potentially at the β5/β6 interface, blocking access of substrates to the β5 pocket. The mutation may prevent binding of PKS21004 to the β5 S1 side pocket and S3 pockets, which lie adjacent to the β6 subunit.
A follow-up study evolved resistance over a period of one week to another AsnEDA, TDI4258 [Citation83] ()). Decreased sensitivity (>500-fold) was observed, with cross-resistance to bortezomib and PKS21004. Whole genome sequencing revealed an Ala49Ser mutation in the S1 pocket of the β5 subunit. Ala49 polymorphisms have been reported in cancer cell lines associated with resistance to bortezomib [Citation84]. Further structural data are required to assess how the β5Ala49Ser mutation restricts binding of the AsnEDA inhibitors. Further studies will also help assess whether different chemotypes induce different levels of resistance.
8.5. Structural analysis of the plasmodium proteasome
Efforts to achieve selective inhibition of enzyme targets are greatly facilitated by structure-guided design. High-resolution structures of the P. falciparum 20S have been determined using cryo-electron microscopy (cryo-EM) and single particle analysis. The structure of Pf20S with bound WLW-vs (PDB: 5 fmg) was solved at 3.6 Å [Citation35]. The structure of Pf20S without bound ligand (PDB: 6muw; 3.6 Å) is also available [Citation34]. The latter study reported the structure of Pf20S in complex with the activator, PfPA28. CryoEM offers the exciting possibility of imaging different conformational states of a protein complex, and it was revealed that PfPA28 binds the core particle asymmetrically – strongly engaging subunits only on one side of the core. The PfPA28 cap undergoes a rocking motion on the 20S barrel, opening and closing a gap at the PfPA28/Pf20S interface, potentially facilitating release of peptide products [Citation34].
Close analysis of residues within the different 20S active sites helps explain substrate preferences. For example, the S1 pocket in β1 is positively charged due to the presence of the guanidinium group of Arg45. The binding domain of the β2 subunit includes residue Glu51, which gives the region an acidic character, leading to the preference for basic residues. Gly45 at the base of the β2 subunit S1 pocket makes it more spacious than the β5 and β1 S1 pockets. Met45 protrudes into the S1 pocket at the base of the β5 active site, allowing it to accommodate hydrophobic residues.
The P. falciparum β2 and β5 subunits are 53% and 55% identical to the corresponding human constitutive 20S orthologues, respectively. The active site residues are well conserved and the overall charge on the residues lining the active sites is similar (), consistent with similar substrate specificities and highlighting the challenges associated with designing specific inhibitors. For example, the β5 active site is lined by the identical Gly47, Ala49 and Ala50 residues that are involved in bortezomib binding in the human enzyme.
Figure 3. Structural analysis of the proteasome reveals the basis of inhibition. (a and b) Overlays of the Pf20S (PDB 6muw) and human 20S (PDB 5le5) Pf20S β2 (a) and β5 (b) active sites. The active site threonine (Thr1) is shown in purple. The P. falciparum 20S β2 active site is open and can accommodate bulkier substrates. The β5 P3 pocket is narrower due to Met22 replacing the equivalent Ala22 in human β5. (c) CryoEM structure of P. falciparum 20S β2 with bound WLW-vs (PDB 5fmg). The active site threonine is shown in red. (d) The human 20S β2 (PDB 5le5) active site with docked WLW-vs illustrating the poor fit. (e) CryoEM structure of the L. donovani 20S β5 active site (PDB 6qm7). Compound 8 (cpd8) binds to the space between Ld20S β4 and β5 subunits and close to the active site threonine (red). (f) Overlay of Ld20S/cpd8 (PDB 6qm7; beige) with human 20S structure/bortezomib (PDB 5lf3; purple). Relative binding positions of cpd8 and bortezomib are depicted.
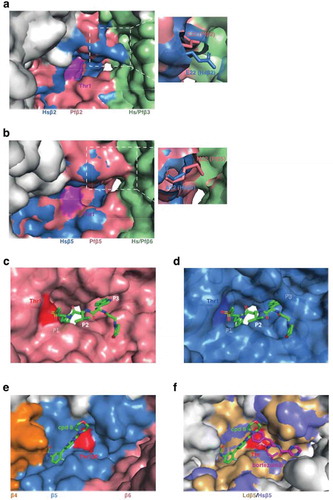
Nonetheless, some of the amino acids surrounding the substrate-binding pockets are different, suggesting that inhibitors could be developed that preferentially bind the Plasmodium enzymes. For example, the P. falciparum 20S β2 active site has an unusually open substrate-binding pocket that can better accommodate the bulky side chain of tryptophan at the P1 and P3 positions, while the human β2 active site cannot ()). Similarly, the P1 pocket is larger in P. falciparum β5 and could accommodate bulkier substituents, while the P3 pocket is narrower due to Met22 replacing the equivalent Ala22 in human β5 ()).
The difference in the architecture of the P. falciparum β2 active site was exploited in the design of a Plasmodium inhibitor that specifically inhibits the P. falciparum β2 active site [Citation35]. A cryoEM structure confirmed binding of WLW-vs to the P. falciparum β2 active site ()) and a model ()) showed that the human β2 active site cannot accommodate the P1 and P3 tryptophan side chains of the inhibitor.
Resistance evolution studies suggest that PKS21004 occupies a site at the β5/β6 interface. There are a number of different residues lining this region that might be explored for P. falciparum selectivity. For example, surface-exposed P. falciparum β6 residues, Asn151 and Cys159, are replaced by Ser123 and Gln131 in human β6. It is predicted that Asp153 and Ser157 in P. falciparum β6 directly interact with PKS21004 [Citation68]. Even though these directly interacting residues are conserved in human β6, substitutions in the neighboring residues Asn151 and Cys159 may alter the properties of the binding pocket and hence provide selectivity.
9. Targeting the kinetoplastid proteasome
While the kinetoplastid proteasome has received much less attention than the P. falciparum proteasome, better progress has been made. In a heroic effort, a library of 3 million compounds was screened by the Genomics Institute of the Novartis Research Foundation (GNF) for activity against L. donovani amastigotes proliferating within primary mouse macrophages, bloodstream T. brucei trypomastigotes and T. cruzi amastigotes proliferating in 3T3 fibroblast cells. One of the hits identified was GNF5343, an azabenzoxazole [Citation85] that was optimized to give GNF6702 ()), which inhibits proliferation of L. donovani amastigotes with an EC50 value of 18 nM. Oral twice-daily dosing over eight days with 10 mg/kg of GNF6702 achieved a 90% reduction in parasite burden in a mouse model of visceral leishmaniasis and a five-fold reduction in a mouse model of cutaneous leishmaniasis. GNF6702 also proved effective in mouse models of T. brucei and T. cruzi [Citation85].
Evolution of resistance to GNF6702 in T. cruzi epimastigotes yielded clones with ~40-fold decreased sensitivity. Whole genome sequencing revealed a mutation (Ile29Met or Phe24Leu) in the proteasome β4 subunit. Interestingly, bortezomib and carfilzomib retained activity against the resistant lines, suggesting a non-overlapping binding site.
T. cruzi epimastigote or T. brucei lines engineered to over-express the Phe24Leu-mutated β4 subunit exhibited 10- and 70-fold reduced sensitivity to GNF6702, confirming the proteasome as the target. Biochemical analyses confirmed that GNF6702 specifically inhibits the activity of the T. cruzi β5 subunit and that GNF6702 exerts its inhibitory activity via a noncompetitive mode. This suggests that GNF6702 disrupts β5 activity without preventing binding of the substrate. Homology modeling was used to identify potential allosteric inhibitor binding sites. The authors proposed a pocket at the β4/β5 interface as a potential binding pocket.
In an independent parallel effort, GSK and Dundee Drug Discovery identified a similar azabenzoxazole hit from a phenotypic screen of a 15,659-compound diversity library against T. cruzi, which they refer to as compound 1. Their medicinal chemistry efforts, which focused on improving potency, bioavailability and mitigating genotoxicity risk of an embedded amine ultimately yielded compound 8 (GSK3494245/DDD01305143), which possesses similar architecture as GNF6702 ()). Compound 8 inhibits proliferation of intramacrophage L. donovani amastigotes with an EC50 value of 1.6 μM and exhibits good solubility and metabolic stability [Citation86]. In a mouse model of visceral leishmaniasis compound 8 elicited a > 95% reduction of parasite load, when dosed at 3 mg/kg orally twice a day for 10 consecutive days, with no evident toxicity. GSK3494245/DDD01305143 is in the DNDi Leishmania portfolio and the feasibility of proceeding to a Phase I study is being assessed [Citation87].
Evolution of resistance in L. donovani cell lines to a related azabenzoxazole yielded clones with >100-fold decreased sensitivity. DNA sequencing revealed mutations within the genes encoding β4 (Thr30Ala) and β5 (Gly197Cys or Gly197Ser). Engineering of L. donovani to overexpress the β5 subunit bearing the Gly197Cys or Gly197Ser mutations reduced sensitivity to the azabenzoxazole inhibitors, again confirming the proteasome as the target. As for the GNF6702-resistant lines, bortezomib and carfilzomib retained activity.
Biochemical analyses in the luminescence assay format (Z-nLPnLD-aminoluciferin for β1, Z-LRR-aminoluciferin for β2 and Suc-LLVY-aminoluciferin for β5) confirmed that compound 8 specifically inhibits the activity of L. donovani β5 and leads to the accumulation of polyubiquitinated proteins. The activity of compound 8 against human β5 is >100-fold lower. The authors purified the 20S proteasome from the nonpathogenic Leishmania tarentolae and prepared the apo and compound 8-bound forms of the proteasome for single particle cryoEM analysis. Compound 8 binds in an open pocket between the β4 and β5 proteasome subunits proximal to the catalytic threonine residue ()). A network of hydrogen bonds to the side chain of Tyr212 and to the backbone amides of Gly228 and Ser229 anchor the molecule within the β5 pocket. The α carbon of Gly197 forms part of the floor of the ligand binding pocket, providing a structural rationale for the decreased sensitivity of the β5 Gly197Cys and Gly197Ser mutants.
Given the structural similarity of compound 8 and GNF6702 and the fact that both are noncompetitive inhibitors, it seems likely that they bind in similar poses. The binding mode proposed for GNF6702 suggests a different orientation compared with compound 8, but this may need to be revised in view of the structural data for compound 8.
The pocket between the β4 and β5 proteasome subunits is more open in the human β5 structure than in the Leishmania proteasome ()), likely underpinning the weaker inhibition of the human proteasome. Unfortunately, the relevant β4/β5 interface pocket is much more restricted in P. falciparum, indicating that these compounds will not have activity against malaria parasites.
In contrast to the compound 8 binding site, bortezomib binds fully within the β5 active site and an overlay of the human 20S/bortezomib and Ld 20S/compound 8 structures confirms that there is little overlap of the binding sites for these compounds ()).
A recent report used a similar luminescence-based assay to screen a library of 18,098 structurally diverse compounds against the T. cruzi proteasome identifying 39 hits of interest that are currently under further investigation [Citation77].
10. Conclusion
Potent inhibitors of P. falciparum and kinetoplastid proteasomes have been identified by screening focused proteasome inhibitor libraries against parasite cultures and by screening larger libraries against purified proteasome. Substrate profiling studies have also contributed to inhibitor design. Counter screens against purified human proteasome (both constitutive and immunoproteasome) and human cell lines have been used to assess selectivity.
Four important classes of plasmodium proteasome inhibitors have so far emerged, three exhibiting covalent binding. Whilst, high selectivity over the human proteasome is shown to be possible and there is promising evidence of in vivo efficacy in mouse models of malaria, it is clear that significant further optimization of pharmacokinetics, in particular, will be required to meet the target product profile for a single dose malaria treatment (TPP1). Improvements in bioavailability and metabolic stability would also facilitate progress of these series. Ideally, novel more drug-like series will be identified that do not carry the liabilities of the current (poly)peptidic series. Moreover, further work is needed to assess the risk of development of resistance [Citation88].
Structure-activity-relationship analyses have revealed that potency against P. falciparum and kinetoplastids correlates with inhibition of the β5 subunit activity. Structural studies reveal subtle differences in the β5 subunit active site that could be exploited for further drug design. While complete specificity of binding would be preferred, it appears that low toxicity against mammalian cell lines can be achieved with compounds that exhibit very low activity against the human β2 subunit.
Two separate studies identified a similar proteasome inhibitor series with excellent potency and selectivity against kinetoplastid parasites, both in vitro and in vivo. Structural studies revealed the binding site in an open pocket between the β4 and β5 proteasome subunits.
Rapid progress in the field over the past few years gives increased confidence that the proteasome can be successfully targeted for neglected diseases and that proteasome inhibitors will be taken forward into clinical studies.
11. Expert opinion
Malaria and kinetoplastid diseases remain a major health burden. In the absence of sterilizing vaccines, and in the face of inadequate chemotherapy regimens and emerging resistance, there is an urgent need for development of new drug candidates. Proteasome inhibitors exhibit potent activity against multiple stages of kinetoplastids and all stages of growth of P. falciparum, as well as exhibiting synergistic interactions with artemisinin antimalarials, making the proteasome a very desirable target.
But, the proteasome is a critical enzyme in every eukaryotic cell and the challenge is to find inhibitors that exhibit sufficient selectivity to enable their use for the treatment of infectious disease. Selectivity can be achieved based on either biochemical differences, i.e. targeting differences in active site architecture, or on differences in cell biology, i.e. exploiting differences in reliance on the proteasome system.
The application of cryoEM is providing the structural information needed to exploit differences in active site architecture; however, a comparison of the plasmodium and human β5 proteasome active sites reveals a high level of conservation. Parasite specificity achieved to date derives partly from selective targeting of plasmodium β5 activity over host β5, but also by minimizing activity against human β2 – which contributes to host cell toxicity.
Safe and effective treatments can be achieved by exploiting biological differences. This strategy underpins the use of proteasome inhibitors in the clinical treatment of malignant myeloma. Cancerous white blood cells overproduce immunoglobulins, making them particularly reliant on their proteasomes. Other cells can withstand partial loss of proteasome function. For example, ixazomib is well tolerated when delivered orally, in a fixed dose, once per week. Thus, proteasome inhibitors with incomplete biochemical specificity may still have an acceptable tolerance profile.
Progress in the development of inhibitors that target kinetoplastid proteasomes is very promising. The front-runner inhibitors exploit a pocket in the kinetoplastid proteasome between the β4 and β5 proteasome subunits. While this pocket is much shallower in the P. falciparum proteasome, preventing cross-species activity of front-runner azabenzoxazoles, noncompetitive inhibitors of the P. falciparum proteasome have been identified. These likely bind to the same β4/β5 interface, providing hope that this mode of inhibition can be further exploited.
P. falciparum acquires resistance (under selection) to covalent proteasome inhibitors only slowly and the levels of resistance achieved are low, while resistance develops more readily to non-covalent inhibitors. The MMV has defined a standard in vitro protocol to measure the resistance selection frequency. Further analysis of the minimal inoculum for resistance (MIR) for each of the proteasome classes under investigation will be needed to assess the risk of developing resistance to candidate proteasome inhibitors.
MMV’s discovery portfolio aims to deliver two new preclinical candidates per year. Currently, no proteasome inhibitor has been declared a preclinical candidate for malaria, and further work is required to determine whether proteasome inhibitors can meet MMV criteria for further development. Encouragingly, kinetoplastid proteasome inhibitors are further progressed and are expected to enter phase 1 trials for leishmaniasis.
Given their potential to deliver fast, effective, single-dose treatment of all stages, strains and species of plasmodium and kinetoplastids, proteasome inhibitors offer significant promise as next-generation antiparasitic agents.
Article highlights
Proteasome inhibitors exhibit potent activity against multiple stages of kinetoplastids and all stages of growth of P. falciparum, as well as exhibiting synergistic interactions with artemisinin antimalarials, making the proteasome a very desirable target.
Proteasome inhibitors are structurally diverse and fall into two large groups based on whether they have a warhead that forms a covalent bond with the active site threonines or inhibit via non-covalent interactions.
Selectivity can be achieved based on either biochemical differences i.e. targeting differences in active site architecture, or on differences in cell biology, i.e. exploiting differences in reliance on the proteasome system.
Non-covalent inhibitors with high selectivity for the kinetoplastid or P. falciparum proteasome and over the human proteasome have been achieved.
Recent cryoEM structures reveal the active site architecture of P. falciparum and Leishmania 20S, providing opportunities for improved inhibitor design.
There is promising evidence of in vivo efficacy in mouse models of infection, however, optimisation of pharmacokinetics will be required to meet the target product profile for a single dose malaria treatment.
This box summarizes key points contained in the article.
Declaration of interest
A Gould is employed by Takeda Pharmaceuticals and S. Brand is employed by the Medicines for Malaria Venture. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Additional information
Funding
References
- Hotez PJ, Pecoul B, Rijal S, et al. Eliminating the neglected tropical diseases: translational science and new technologies. PLoS Negl Trop Dis. 2016 Mar;10(3):e0003895.
- World_Health_Organisation. World Malaria Report 2018. 2018.
- Rogerson SJ, Desai M, Mayor A, et al. Burden, pathology, and costs of malaria in pregnancy: new developments for an old problem. Lancet Infect Dis. 2018 Jan 30;18:e107–e118.
- Teixeira AR, Nascimento RJ, Sturm NR. Evolution and pathology in chagas disease–a review. Mem Inst Oswaldo Cruz. 2006 Aug;101(5):463–491.
- Corbett CE, Ribeiro U Jr., Prianti MG, et al. Cell-mediated immune response in megacolon from patients with chronic Chagas’ disease. Dis Colon Rectum. 2001 Jul;44(7):993–998.
- Rassi A Jr., Rassi A, Little WC. Chagas’ heart disease. Clin Cardiol. 2000 Dec;23(12):883–889.
- Global_Health_Observatory. Human African trypanosomiasis. 2018. [cited 2019 Oct 31]. Available from: https://www.who.int/gho/neglected_diseases/human_african_trypanosomiasis/en/
- Mpanya A, Hendrickx D, Vuna M, et al. Should I get screened for sleeping sickness? A qualitative study in Kasai province, democratic republic of Congo. PLoS Negl Trop Dis. 2012 Jan;6(1):e1467.
- RTSS_Clinical_Trials_Partnership. Efficacy and safety of RTS,S/AS01. Malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet. 2015 Jul 4;386(9988):31–45.
- Gosling R, von Seidlein L. The future of the RTS,S/AS01 malaria vaccine: an alternative development plan. PLoS Med. 2016 Apr;13(4):e1001994.
- von Seidlein L.The Advanced Development Pathway of the RTS,S/AS01 Vaccine. Methods Mol Biol. 2019;2013:177–187. DOI: 10.1007/978-1-4939-9550-9_13. PMID: 31267502.
- Matuschewski K. Vaccines against malaria-still a long way to go. FEBS J. 2017 Aug;284(16):2560–2568.
- Noedl H, Se Y, Schaecher K, et al. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008 Dec 11;359(24):2619–2620.
- Hamilton WL, Amato R, van der Pluijm RW, et al. Evolution and expansion of multidrug-resistant malaria in southeast Asia: a genomic epidemiology study. Lancet Infect Dis. 2019Sep;19(9):943–951.
- van der Pluijm RW, Imwong M, Chau NH, et al. Determinants of dihydroartemisinin-piperaquine treatment failure in Plasmodium falciparum malaria in Cambodia, Thailand, and Vietnam: a prospective clinical, pharmacological, and genetic study. Lancet Infect Dis. 2019 Sep;19(9)952–961.
- Amaratunga C, Lim P, Suon S, et al. Dihydroartemisinin-piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Dis. 2016 Mar;16(3):357–365.
- Spring MD, Lin JT, Manning JE, et al. Dihydroartemisinin-piperaquine failure associated with a triple mutant including kelch13 C580Y in Cambodia: an observational cohort study. Lancet Infect Dis. 2015 Jun;15(6):683–691.
- Cullen DR, Mocerino M. A brief review of drug discovery research for human African trypanosomiasis. Curr Med Chem. 2017;24(7):701–717.
- Rao SPS, Barrett MP, Dranoff G, et al. Drug discovery for kinetoplastid diseases: future directions. ACS Infect Dis. 2019 Feb 8;5(2):152–157.
- Bilbe G. Infectious diseases. Overcoming neglect of kinetoplastid diseases. Science. 2015 May 29;348(6238):974–976.
- Alves F, Bilbe G, Blesson S, et al. Recent development of visceral Leishmaniasis treatments: successes, pitfalls, and perspectives. Clin Microbiol Rev. 2018;31:4.
- Burrows JN, van Huijsduijnen RH, Mohrle JJ, et al. Designing the next generation of medicines for malaria control and eradication. Malar J. 2013;12:187.
- Burrows JN, Duparc S, Gutteridge WE, et al. New developments in anti-malarial target candidate and product profiles. Malar J. 2017 Jan 13;16(1):26.
- Lupas A, Zwickl P, Wenzel T, et al. Structure and function of the 20S proteasome and of its regulatory complexes. Cold Spring Harb Symp Quant Biol. 1995;60:515–524.
- Löwe J, Stock D, Jap B, et al. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 angstrom resolution. Science. 1995;268:533–539.
- Groll M, Berkers CR, Ploegh HL, et al. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006 Mar;14(3):451–456.
- Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol. 2012 Jan 27;19(1):99–115.
- Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068.
- Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Best Pract Res Clin Haematol. 2017 Dec;30(4):341–355.
- Eisele MR, Reed RG, Rudack T, et al. Expanded coverage of the 26S proteasome conformational landscape reveals mechanisms of peptidase gating. Cell Rep. 2018;24:1301–15.e5.
- Zhu Y, Wang WL, Yu D, et al. Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat Commun. 2018;9:1–12.
- de la Pena A, Goodall EA, Gates SN, et al. Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science. 2018.
- Mott JD, Pramanik BC, Moomaw CR, et al. PA28, an activator of the 20 S proteasome, is composed of two nonidentical but homologous subunits. J Biol Chem. 1994 Dec 16;269(50):31466–31471.
- Xie SC, Metcalfe RD, Hanssen E, et al. The structure of the PA28-20S proteasome complex from Plasmodium falciparum and implications for proteostasis. Nat Microbiol. 2019;4:1990–2000.
- Li H, O’Donoghue A, van der Linden WA, et al. Structure and function based design of plasmodium-selective proteasome inhibitors. Nature. 2016;530:233–236.
- Xie SC, Gillett DL, Spillman NJ, et al. Target validation and identification of novel boronate inhibitors of the Plasmodium falciparum proteasome. J Med Chem. 2018 Nov 21;61(22):10053–10066.
- LaMonte GM, Almaliti J, Bibo-Verdugo B, et al. Development of a potent inhibitor of the plasmodium proteasome with reduced mammalian toxicity. J Med Chem. 2017 Aug 10;60(15):6721–6732.
- Adams J, Behnke M, Chen S, et al. Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg Med Chem Lett. 1998 Feb 17;8(4):333–338.
- Kupperman E, Lee EC, Cao Y, et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010 Mar 1;70(5):1970–1980.
- Vinitsky A, Michaud C, Powers JC, et al. Inhibition of the chymotrypsin-like activity of the pituitary multicatalytic proteinase complex. Biochemistry. 1992 Oct 6;31(39):9421–9428.
- Groll M, Bochtler M, Brandstetter H, et al. Molecular machines for protein degradation. Chembiochem. 2005 Feb;6(2):222–256.
- Yoo E, Stokes BH, de Jong H, et al. Defining the determinants of specificity of Plasmodium proteasome inhibitors. J Am Chem Soc. 2018 Sep 12;140(36):11424–11437.
- Chen P, Hochstrasser M. Autocatalytic subunit processing couples active site formation in the 20S proteasome to completion of assembly. Cell. 1996 Sep 20;86(6):961–972.
- Heinemeyer W, Fischer M, Krimmer T, et al. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J Biol Chem. 1997 Oct 3;272(40):25200–25209.
- Geurink PP, van der Linden WA, Mirabella AC, et al. Incorporation of non-natural amino acids improves cell permeability and potency of specific inhibitors of proteasome trypsin-like sites. J Med Chem. 2013 Feb 14;56(3):1262–1275.
- Mirabella AC, Pletnev AA, Downey SL, et al. Specific cell-permeable inhibitor of proteasome trypsin-like sites selectively sensitizes myeloma cells to bortezomib and carfilzomib. Chem Biol. 2011 May 27;18(5):608–618.
- Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017 July;14(7):417–433.
- Karin M, Delhase M. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin Immunol. 2000 Feb;12(1):85–98.
- Kreidenweiss A, Kremsner PG, Mordmuller B. Comprehensive study of proteasome inhibitors against Plasmodium falciparum laboratory strains and field isolates from Gabon. Malar J. 2008;7:187.
- Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol. 2014 Dec;15(13):1503–1512.
- Demo SD, Kirk CJ, Aujay MA, et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007 July 1;67(13):6383–6391.
- Britton M, Lucas MM, Downey SL, et al. Selective inhibitor of proteasome’s caspase-like sites sensitizes cells to specific inhibition of chymotrypsin-like sites. Chem Biol. 2009 Dec 24;16(12):1278–1289.
- Smolewski P, Rydygier D. Ixazomib: an investigational drug for the treatment of lymphoproliferative disorders. Expert Opin Investig Drugs. 2019 May;28(5):421–433.
- Hungria VTM, Crusoe EQ, Bittencourt RI, et al. New proteasome inhibitors in the treatment of multiple myeloma. Hematol Transfus Cell Ther. 2019 Jan–Mar;41(1):76–83.
- Ishii T, Tanaka Y, Kawakami A, et al. Multicenter double-blind randomized controlled trial to evaluate the effectiveness and safety of bortezomib as a treatment for refractory systemic lupus erythematosus. Mod Rheumatol. 2018 Nov;28(6):986–992.
- Ladi E, Everett C, Stivala CE, et al. Design and evaluation of highly selective human immunoproteasome inhibitors reveal a compensatory process that preserves immune cell viability. J Med Chem. 2019 July 29;62:7032–7041.
- Huber EM, Basler M, Schwab R, et al. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell. 2012;148:727–738.
- Harshbarger W, Miller C, Diedrich C, et al. Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure. 2015;23:418–424.
- Groll M, Ditzel L, Lowe J, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997 Apr 3;386(6624):463–471.
- Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev. 2007 Mar;107(3):687–717.
- Schrader J, Henneberg F, Mata RA, et al. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science. 2016 Aug 5;353(6299):594–598.
- Becker K, Tilley L, Vennerstrom JL, et al. Oxidative stress in malaria parasite-infected erythrocytes: host-parasite interactions. Int J Parasitol. 2004 Feb;34(2):163–189.
- Zhang M, Wang C, Otto TD, et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science. 2018;360:6388.
- Wang J, Zhang C-J, Chia W, et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun. 2015 Dec 22;6:10111. Online.
- Chen MZ, Moily NS, Bridgford JL, et al. A thiol probe for measuring unfolded protein load and proteostasis in cells. Nat Commun. 2017 Sep 7;8(1):474.
- Bridgford JL, Xie SC, Cobbold SA, et al. Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat Commun. 2018 Sep 18;9(1):3801.
- Dogovski C, Xie SC, Burgio G, et al. Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol. 2015 Apr;13(4):e1002132.
- Kirkman LA, Zhan W, Visone J, et al. Antimalarial proteasome inhibitor reveals collateral sensitivity from intersubunit interactions and fitness cost of resistance. Proc Natl Acad Sci U S A. 2018 July 17;115(29):E6863–E70.
- Hunt P, Afonso A, Creasey A, et al. Gene encoding a deubiquitinating enzyme is mutated in artesunate- and chloroquine-resistant rodent malaria parasites. Mol Microbiol. 2007 July;65(1):27–40.
- Henriques G, Hallett RL, Beshir KB, et al. Directional selection at the pfmdr1, pfcrt, pfubp1, and pfap2mu loci of Plasmodium falciparum in Kenyan children treated with ACT. J Infect Dis. 2014 Dec 15;210(12):2001–2008.
- Cerqueira GC, Cheeseman IH, Schaffner SF, et al. Longitudinal genomic surveillance of Plasmodium falciparum malaria parasites reveals complex genomic architecture of emerging artemisinin resistance. Genome Biol. 2017 Apr 28;18(1):78.
- Li H, Ponder EL, Verdoes M, et al. Validation of the proteasome as a therapeutic target in Plasmodium using an epoxyketone inhibitor with parasite-specific toxicity. Chem Biol. 2012 Dec 21;19(12):1535–1545.
- Tschan S, Brouwer AJ, Werkhoven PR, et al. Broad-spectrum antimalarial activity of peptido sulfonyl fluorides, a new class of proteasome inhibitors. Antimicrob Agents Chemother. 2013 Aug;57(8):3576–3584.
- Ng CL, Fidock DA, Bogyo M. Protein degradation systems as antimalarial therapeutic targets. Trends Parasitol. 2017 Sep;33(9):731–743.
- Reynolds JM, El Bissati K, Brandenburg J, et al. Antimalarial activity of the anticancer and proteasome inhibitor bortezomib and its analog ZL3B. BMC Clin Pharmacol. 2007;7:13.
- Li H, Tsu C, Blackburn C, et al. Identification of potent and selective non-covalent inhibitors of the Plasmodium falciparum proteasome. J Am Chem Soc. 2014 Oct 1;136(39):13562–13565.
- Zmuda F, Sastry L, Shepherd SM, et al. Identification of novel Trypanosoma cruzi proteasome inhibitors using a luminescence-based high-throughput screening assay. Antimicrob Agents Chemother. 2019;63(9):Sep.
- Cowell AN, Istvan ES, Lukens AK, et al. Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science. 2018 Jan 12;359(6372):191–199.
- Flannery EL, Fidock DA, Winzeler EA. Using genetic methods to define the targets of compounds with antimalarial activity. J Med Chem. 2013 Oct 24;56(20):7761–7771.
- Franke NE, Niewerth D, Assaraf YG, et al. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia. 2012 Apr;26(4):757–768.
- Barrio S, Stuhmer T, Da-Via M, et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia. 2018 Feb;33(2):447–456.
- Stokes BH, Yoo E, Murithi JM, et al. Covalent Plasmodium falciparum-selective proteasome inhibitors exhibit a low propensity for generating resistance in vitro and synergize with multiple antimalarial agents. PLoS Pathog. 2019 June;15(6):e1007722.
- Zhan W, Visone J, Ouellette T, et al. Improvement of asparagine ethylenediamines as anti-malarial Plasmodium-selective proteasome inhibitors. J Med Chem. 2019 Jul 11;62(13):6137–6145.
- Oerlemans R, Franke NE, Assaraf YG, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008 Sep 15;112(6):2489–2499.
- Khare S, Nagle AS, Biggart A, et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature. 2016 Sep 8;537(7619):229–233.
- Wyllie S, Brand S, Thomas M, et al. Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc Natl Acad Sci U S A. 2019 May 7;116(19):9318–9323.
- Wasunna M. Drug therapy in Africal visceral leishmaniasis. 28th ECCMID Conference; 2018 Apr 21; Madrid, Spain; 2018. Available from: https://www.escmid.org/escmid_publications/escmid_elibrary/material/?mid=64953
- Ding XC, Ubben D, Wells TN. A framework for assessing the risk of resistance for anti-malarials in development. Malar J. 2012 Aug;22(11):292.