
1. Introduction
Mitochondria are essential cellular organelles for energy production and calcium (Ca2+) critically regulates their function. At physiological levels, mitochondrial calcium (mCa2+) uptake stimulates dehydrogenases and promotes bioenergetics. In pathological conditions such as ischemia-reperfusion (IR) injury, heart failure, skeletal muscle dystrophy, and neurodegenerative diseases, cell signaling causes sustained elevations in cCa2+ levels by many mechanisms [Citation1]. In these conditions, mitochondria become overloaded with Ca2+ resulting in metabolic derangement, activation of Ca2+-dependent proteases, and excessive reactive oxygen species (ROS) generation; all mechanisms that contribute to cellular demise. Mechanistically, Ca2+ and ROS are prominent sensitizers of the mitochondrial permeability transition pore (mPTP), a large channel at the inner mitochondrial membrane (IMM) which permits ions and solutes up to 1.5 kD to diffuse causing matrix swelling, membrane reorganization, mitochondrial membrane potential collapse, and loss of the electrochemical gradient, thus disrupting oxidative phosphorylation [Citation2]. Cristae remodeling and outer mitochondrial membrane permeability allows apoptogen release initiating the caspase-mediated apoptotic cascade [Citation3,Citation4,]. Additionally, if ATP is depleted due to loss of mitochondrial function, mPTP activation results in necrotic cell death [Citation5]. Therefore, preventing mCa2+ overload is an attractive therapeutic target for a wide range of diseases, yet to date, no entities targeting mCa2+ exchange have entered clinical trials. This lag in therapeutic application can be explained by the fact that the molecular components mediating mCa2+ exchange were only recently identified [Citation6]. In this editorial, we will discuss current strategies to prevent mCa2+ overload and highlight MCUB (mitochondrial calcium uniporter dominant negative β), formerly known as coiled-coil domain-containing 109B (CCDC109B), as a molecular target to modulate mCa2+ uptake and reduce mCa2+ overload in the context of disease [Citation7].
2. Targeting mCa2+ uptake – the Mitochondrial Calcium Uniporter channel (mtCU)
The genetic identification of the mtCU quickly led to the realization of considerable complexity due to its macromolecular nature. The precise stoichiometry and molecular weight (MW) of the uniporter remain unknown, but in general, blue native page and size-exclusion chromatography suggests a complex of ~600–800 kD in size [Citation7,Citation8,]. MCU (Mitochondrial Calcium Uniporter), the pore-forming subunit necessary for Ca2+ permeation, likely predominantly exists as a tetramer in a ‘dimer of dimers’ configuration [Citation9]. The N- and C-terminus face the matrix, with two transmembrane domains forming a hydrophilic channel containing a highly conserved DIME sequence necessary for Ca2+ selectivity. MICU1 and MICU2 (Mitochondrial Calcium Uptake 1/2) contain EF-hand motifs to mediate Ca2+ binding to regulate channel opening. They inhibit channel activity at low cCa2+ concentrations and sense elevations in cytosolic Ca2+ levels to increase open probability (gatekeeping function) [Citation10–Citation12]. EMRE (Essential MCU Regulator) is another mtCU subunit necessary for uniporter function, which is a conduit for the interaction between MCU and MICU1 [Citation13,Citation14,]. MCUB, a gene paralog of the pore-forming subunit, was originally proposed to exert a dominant negative effect on uniporter function [Citation15]. MCUB shares ~50% amino acid sequence identity with MCU and contains an identical DIME sequence shown to be critical for Ca2+ pore selectivity calling into question MCUBs true mechanism of action [Citation15].
The mtCU has been implicated in various disease conditions since it is the primary conduit for mCa2+ overload. In humans, MICU1 loss-of-function mutations result in neurological disorders and skeletal muscle myopathies, presumably due to loss of gatekeeping and mCa2+ overload [Citation16]. Mutations in the mitochondrial m-AAA proteases, which are associated with spinocerebellar ataxia and hereditary spastic paraplegia, were recently proposed to result in the accumulation of hyperactive mtCUs resulting in mCa2+ overload [Citation17]. This work mechanistically linked the failure to degrade EMRE to the accumulation of constitutively active uniporter channels. This directly links mtCU stoichiometry and assembly as critical to both proper function and disease.
Recent work in several disease models has revealed that targeting mtCU can indeed be protective against stressors and disease states featuring mCa2+ overload. Reducing mCa2+ uptake by MCU knockdown prevented glutamate excitotoxicity in primary neurons [Citation18]. mcu deletion prevented mCa2+ overload and dopaminergic neuronal loss in zebrafish models of Parkinson’s [Citation19]. Our group and others showed that conditional Mcu deletion in the adult mammalian heart (Mcu cKO) improved resistance to Ca2+ induced mitochondrial permeability transition and was sufficient to reduce infarct size and cardiac pathology following ischemia-reperfusion injury [Citation20,Citation21]. These reports suggest that targeting the mtCU to reduce mCa2+ overload has therapeutic potential. However, loss of MCU did diminish contractile responsiveness to isoproterenol (adrenergic signaling), suggesting mtCU-dependent Ca2+ uptake is necessary for cellular bioenergetics during stress [Citation20,Citation21]. This indicates that targeting MCU expression itself, or completely blocking mCa2+ uptake, is likely to come with negative consequences. Given the presumed function of MCUB to negatively regulate the mtCU, it presented as an attractive target to reduce mCa2+ overload without completely ablating the physiological/signaling function of matrix Ca2+. To discern how MCUB modulates the mtCU and examine if it was amendable to therapeutic manipulation, we recently generated gain- and loss-of-function models to explore MCUB’s mechanism of action and relevance to cardiac physiology [Citation7].
3. New insights regarding MCUB function in physiology and disease
Our first evidence that mechanisms were at play to modulate the assembly of the mtCU was that FPLC size-exclusion chromatography in MCUB−/- cells revealed an overall increase in high-molecular weight (MW) complexes containing MCU, MICU1, and MICU2 [Citation7]. Conversely, overexpression of MCUB in cardiomyocytes decreased the levels of MCU, MICU1, and MICU2 in the mtCU complex [Citation7]. These findings, coupled with the discovery that unlike MCU, MCUB does not directly interact with MICU1 or MICU2 suggests that the incorporation of MCUB into the uniporter channel displaces MCU and MCU-bound MICU1/2 [Citation7]. Physiologically, the incorporation of MCUB appears to be a stress-responsive mechanism to alter mtCU gating and limit cooperative activation of the channel. In support of this notion, we found that MCUB was not expressed in the high-MW uniporter complex of the heart at baseline, but only after stress or ischemic injury [Citation7]. Functionally, we found that loss of MCUB increased channel capacitance and amplified cooperative activation resulting in enhanced mCa2+ uptake at a given cytosolic Ca2+ level [Citation7]. Conversely, forced expression of MCUB reduced mCa2+ uptake and mitochondrial swelling in response to calcium challenge [Citation7]. Altogether, these results paint a picture that the induction of MCUB is a compensatory mechanism to reduce mCa2+ uptake and overload by altering the stoichiometry of regulatory subunits in the uniporter complex.
The controlled expression of MCUB in cardiomyocytes (flox-stop transgenic approach) in the adult heart revealed some interesting physiological findings. First, MCUB expression was sufficient to reduce mCa2+ transients on a beat-to-beat basis in slowly paced cardiomyocytes [Citation7]. It should be noted that we employed a very slow pacing frequency (0.1 Hz) to allow the assessment individual transients, as our group [Citation22] and others [Citation23,Citation24,] have shown a summation of transients at higher pacing frequency due to slower efflux, such that resolving and interpreting uptake dynamics becomes difficult. The MCUB-mediated reduction in mCa2+ uptake caused a huge reduction in contractile responsiveness to the beta-adrenergic agonist, isoproterenol [Citation7]. Surprisingly, this effect was even greater than what we observed with the deletion of Mcu [Citation20,Citation21,], which we think is due to temporal changes in compensatory mechanisms that will be addressed later. Our data suggest that contractile deficiencies were likely due to a loss of Ca2+-dependent activation of mitochondrial dehydrogenases, since we observed a significant increase in phosphorylated-pyruvate dehydrogenase (p-PDH), both at baseline and during adrenergic stimulation [Citation7]. (Dephosphorylation of PDH at the E1α subunit is mediated by matrix Ca2+ activation of PDH phosphatase and results in increased PDH activity). In agreement, the assessment of oxygen consumption rates of adult cardiomyocytes revealed a substantial decrease in respiratory reserve capacity suggesting an overall impairment in cellular bioenergetics [Citation7]. This energetic compromise was so significant that our first experiments of in vivo ischemia-reperfusion (IR) 1 week after the induction of MCUB expression resulted in lethality within the first 10 min of ischemia [Citation7]. Our interpretation is that mtCU-Ca2+ uptake is needed to activate mitochondrial energetics in the non-ischemic/remote zone to maintain contractility and cardiac output in the severely compromised heart. Further investigation found that the expression of MCUB alone, elicited a transient decrease in cardiac function 1 week following MCUB expression, which was resolved 3 weeks later [Citation7]. Our data suggest compensatory changes in mitochondrial energetics 1 month following MCUB expression was sufficient to restore oxidative reserve capacity and contractile function in response to adrenergic stimulation. We hypothesize that MCUB-expressing mitochondria activated calcium-independent mechanisms to regulate energetics since the phosphorylation of PDH at baseline and in response to isoproterenol was restored to control levels, even though mCa2+ uptake was still blunted. In fact, a second IR study 1 month after MCUB expression found a significant reduction in infarct size and Ca2+-overload induction of mPTP and mitochondrial swelling [Citation7]. The contrasting temporal phenotype of MCUB expression highlights the strong compensatory mechanisms that are induced in response to impaired mCa2+ uptake and may explain the divergent phenotypes in various mtCU genetic models, particularly those with constitutive or germline alterations in expression [Citation25–Citation27]. It is likely that key mitochondrial calcium control points are rewired to maintain energetics by other means. For example, PDH activity is not only regulated by Ca2+ activation of the phosphatase, but also by Acetyl-CoA, NAD(H), pyruvate, and fructose-1,6-bisphosphate levels. Thereby, compensatory alterations in the generation and consumption of metabolites are likely employed to counteract the loss of mCa2+ uptake.
No effective therapeutics exist to prevent or reduce mCa2+ overload. The only current strategy to block mCa2+ uptake is Ruthenium Red derivatives, such as Ru360, which directly bind MCU to inhibit channel function [Citation28,Citation29,]. Ruthenium Red derivatives lack specificity [Citation30–Citation32] and Ru360 is not very cell permeable [Citation29] making these compounds unsuitable for clinical translation. However, significant advances in drug design are generating more selective, cell permeable MCU inhibitors that may have in vivo potential [Citation29]. Further, the tissue-dependent differences in mtCU density and composition [Citation33,Citation34,] suggest the use of a general MCU inhibitor may have substantial on-target deleterious effects. The complexity of channel formation, stoichiometry, and regulation suggest more basic research is needed to define the mechanistic targets with greatest therapeutic potential. MCUB represents an exciting alternative for regulating mtCU function to limit mCa2+ overload. MCUB contains the exact same DIME sequence as MCU, which is critical for creation of the Ca2+ permeable pore. This calls into question exactly how MCUB limits mCa2+ uptake. Does MCUB alter the structure of the pore or is the majority of regulation by way of altering channel composition and gating as we propose here? Previous work by the Rizzuto group [Citation15] suggests that MCUB cannot form a Ca2+ permeable channel independent of MCU, but the evidence for this negative result is somewhat limited. In fact, the addition of excess MCUB did not alter the electrophysiological properties of the uniporter and had a negligible effect on channel open probability in their experiments [Citation15]. Further, MCUB alone could give rise to a functional channel permissive to Na+ conduction.
4. Expert opinion on MCUB
We used Modeler [Citation35] to align human MCU to the fungal MCU crystallography data reported by Fan et al. [Citation36] and used this theoretical framework for the structural prediction of channel formation by human MCUB (–). It should be noted that using Modeler has the caveat of lacking significant energy minimization, so the modeled structures may not represent the lowest energy state found in cells. Interestingly, there is striking similarity between the predicted MCU and MCUB structure both at the monomer () and tetramer () configurations particularly in the configuration of the pore where minor changes in pore size are observed (). It can also be appreciated that structurally the N- and C- termini of MCU and MCUB are more divergent. Altogether, this argues against the mechanism of action being solely due to alterations in Ca2+ permeation. Perhaps the mtCU is still capable of some minimal Ca2+ conduction when MCUB is present. It is even possible that MCUBs mechanism of action is independent of modulating the actual channel or pore region and is a direct result of altered gating/MICU interacting domains.
Figure 1. Structural modeling of MCUB and the mitochondrial calcium uniporter (mtCU). Using Modeler [Citation35], a program for comparative three-dimensional protein structure analysis [Citation37], we uploaded the structural data for fungal MCU (PDB 6D7 W) reported in Fan et al. [Citation36] and predicted the structure of human MCU followed by a subsequent prediction of human MCUB. (a) Comparison of monomeric structure. (b) Comparison of human MCU and MCUB homo-oligomeric tetrameric structure. (c) Zoom view of the mouth of the hydrophilic ionic pore formed by MCU tetramer, MCUB homo-oligomeric tetramer, and a hetero-oligomeric tetramer (1 MCUB: 3 MCU). Red sphere represents a calcium (Ca2+) ion and purple and orange highlight the DIME motif. The dashed white line is a theoretic measurement in angstroms (a) between Aspartate-261 MCU residues or Aspartate-322 MCUB residues. (d) Zoom of the sideview of opposing DIME motifs of MCU, MCUB, and MCU/MCUB hetero-oligomeric structure.
![Figure 1. Structural modeling of MCUB and the mitochondrial calcium uniporter (mtCU). Using Modeler [Citation35], a program for comparative three-dimensional protein structure analysis [Citation37], we uploaded the structural data for fungal MCU (PDB 6D7 W) reported in Fan et al. [Citation36] and predicted the structure of human MCU followed by a subsequent prediction of human MCUB. (a) Comparison of monomeric structure. (b) Comparison of human MCU and MCUB homo-oligomeric tetrameric structure. (c) Zoom view of the mouth of the hydrophilic ionic pore formed by MCU tetramer, MCUB homo-oligomeric tetramer, and a hetero-oligomeric tetramer (1 MCUB: 3 MCU). Red sphere represents a calcium (Ca2+) ion and purple and orange highlight the DIME motif. The dashed white line is a theoretic measurement in angstroms (a) between Aspartate-261 MCU residues or Aspartate-322 MCUB residues. (d) Zoom of the sideview of opposing DIME motifs of MCU, MCUB, and MCU/MCUB hetero-oligomeric structure.](/cms/asset/b12ee0b7-0d25-405c-a934-eea7d27c1054/iett_a_1732926_f0001_oc.jpg)
Figure 2. Mitochondrial calcium uniporter (mtCU) electrostatic interactions and transmembrane domains. (a) Electrostatic map of MCU and MCUB tetramers and monomers. (b) MCU and MCUB transmembrane domain interactions of MCU homo-oligomeric tetramer vs. hetero-oligomeric tetramer. Critical amino acid residues likely mediating hydrophobic interactions are highlighted.
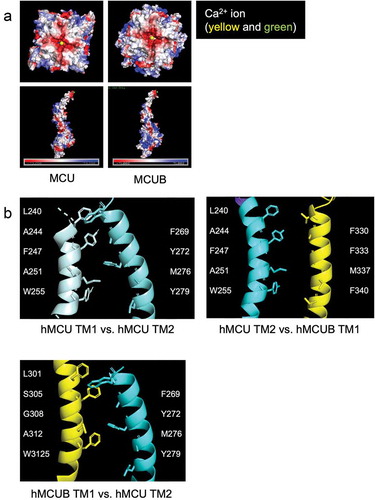
Notably, Raffaello et al. identified two key amino acid residues within MCUB that differ from MCU, (MCU 251R, 256E vs. MCUB 251W, 256V), which they proposed may influence Ca2+ permeation of the pore [Citation15]. Our structural models () indicate MCU and MCUB transmembrane domains vary in several amino acid residues which may alter Ca2+ binding capacity and pore structure, assuming a tetramer for discussion purposes. Transmembrane domain-1 (TM1) of MCU contains the hydrophobic residues Leu, Ala, Phe, Ala, and Trp, which interact with hydrophobic and polar-neutral amino acids Phe, Thr, Met, and Tyr of TM2 in the neighboring MCU subunit. The insertion of MCUB into the mtCU, may disrupt these interactions, since TM1 of MCUB contains a polar-neutral Ser and a Gly in place of the MCU hydrophobic Ala and Phe residues. TM2 of MCUB contains a hydrophobic aromatic Phe residue in place of MCU’s polar neutral Thr. Altogether, our minimalist modeling suggests that these differing interactions result in a slight widening of the initial opening of the ionic pore.
Our proteomic data, indicate that MCUB does not interact with MICU1. Interestingly, the newly discovered DIME interacting-domain of MCU is proposed to be a critical interface of MICU1 and MCU [Citation14]. The original amino acids Raffaello et al. [Citation15] identified in MCUB lie in very close proximity to this DIME interacting-domain and it is of significant interest to investigate if these amino acid differences are not only responsible for MCUBs lack of interaction with MICU1, but also for MCUBs mechanism of action.
Our study suggests a major molecular mechanism for MCUB regulation appears to be alterations in overall mtCU assembly. This is unique because it suggests alternatives to drug strategies that only target the pore and instead proposes that targeting the assembly or subunit stoichiometry is a new strategy to reduce mCa2+ overload (). Further research is needed to determine the exact amino acids responsible for MCUBs differential function in comparison to MCU. This insight may guide the generation of small peptides to modulate mtCU function in a cell-specific manner. In summary, even with the plethora of new studies on mtCU structure and function, much remains to be discovered about the molecular mechanisms governing the mtCU to aid the development of novel therapeutics to prevent the mCa2+ overload that occurs in numerous disease states.
Figure 3. Mitochondrial calcium uniporter (mtCU) structure and function amendable to therapeutic targeting. (a) Ruthenium red derivatives, such as Ru360, currently are the only major class of inhibitors of mitochondrial calcium (mCa2+) uptake. (b) The mitochondrial AAA+ proteases are localized to the inner mitochondrial membrane and functionally target proteins for degradation. Lack of Afgl32 and Spg7 lead to excessive EMRE accumulation resulting in overactive MCU, mitochondrial calcium overload, and neurodegeneration which is implicated in the conditions of spinocerebellar ataxia and hereditary spastic paraplegia. This directly implicates the regulation of mtCU assembly in health and disease and suggests protease activity may be a potential therapeutic target. (c) MICU1/2/3 (mitochondrial calcium uptake 1/2/3) are paralogs with functional distinction, as each member sets a different calcium threshold for entry and uniquely influences cooperative activation of mtCU. Therefore, modulating the expression or localization of MICUs may present a novel way to mitigate mCa2+ overload. (d) MCUB displaces MCU and MCU-bound MICU1/2 from mtCU, thus altering stoichiometry and assembly. Functionally, MCUB reduces mCa2+ uptake and critically can prevent mCa2+ overload during stress. (e) Post-translational modifications such as phosphorylation [Citation38,Citation39,] or glutathionylation [Citation40] have been reported to modulate the uniporter, opening the door to therapeutic targeting of the kinome, etc.
![Figure 3. Mitochondrial calcium uniporter (mtCU) structure and function amendable to therapeutic targeting. (a) Ruthenium red derivatives, such as Ru360, currently are the only major class of inhibitors of mitochondrial calcium (mCa2+) uptake. (b) The mitochondrial AAA+ proteases are localized to the inner mitochondrial membrane and functionally target proteins for degradation. Lack of Afgl32 and Spg7 lead to excessive EMRE accumulation resulting in overactive MCU, mitochondrial calcium overload, and neurodegeneration which is implicated in the conditions of spinocerebellar ataxia and hereditary spastic paraplegia. This directly implicates the regulation of mtCU assembly in health and disease and suggests protease activity may be a potential therapeutic target. (c) MICU1/2/3 (mitochondrial calcium uptake 1/2/3) are paralogs with functional distinction, as each member sets a different calcium threshold for entry and uniquely influences cooperative activation of mtCU. Therefore, modulating the expression or localization of MICUs may present a novel way to mitigate mCa2+ overload. (d) MCUB displaces MCU and MCU-bound MICU1/2 from mtCU, thus altering stoichiometry and assembly. Functionally, MCUB reduces mCa2+ uptake and critically can prevent mCa2+ overload during stress. (e) Post-translational modifications such as phosphorylation [Citation38,Citation39,] or glutathionylation [Citation40] have been reported to modulate the uniporter, opening the door to therapeutic targeting of the kinome, etc.](/cms/asset/6f442fa4-44ac-48e4-93db-70d16d37db6b/iett_a_1732926_f0003_oc.jpg)
Declaration of Interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Karch J, Bround MJ, Khalil H, et al. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci Adv. 2019;5(8):eaaw4597.
- Bernardi P, Colonna R, Costantini P, et al. The mitochondrial permeability transition. Biofactors. 1998;8(3–4):273–281.
- Zamzami N, Susin SA, Marchetti P, et al. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996 Apr 1;183(4):1533–1544.
- Frezza C, Cipolat S, Martins de Brito O, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006 [2006 July 14];126(1):177–189.
- Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J. 2013;77(5):1111–1122.
- Perocchi F, Gohil VM, Girgis HS, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010 Sep 16;467(7313):291–296.
- Lambert JP, Luongo TS, Tomar D, et al. MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation. 2019;140(21):1720–1733.
- Sancak Y, Markhard AL, Kitami T, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science (New York, NY). 2013 Dec 13;342(6164):1379–1382.
- Yoo J, Wu M, Yin Y, et al. Cryo-EM structure of a mitochondrial calcium uniporter. Science (New York, NY). 2018 Aug 3;361(6401):506–511.
- Csordas G, Golenar T, Seifert EL, et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013 June 4;17(6):976–987.
- Payne R, Hoff H, Roskowski A, et al. MICU2 restricts spatial crosstalk between InsP3R and MCU channels by regulating threshold and gain of MICU1-mediated inhibition and activation of MCU. Cell Rep. 2017 Dec 12;21(11):3141–3154.
- Mallilankaraman K, Doonan P, Cárdenas C, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell. 2012;151(3):630–644.
- Wang Y, Nguyen NX, She J, et al. Structural mechanism of EMRE-dependent gating of the human mitochondrial calcium uniporter. Cell. 2019 [2019 May 16];177(5):1252–61.e13.
- Paillard M, Csordas G, Huang KT, et al. MICU1 interacts with the D-ring of the MCU pore to control its Ca2+ flux and sensitivity to Ru360. Mol Cell. 2018 Nov 15;72(4):778–85.e3.
- Raffaello A, De Stefani D, Sabbadin D, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013 Aug 28;32(17):2362–2376.
- Logan CV, Szabadkai G, Sharpe JA, et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet. 2013 Dec 15;46:188. online.
- König T, Tröder Simon E, Bakka K, et al. The m-AAA protease associated with neurodegeneration limits MCU activity in mitochondria. Mol Cell. 2016;64(1):148–162.
- Qiu J, Tan Y-W, Hagenston AM, et al. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun. 2013;4:2034.
- Soman SK, Bazała M, Keatinge M, et al. Restriction of mitochondrial calcium overload by mcu inactivation renders a neuroprotective effect in zebrafish models of Parkinson’s disease. Biol Open. 2019;8(10):bio044347.
- Luongo TS, Lambert JP, Yuan A, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015 July 7;12(1):23–34.
- Kwong JQ, Lu X, Correll RN, et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015 July 7;12(1):15–22.
- Luongo TS, Lambert JP, Gross P, et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca(2+) homeostasis and viability. Nature. 2017 May 4;545(7652):93–97.
- Lu X, Ginsburg KS, Kettlewell S, et al. Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca2+ release. Circ Res. 2013;112(3):424–431.
- Liu T, Takimoto E, Dimaano Veronica L, et al. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a guinea pig model of heart failure. Circ Res. 2014 [2014 June 20];115(1):44–54.
- Pan X, Liu J, Nguyen T, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013 Dec;15(12):1464–1472.
- Liu JC, Liu J, Holmstrom KM, et al. MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 2016 Aug 9;16(6):1561–1573.
- Wu Y, Rasmussen TP, Koval OM, et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun. 2015 [2015 Jan 20];6(1):6081.
- Baughman JM, Perocchi F, Girgis HS, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–345.
- Woods JJ, Nemani N, Shanmughapriya S, et al. A selective and cell-permeable mitochondrial calcium uniporter (MCU) inhibitor preserves mitochondrial bioenergetics after hypoxia/reoxygenation injury. ACS Cent Sci. 2019 Jan 23;5(1):153–166.
- Xu L, Tripathy A, Pasek DA, et al. Ruthenium red modifies the cardiac and skeletal muscle Ca2+ release channels (ryanodine receptors) by multiple mechanisms. J Biol Chem. 1999;274(46):32680–32691.
- Pessah IN, Francini AO, Scales DJ, et al. Calcium-ryanodine receptor complex. Solubilization and partial characterization from skeletal muscle junctional sarcoplasmic reticulum vesicles. J Biol Chem. 1986;261(19):8643–8648.
- Emerson J, Clarke MJ, Ying WL, et al. The component of “ruthenium red” responsible for inhibition of mitochondrial calcium ion transport. Spectra, electrochemistry, and aquation kinetics. Crystal structure of .mu.-O-[(HCO2)(NH3)4Ru]2Cl3. J Am Chem Soc. 1993 [1993 Dec 1];115(25):11799–11805.
- Paillard M, Csordas G, Szanda G, et al. Tissue-specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep. 2017 Mar 7;18(10):2291–2300.
- Fieni F, Lee SB, Jan YN, et al. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun. 2012;3:1317.
- Eswar N, Webb B, Marti-Renom MA, et al. Comparative protein structure modeling using modeller. Curr Protoc Bioinf. 2006;15:5–6. Chapter 5:Unit-5.6.
- Fan C, Fan M, Orlando BJ, et al. X-ray and cryo-EM structures of the mitochondrial calcium uniporter. Nature. 2018;559(7715):575–579.
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993 Dec 5;234(3):779–815.
- Zhao H, Li T, Wang K, et al. AMPK-mediated activation of MCU stimulates mitochondrial Ca2+ entry to promote mitotic progression. Nat Cell Biol. 2019 [2019 Apr 1];21(4):476–486.
- Joiner ML, Koval OM, Li J, et al. CaMKII determines mitochondrial stress responses in heart. Nature. 2012 Nov 8;491(7423):269–273.
- Dong Z, Shanmughapriya S, Tomar D, et al. Mitochondrial Ca(2+) uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell. 2017 Mar 16;65(6):1014–28.e7.