
ABSTRACT
Introduction
Activin A is involved in the regulation of a surprisingly broad number of processes that are relevant for cancer development and treatment; it is implicated in cell autonomous functions and multiple regulatory functions in the tumor microenvironment.
Areas covered
This article summarizes the current knowledge about activin A in cell growth and death, migration and metastasis, angiogenesis, stemness and drug resistance, regulation of antitumor immunity, and cancer cachexia. We explore the role of activin A as a biomarker and discuss strategies for using it as target for cancer therapy. Literature retrieved from Medline until 25 June 2020 was considered.
Expert opinion
While many functions of activin A were investigated in preclinical models, there is currently limited experience from clinical trials. Activin A has growth- and migration-promoting effects, contributes to immune evasion and cachexia and is associated with shorter survival in several cancer types. Targeting activin A could offer the chance to simultaneously limit tumor growth and spreading, improve drug response, boost antitumor immune responses and improve cancer-associated or treatment-associated cachexia, bone loss, and anemia. Nevertheless, defining which patients have the highest likelihood of benefiting from these effects is challenging and will require further work.
1. Introduction – activins and inhibins
Activin A is the most prominent member of the activin/inhibin branch of the transforming growth factor-β (TGF-β) superfamily of growth, death, and differentiation factors. Activins and inhibins have originally been defined as factors activating and inhibiting follicle-stimulating hormone (FSH) release from the pituitary [Citation1]. Protein and gene identification revealed that all mammalian activins are dimers made from four different β subunits (βA, βB, βC, and βE, encoded by four genes: INHBA, INHBB, INHBC, and INHBE) [Citation2]. The β subunits can also dimerize with one α subunit (encoded by the INHA gene) to form inhibins. Activins exert their biological activities via binding to activin receptors, while inhibins can block activation of these receptors [Citation3]. Activins are either homodimers or heterodimers, but the evidence for in vivo occurrence of heterodimeric activins other than activin AB has remained scant [Citation2]. Activin A, the homodimer of two βA subunits, is by far the most extensively characterized activin followed at some distance by activin B. βA subunits are initially produced as larger pro-proteins of around 45 kDa. The pro-domains are required for dimerization and after cleavage from the 15 kDa mature domains by furin-like proteases, the pro-domains remain non-covalently associated with the dimeric activin A proteins during and after secretion [Citation4].
In the current article, we review the different functions of activin A in cancer as a basis for discussing the potential and the remaining challenges for using activin A as a therapeutic target in cancer patients. We have performed electronic literature searches in Medline, Google Scholar, and clinicaltrials.gov using the search term activin in combination with additional search terms including cancer, immunity, cachexia, treatment, and others. We have considered only literature that was published before June 25, 2020.
2. Activin A signaling
Like most members of the TGF-β superfamily, activins bind to and activate transmembrane serine/threonine kinase receptors. TGF-β family signal transduction via Smad proteins was worked out more than a decade ago and has been reviewed in detail, for instance by Weiss and Attisano [Citation5]. Of the seven type 1 and five type 2 receptors of the mammalian receptor serine/threonine kinase family, activin A was shown to use predominantly the type 1 receptor activin receptor type 1B (ActRIB or ACVR1B, also known as activin like kinase 4 or ALK4) and the type 2 receptors activin receptor 2A and 2B (ActRIIA, ACVR2A; ActRIIB, ACVR2B) [Citation2]. The closely related type 1 receptor ActRIC (ALK7), which is involved in regulation of insulin secretion from pancreatic β cells and was recently proposed as a tumor and metastasis suppressor in a mouse model, is used by activin B and AB but not by activin A [Citation6–8]. Target genes regulated by activin A include developmental genes like Pax-6 [Citation9], hormones like FSH [Citation10], cell cycle regulators like p21 [Citation11], and p15 [Citation12] as well as its own antagonists follistatin [Citation13] and Smad7 [Citation14]. An overview of activin A signal transduction is shown in . Importantly, there is a high degree of promiscuity in signaling mechanisms of the TGF-β superfamily, since other ligands such as myostatin and growth and differentiation factor (GDF) 11 signal via an overlapping set of receptors and, moreover, TGF-β receptors are coupled to the same Smad proteins as activin receptors [Citation15]. Despite this overlap in some components of the signaling pathway, the functional redundancy between members of the TGF-β superfamily is limited by different spatiotemporal expression patterns of ligands and receptors, differences in signaling dynamics as well as by a number of regulatory proteins that modulate the response at different steps [Citation16,Citation17]. In addition to the canonical signaling pathway, non-canonical signaling via p38, Erk and Akt has also been reported for activin A in normal and malignant cells [Citation18–20]. Non-canonical activin A signals still depend on activin receptors but are Smad-independent.
Figure 1. Expression and signaling mechanisms of activin A and intervention points for targeting approaches. The synthesis of activin subunits and activin receptors, the regulation of activin receptor binding at the plasma membrane as well as activin A-induced signaling routes via Smad-dependent and Smad-independent pathways. Intervention points that have been used for targeting activin A signaling in preclinical models or clinical trials are highlighted
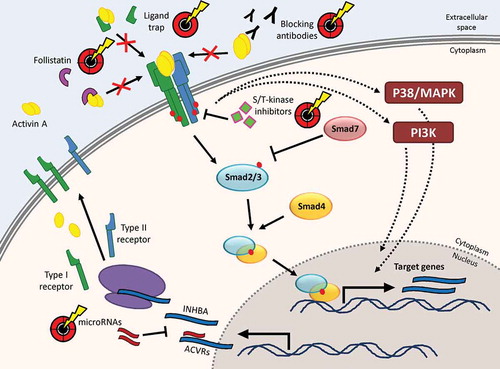
3. Regulation of activin A activity
Like most other potent signaling molecules, activin A is regulated at multiple levels. Expression of activin A was shown to be controlled by several other cytokines and hormones including IL-1, TNF-α, and glucocorticoids in the hematopoietic system [Citation21,Citation22] and by N-myc in osteosarcoma [Citation23]. Since the βA subunit is not only required for formation of activin A but also for inhibin A as well as for heterodimeric activins like activins AB or AC, the relative abundance of potential dimerization partners will influence the amount of activin A formed from the cellular pool of activin/inhibin subunits. Indeed, inhibin A is an important antagonist of activin A signals in the pituitary [Citation24], but whether – in the presence of several possible dimerization partners – some dimers are preferred over others and whether that may be a regulated process, remains to be explored. The influence of heterodimeric activins on signal transduction is also sparsely investigated, but at least for activin AC an antagonistic function with respect to receptor activation by activin A has been reported [Citation25]. Downstream from the receptors, Smad7 acts as an important intracellular feedback inhibitor of Smad-dependent activin A signal transduction by interfering with activation of Smad2/3 through activin receptors [Citation14].
A very relevant extracellular regulator of activin A after its secretion is follistatin. Follistatin is a monomeric secreted protein that binds activin A with high affinity and thereby blocks its capacity to activate receptor signaling [Citation26]. Follistatin is present in a circulating and in a cell membrane-associated form [Citation27] and a feedback loop upregulating follistatin in response to activin A signals has been described [Citation13]. In addition to activin A, follistatin can also bind myostatin and some bone morphogenetic proteins (BMP) [Citation28,Citation29]. A number of follistatin-related proteins exist but of these only follistatin-like 3 (also known as FLRG) can bind and antagonize activin A [Citation30]. While Smad7 and follistatin have been recognized as endogenous activin A-antagonizing factors for many years, an increasing number of studies show that both activin A itself as well as activin receptors are also subject to regulation by microRNAs. Examples for this are the targeting of ActRIIA by miR-15/16 [Citation31] and of the βA subunit by miR-146a [Citation32]
4. Activin A expression in normal and malignant cells
Activin βA mRNA is widely expressed in normal human tissues with high expression for instance in placenta, cartilage tissue, and testis according to the Bgee database of the University of Lausanne (https://bgee.org/?page=gene&gene_id=ENSG00000122641, accessed on 25 July 2020). With respect to activin A expression and activity in malignant cells compared to normal cell counterparts, both increases and decreases have been found. Cancers with a reported increase in activin A signaling include lung adenocarcinoma [Citation33], esophageal carcinoma [Citation34] or malignant pleural mesothelioma (MPM) [Citation35], whereas in breast cancer [Citation36], hepatocellular carcinoma (HCC) [Citation37] or neuroblastoma [Citation23] decreased activin signaling was proposed, often mediated by upregulation of follistatin or follistatin-related protein. Notably, overexpression of activin A by tumor cells can affect the tumor microenvironment, while the tumor cells themselves could be non-responsive to activin A by loss-of-function mutations in receptor molecules or Smad proteins. This was demonstrated, for instance, in microsatellite instable colorectal carcinoma [Citation38]. Cells of the tumor microenvironment like fibroblasts, endothelial cells and macrophages, known to influence tumor development, can also express and secrete activin A. The functional implications of activin A signals for tumor cell behavior and interaction with the tumor microenvironment will be reviewed in sections 5 to 10, the prognostic and predictive value of altered activin A levels in section 11.
5. Activin A in cell growth and death
In contrast to some cytokines that predominantly induce cell growth (like EGF) or cell death (like Fas ligand), activin A was shown to induce apoptosis and growth suppression in some contexts and increased proliferation in others. TGF-β, which has structural similarity with activin A and shares significant parts of the signal transduction pathway, has been assigned a dual role in carcinogenesis [Citation39]. It was described that TGF-β acts as a tumor suppressor in early stages of tumorigenesis and can switch to a protumorigenic role later on by stimulating epithelial to mesenchymal transition (EMT) and metastasis. Many of the same capabilities described for TGF-β were also reported for activin A, but the dependency on the stage of tumorigenesis appears less obvious. Instead, the cell type often seems to play a dominant role in determining the response to activin A. Exposure to activin A induces growth inhibition and apoptosis in normal hepatocytes [Citation40] but also in HCC cell lines [Citation41,Citation42]. Likewise, growth-limiting activity of activin A was shown for cell lines from epithelial tumors like prostate cancer [Citation43], breast cancer [Citation36], or colon cancer [Citation44] but also for diffuse large B-cell lymphoma [Citation45] and endothelial cells [Citation11,Citation46]. Stimulation of cell growth and survival by activin A in contrast, was demonstrated in lung fibroblasts [Citation47] and cell lines from lung cancer [Citation33] and MPM [Citation35]. Thus, based on the available evidence, neither the distinction between normal and malignant cells nor the epithelial versus mesenchymal origin of the cells alone is sufficient to predict the outcome of activin signals with respect to cell growth. While unresponsiveness of cells to activin A can be explained by a loss of signaling components like activin receptors or Smad proteins, or the overexpression of follistatin, the factors that decide between growth stimulation and growth inhibition need further analysis. For TGF-β, it was shown in cell models from different epithelial tumors that the cooperation with hyperactivated Ras can switch the response from apoptosis induction to stimulation of EMT [Citation48]. Whether this also applies to activin A has not been clarified. Thus, identifying molecular factors that determine the direction of the activin response with respect to cell growth is one of the unresolved issues in the activin field. Nevertheless, for a number of cancer types, there is sufficient evidence that inhibiting activin A signals could be used to impair growth and survival of tumor cells.
6. Activin A in migration, invasion and metastasis
While the response of normal and tumor cells to activin A with respect to cell growth is mixed, there is plenty of evidence that activin A stimulates cell migration in various cell types including some in which it inhibits proliferation like intestinal epithelial cells [Citation49] and colon cancer cells [Citation44]. In addition to migration, also invasion and metastasis were reported to be enhanced by activin A in several tumor entities including prostate [Citation50] and breast cancer [Citation51], oral squamous cell carcinoma [Citation52] and MPM [Citation20]. Enhancement of cell migration and invasion by activin A was linked to the activation of distinct signaling molecules. In mesothelioma cell lines, it was associated with a switch from canonical Smad signaling to non-canonical Erk signaling [Citation20] and activin A-mediated activation of Akt, Erk, and Rac1 was required to increase cell migration in ovarian cancer [Citation19]. In another report on ovarian tumor cells, however, activin A was shown to increase migration and suppress E-cadherin by upregulating the transcription factors Snail and Slug through Smad-dependent signaling [Citation53]. Loss of E-cadherin represents a hallmark of EMT, which is frequently associated with increased cell invasion, and also contributes to activin A-induced invasion in breast cancer cells [Citation54]. In a separate study, knockdown of activin A in breast cancer cells was shown to suppress migration in vitro and inhibit lung metastasis in vivo by inducing interleukin 13 receptor alpha 2 (IL13Rα2) overexpression [Citation51].
Activin A impacts migratory, invasive and metastatic capacities of tumor cells also via the tumor microenvironment [Citation55]. In liver metastasis, for instance, Activin A produced by the target organ was reported to promote metastasis formation [Citation56]. In the bone marrow niche, activin A production by stromal cells was induced by B-cell precursor-acute lymphoblastic leukemic cells and provided a migratory advantage for leukemic cells over healthy hematopoiesis [Citation57]. Overall, the great majority of studies conclude that activin A has a promigratory and prometastatic function and its inhibition could be used to prevent or reduce metastatic spreading.
7. Activin A in angiogenesis
Angiogenesis is an important process during embryonic development, but also critical for tissue regeneration after injury. Cancer cell-induced neoangiogenesis is a hallmark of tumor progression and antiangiogenic treatments have been included in the fight against cancer since the early development of vascular endothelial growth factor (VEGF)-targeting antibodies and VEGF receptor inhibitors [Citation58]. Several studies support the view that activin A has primarily antiangiogenic effects. Activin A inhibits the proliferation of endothelial cells in vitro and suppresses angiogenesis in the chicken chorioallantoic membrane (CAM) assay [Citation23], while angiogenesis is induced by the activin antagonist follistatin [Citation59]. Cancer-induced stimulation of angiogenesis results from a cooperation of angiostimulatory and angioinhibitory signals, which can be modulated by oncogenes in cancer cells. In neuroblastoma, overexpression of the oncogene N-myc is associated with a downregulation of endothelial cell proliferation inhibitors including activin A [Citation46]. Consequently, neuroblastoma cells with restored activin A expression exhibited decreased tumor growth and reduced vascularity, suggesting a tumor-suppressing function of activin A via reducing angiogenesis. Likewise, activin A expression in gastric cancer inhibits proliferation of vascular endothelial cells by inducing the cell cycle inhibitor p21 and hence, functions as suppressor of angiogenesis and tumor growth [Citation11]. In contrast to these effects of activin A on endothelial cell proliferation, tube formation by bovine aortic endothelial cells (BAEC) was stimulated by activin A through upregulation of VEGF and its receptors [Citation60]. Inhibition of activin A blocked VEGF activity, indicating that activin A represents a critical mediator of VEGF-induced angiogenesis in this model. In mammary carcinoma cells, however, activin A inhibited FGF2-induced sprouting angiogenesis [Citation61]. Activin A-expressing tumors exhibited a lower microvessel density, yet they grew faster than follistatin-expressing tumors that had a dense capillary network but were more susceptible to apoptosis. This indicates a dissociation of angiogenesis and tumorigenesis during tumor progression when tumor growth may not be limited by angiogenesis as long as a minimal intratumoral microvessel density is maintained. Comparable findings were described for activin A with respect to lymphangiogenesis. Despite a clear antilymphangiogenic effect of activin A in vitro and in vivo, activin A-overexpressing melanoma cells did not show reduced metastasis due to the migration-enhancing effects of activin A on the melanoma cells [Citation62]. It seems thus, that in several animal models the antimetastatic effect of targeting activin A predominates over stimulating effects on angiogenesis.
8. Activin A in antitumor immune response
Both activin A and its antagonist follistatin are increased during systemic inflammation and activin A has been implicated in conditions of chronic inflammation in the lung, gut, and liver [Citation63]. The majority of immune cells, including T and B lymphocytes, neutrophils, macrophages, dendritic cells and natural killer cells are capable of producing and responding to activin A. The response differs between cell types and displays variability depending on cell maturation, cell activation status, as well as on the spatial and temporal context. Hence, activin A can exert a broad range of pro- and antiinflammatory functions in infections, allergy and autoimmunity [Citation64]. The effects of activin A in cancer immunology can be equally divergent and depend on the tumor entity as well as the context of the immune response. In non-melanoma skin cancer, activin A overexpression in keratinocytes has been shown to promote chemically induced development of skin tumors in mice by inhibiting proliferation and epidermis-infiltration of epidermal γδ T cells, which confer protection against skin carcinogenesis [Citation65]. In the same study, activin A also mediated increased accumulation of tumor-promoting Langerhans cells and regulatory T cells (Treg) in the epidermis. Furthermore, increased levels of activin A were demonstrated to correlate with activation of a protumorigenic program in macrophages, leading to facilitated progression of skin carcinogenesis [Citation66]. In melanoma, tumor growth-promoting effects of activin A were observed in immunocompetent but not in immunocompromised mice and depended on activin A-induced reduction of T cell-mediated antitumor immunity [Citation67]. In another study in melanoma, it was shown that YAP, a coactivator of the Hippo pathway, cooperates with activin signaling in Treg-mediated repression of antitumor immunity [Citation68]. Consequently, activin blockade decreased tumor growth and boosted the effect of the antitumor vaccine GM-Vac. In pancreatic ductal adenocarcinoma, activin A contributed to the tumor-promoting interaction between cancer stem cells and tumor-associated macrophages by inducing the secretion of the immunomodulatory antimicrobial peptide 18/LL37 [Citation69]. In summary, inhibition of antitumor immune responses appears to substantially contribute to the protumorigenic effect of activin A. Stimulation of immune reactions could be an important consequence of targeting activin A that is worth considering for potential combinations with immunotherapeutic approaches.
9. Activin A in stemness and drug resistance
Activin A was described to maintain pluripotency of human embryonic stem cells (hESC) [Citation70] and was also used in protocols to differentiate them into, for instance, insulin-producing cells [Citation71] and hepatocytes [Citation72]. With respect to cancer stem cells – a controversial field in many cancer types – activin A was postulated to drive self-renewal of pancreatic [Citation73] and colorectal [Citation74] cancer stem cells and support cancer stem cell-like features in lung cancer [Citation75,Citation76] and mesothelioma cells [Citation77]. One characteristic feature of stem cells is their high degree of drug resistance. Thus, the impact of activin A on the response to various anticancer drugs has been investigated. Inhibition of activin signaling reversed gemcitabine resistance of pancreatic cancer stem cells [Citation73] and the ability of miR-17-92 to reduce tumorigenicity and chemoresistance depended on targeting this signaling axis [Citation78]. In lung adenocarcinoma, activin A increased resistance to platinum chemotherapy [Citation79]. In this model, inhibition of activin receptors or administration of follistatin not only improved chemotherapy response but also reduced cisplatin-induced nephrotoxicity. In patients suffering from MPM, the change in tumor size after platinum chemotherapy was smaller when they had high circulating activin A levels [Citation80]. No effect of activin A treatment on the response to seven anticancer drugs (cisplatin, carboplatin, doxorubicin, taxol, SN38, terarubicin and etoposide) was found, in contrast, in an ovarian adenocarcinoma cell line, but a cisplatin-resistant derivative showed decreased activin A sensitivity by loss of activin receptor type 2 expression [Citation81]. In diffuse large B-cell lymphoma, ectopic expression of activin A resulted in reduced cell growth and enhanced chemosensitivity [Citation45], and in the Bcr-Abl-positive chronic myeloid leukemia cell line K562, activin A-induced differentiation sensitized the cells to imatinib treatment [Citation82]. Apart from the K562 cell model, however, the impact of activin A on the response to kinase inhibitors or other targeted cancer therapeutics has remained largely unexplored so far. Overall, activin A seems to contribute to stemness and chemotherapy resistance especially in those cancer types, in which it enhances tumor cell growth. More data on its role in the response to molecularly targeted anticancer compounds would be urgently needed.
10. Activin A in cancer cachexia, bone loss and anemia
Early evidence for a role of activin A in cancer cachexia came from the study of inhibin knock-out mice. These mice develop gonadal tumors and, due to the lack of the inhibin α subunit, showed strongly elevated circulating levels of activin A that were linked to liver damage and weight loss [Citation83,Citation84]. Only more than a decade later, however, the link between inhibition of the activin receptor ActRIIB, reduction, and even reversal of cachexia and prolonged survival was firmly established [Citation85]. Since then, multiple studies have confirmed a critical role for activin A and the activin signaling axis in experimental models of cancer-induced weight loss and decrease of muscle mass [Citation86–88]. Moreover, a link between high activin A levels and cachexia was found in patients suffering from colorectal cancer, lung cancer, and MPM [Citation80,Citation89]. Toxicity from chemotherapeutic agents can contribute to cachexia and blocking activin receptors was found to protect from cachexia-enhancing effects of cisplatin [Citation90] and the FOLFIRI (leucovorin, fluorouracil, irinotecan) regimen [Citation91]. In addition to its role in muscle, activin A was shown to be a critical mediator of cancer-induced bone loss in multiple myeloma [Citation92] and contribute to chemotherapy-induced bone degeneration [Citation91]. Moreover, activin A and related TGF-β family ligands are also important regulators of erythropoiesis [Citation93] and can contribute to cancer-related anemia [Citation94,Citation95]. Cancer cachexia as well as cancer-related bone loss and anemia are important clinical problems contributing to cancer mortality. Interfering with the activin A signaling axis offers additional opportunities to prevent or reverse these cancer-induced or cancer therapy-induced conditions.
11. Activin A as cancer biomarker
Since activin A is a secreted protein and readily detectable in the circulation of patients by ELISA assays, it offers the chance to be used as noninvasive blood-based biomarker. Elevated activin A levels were found in the blood of patients suffering from HCC [Citation96], breast cancer [Citation97], adenocarcinoma of the lung [Citation98], or MPM [Citation99]. In the latter two studies, high circulating levels of activin A were associated with a worse prognosis. High serum activin A was also connected to poor overall survival (OS) in a cohort of older patients with various metastatic solid tumors [Citation100]. In breast and prostate cancer, activin A levels were elevated in patients with bone metastasis [Citation101]. Due to the contribution of activin A to cancer cachexia (compare above) activin A could be used as a marker for patients at risk of developing cachexia. Indeed, high concentrations of circulating activin A correlated with increased cachexia in patients with pancreatic cancer [Citation86], MPM [Citation80] as well as colorectal and lung cancer [Citation102], where it also proved to be a negative prognostic marker for survival independent of tumor stage or inflammatory markers. A recent study in metastatic melanoma suggested that a high serum concentration of activin A is predictive for objective response to monotherapy with the VEGF-targeting antibody bevacizumab [Citation103].
In addition to studies using detection of activin A in serum or plasma, several studies have tried to correlate mRNA or protein expression of the βA subunit with survival and clinicopathologic parameters of cancer patients. Diffuse large B-cell lymphoma patients with high βA mRNA in the tumor tissue had a significantly better OS than those with low βA levels [Citation45]. In colorectal cancer patients in contrast, and in gastric cancer patients after curative surgery, high βA mRNA correlated with a worse OS [Citation104,Citation105]. Likewise in oral tongue squamous cell carcinoma, high βA protein expression correlated with worse OS and was a predictor of occult lymph node metastasis [Citation106]. Correlation of high βA levels with adverse clinical outcome were also reported for endometrioid adenocarcinomas [Citation107], urothelial carcinoma [Citation108], and acute myeloid leukemia [Citation109]. In summary, in the great majority of investigated cancer types, the available evidence suggests a negative impact of high activin A expression on disease progression and patient survival. So far, however, few confirmatory studies in the same tumor entities have been performed and the benefit of using activin A as biomarker in clinical routine still needs to be proven.
12. Targeting approaches for activin A
There are a number of different strategies that are being used to interfere with activin A signaling (, ). It has to be considered, however, that most of these approaches have limited specificity for activin A and will – to various degrees and depending on the specific agent – affect additional signaling molecules such as activin B, myostatin, GDFs, BMPs, or TGF-β. The most widely used approach to block the interaction between secreted growth factors and their cognate receptors in multiple settings is the use of monoclonal antibodies. Antibodies bind their targets with high specificity and experience from over three decades of clinical application have shown that their off-target effects are limited and manageable. The activin A-specific monoclonal antibody garetosmab (REGN2477) currently represents the most specific approach for targeting activin A. It is being evaluated in clinical trials for fibrodysplasia ossificans progressive (FOP) [Citation110]. In this rare genetic disease activin A induces heterotopic bone formation via a mutated form of ActRI (ACVR1, ALK2). Garetosmab showed no dose-limiting toxicities in a phase I study [Citation110] and promising interim results in a phase II study (NCT03188666) against FOP. No studies in cancer patients have been reported so far. Activin receptors in contrast, are used also by myostatin, GDF11, and additional TGF-β family members. Bimagrumab (BYM338) is a dual-specific antibody that binds both ActRIIA and IIB and leads to muscle hypertrophy in experimental animal models [Citation111]. It was shown to protect mice from cancer cachexia and prolong survival under cisplatin treatment [Citation90]. So far, it has been tested in clinical trials for sarcopenia [Citation112], muscle depletion in chronic obstructive pulmonary disease (COPD) [Citation113], and inclusion body myositis [Citation114], but not in cancer patients.
Table 1. Therapeutic agents with activin signaling inhibition capacity that have been used in clinical trials in cancer patients
A slightly different targeting approach is the design of ligand traps that bind secreted proteins and thereby block their interaction with native receptors. Sotatercept (ACE-011) is a dimeric fusion protein comprised of the extracellular domain of ActRIIA linked to the Fc portion of human IgG1. Like its native counterpart, it can bind activins A and B, myostatin, GDF11, and some BMPs. It has been investigated in clinical trials in patients with thalassemia [Citation115] and myelodysplastic syndromes [Citation116]. With respect to cancer, it was tested in multiple myeloma patients with osteolytic lesions [Citation117] and for treatment of chemotherapy-induced anemia in patients with metastatic cancer [Citation94]. In both indications, an acceptable safety profile and increases in hemoglobin were reported. However, the company is not further pursuing these applications and focuses currently on pulmonary arterial hypertension (PAH), where promising interim results of a phase II study with sotatercept were recently obtained (NCT03496207). Luspatercept (ACE-536) is a ligand trap based on the extracellular domain of ActRIIB and has lower affinity for activin A compared to sotatercept [Citation116]. It has recently become the first activin receptor-based drug to gain regulatory approval by the FDA for the treatment of anemia associated with β-thalassemia. Another ActRIIB-based ligand trap targeting activin A, STM 434, was recently tested in a phase I study with 32 patients suffering from gynecological and other solid tumors [Citation118]. It achieved an increase in total lean body mass and improvements in a walking test indicative of successful treatment of cachexia. Stable disease was the best antitumor response achieved. This was seen in 53% of all patients including 10 of 12 (80%) of patients with granulosa cell ovarian cancer, a tumor type known to frequently express and secrete high levels of activin A [Citation119]. Mucocutaneous bleeding complications were observed during this study and were attributed to an off-target interaction of STM 434 with BMP9, which normally binds to activin receptor-like kinase 1 (ALK1).
Blocking activin A can also be performed by using its physiological binding proteins follistatin or fstl-3/FLRG or derivatives thereof. Fusing follistatin to the Fc domain of human IgG2 (ACE-083) [Citation120] and a gene therapy approach expressing follistatin from an adeno-associated virus (AAV) vector [Citation121] have been tested as potential therapeutics for muscle diseases but not in cancer patients. Follistatin was shown to decrease radiation-induced fibrosis in a mouse model [Citation122], an observation of potential interest for cancer radiotherapy.
Small molecule inhibitors (SMI) targeting the kinase function of activin receptors are an alternative approach for blocking activin A signals. Due to the high degree of conservation within the TGF-β receptor family, engineering kinase inhibitors with high selectivity has been challenging and a number of inhibitors co-target ActRIB (ALK4), ActRIC (ALK7), and TGFBRI (ALK5). Galunisertib is farthest developed in this group and was trialed in pancreatic cancer, HCC, and glioblastoma [Citation123,Citation124]. Although galunisertib inhibits ActRIB at a lower concentration than TGFBRI, it has been used in clinical trials only as a TGF-β antagonist. Finally, miRNAs targeting activin A, activin receptors or other regulators of activin signaling could be used for targeting approaches. The feasibility of using mir-16, which includes ActRIIA among its confirmed targets [Citation31], in cancer patients has been demonstrated [Citation125], but specific application scenarios for activin A signaling have yet to be defined.
13. Conclusion
Activin A functions in the regulation of growth, migration and drug response of cancer cells and in addition controls important aspects of crosstalk within the tumor microenvironment. While studies performed in different cancer types have reported partially contradictory results, the overall view emerges that for the majority of cancers activin A signaling contributes to cancer progression and cancer-associated morbidity and mortality. In consequence, several strategies for blocking activin A signals have been developed and tested in preclinical models. The number of clinical studies targeting activin A is still small, but these studies have demonstrated the safety and feasibility of targeting activin A in clinical settings. Remaining challenges, unresolved issues, and future perspectives will be discussed below.
14. Expert Opinion: Perspectives and challenges for targeting activin A in cancer
Given the multiple and complex roles that have been described for activin A in many aspects of tumor development and progression as outlined above and depicted in , it is obvious that targeting activin A for cancer treatment is challenging. Part of the challenge undoubtedly comes from the multitude of closely related ligands and receptors of the TGF-β family, the tendency of receptors to bind multiple ligands and form complexes with several other members of the receptor family as well as from the shared components of the signaling pathway. Thus, achieving specific inhibition of activin A signals without co-inhibiting other activins, myostatin, GDFs, BMPs, and even TGF-β, would limit the available strategies. Indeed, most agents that have been used for targeting activin A in clinical applications so far, target additional TGF-β family members. On the one hand, this can be beneficial and increase efficacy, because it limits the possibility that other family members compensate the loss of activin A signals. On the other hand, it increases the risk of unwanted side effects as exemplified by the clinical study with the ActRIIB-based ligand trap STM 434, in which mucocutaneous bleeding was most likely caused by interaction with BMP9 [Citation118]. In addition, the lack of specificity of clinically used targeting agents also makes it difficult to dissect how much of an observed response can be attributed to inhibition of activin A versus other related cytokines. Nevertheless, it appears that in the view of pharmaceutical companies the higher chance for achieving meaningful effects by using agents that can block several related signals outweigh the risks of increased adverse reactions and, consequently, less specific agents are farther developed with respect to clinical trials. This may be a parallel to tyrosine kinase inhibitors, where multikinase inhibitors like sorafenib or sunitinib have been more successful than, for instance, more specific fibroblast growth factor (FGF)-receptor and platelet-derived growth factor (PDGF)-receptor inhibitors [Citation126].
Figure 2. The multiple functions of activin A in cancer. Aspects of cancer development and progression for which a regulation by activin A has been demonstrated. Targeting activin A can be used to interfere with these processes
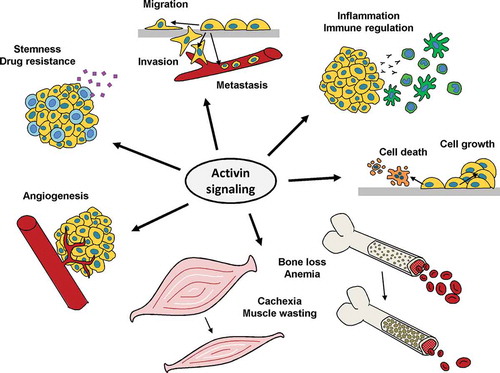
An even bigger challenge is the diversity of activin A-regulated physiological and pathophysiological effects. Unlike many other molecules that are being used – or have been considered – as targets for cancer therapy, activin A is not a clear oncogenic driver. Although it is overexpressed in many cancer types, mutations or gene amplification are very rare and its receptors and downstream signaling mediators show loss-of-function mutations rather than gain-of-function mutations [Citation38,Citation127]. Impairment of tumor cell growth and decreased tumor cell migration and invasion have been achieved by activin A signaling inhibition in experimental settings, but it is still unclear whether blockade of activin A signals alone will be sufficient to achieve tumor shrinkage or reduction of metastasis in patients. In addition, the absence of gain-of-function mutations of either the ligand or the receptor makes it more difficult to identify patients that have an increased likelihood of their tumors being responsive to activin A blockade. More promising opportunities for improving cancer treatment by targeting activin A arise, in our opinion, when interactions with the tumor microenvironment are also taken into account. While ALK1 and its ligands BMP9 and BMP10 have been considered as target for antiangiogenic treatment [Citation128], most of the evidence for activin A suggests that it has antiangiogenic effects itself and thus would not be suitable as target for antiangiogenic therapies. In tumor immunology in contrast, the majority of studies indicate a role of activin A in limiting antitumor immunity. Blocking activin A could therefore contribute to boosting antitumor immune responses and combinations of activin inhibitors with immunotherapeutic approaches such as checkpoint inhibitors could be promising. This strategy is currently being investigated with the TGFBRI/ActRIB inhibitor galunisertib in preclinical models and clinical trials. In a murine colon cancer model, the combination of galunisertib with anti PD-L1 immunotherapy resulted in enhanced tumor growth inhibition and complete regressions [Citation129]. The question whether TGFBRI or ActRIB is the more important target of galunisertib in this setting (or whether inhibition of both targets is required) has not been clarified. Since both activin A and TGF-β regulate immune cell functions at multiple levels (reviewed in detail by Chen and ten Dijke [Citation130]), knock-out animal models or the comparative use of specific antibodies for both factors will be required to answer this question. Regarding the combination of activin A-targeting agents with chemotherapy, an impairment of cisplatin response by activin A was suggested in several cancer types [Citation79,Citation80]. Therefore, the combination of activin A-targeting agents with this widely used chemotherapy drug and potentially with other chemotherapeutic agents should receive increased attention and be investigated further in carefully designed experimental models and eventually clinical trials.
Another area where activin A inhibition could benefit cancer patients is the treatment of cancer-associated cachexia, muscle wasting, bone loss and anemia. Indeed, clinical trials with sotatercept and STM 434 have shown the potential of activin A inhibition to improve anemia and cachexia in patients [Citation94,Citation118]. In recent years, high levels of circulating activin A were shown to predict a shorter overall survival for an increasing number of cancer types. The degree to which tumor cell autonomous effects like enhanced tumor cell growth, migration and drug resistance versus microenvironment-mediated effects like suppressed antitumor immune reactions or more severe cachexia are responsible for the worse prognosis has not been resolved and should be addressed in future studies. Moreover, it still needs to be worked out whether tumor cells are the main contributor to the enhanced activin A levels found in the blood of cancer patients or whether cells of the tumor microenvironment like cancer-associated fibroblasts, macrophages or endothelial cells are a major source of circulating activin A. An important question remains, whether activin A blood concentrations can be used as biomarker for patient selection in clinical studies. The ability to use circulating activin A levels in cancer patients not only as a prognostic marker but also as a predictive marker for benefit from activin A-targeting agents would be an important step forward and could help to individualize treatment with such agents. Analyzing activin A levels could even be incorporated into clinical trials in which galunisertib is used as a TGF-β antagonist. Assessing whether there is a correlation between galunisertib response and activin A blood levels could provide important additional information on potential contributions of activin A antagonism to galunisertib activity in patients.
Overall, it has been fascinating to follow how many new roles of activin A were discovered in the last two decades. It is rather unusual for a molecule to be involved in so many and so diverse functions relevant for tumor biology. Blocking activin A signaling could offer a rare combination of beneficial effects including a decrease in tumor growth and spreading, a reduction of stemness and drug resistance, an enhancement of antitumor immune responses and a decrease in tumor-induced or treatment-induced cachexia. It will be critical however, to define the right scenarios and identify patient collectives where beneficial effects of targeting activin A outweigh risks and potential detrimental effects.
Article highlights
Activin A is a secreted cytokine of the TGF-ß family and can have both pro- and antitumorigenic effects on tumor cells and various cells of the tumor microenvironment.
It contributes to the regulation of cell growth, differentiation and death, cell migration and invasion, antiangiogenesis, stemness and drug resistance, immune evasion as well as to cancer cachexia.
Hence, activin A could serve as a target to improve several aspects of cancer treatment including prevention of metastasis, enhancement of antitumor immune responses and amelioration of cancer-induced muscle wasting, bone loss, and anemia.
Strategies for targeting activin A signaling include the use of monoclonal antibodies, endogenous inhibitors, ligand traps, kinase inhibitors and micro RNAs. The limited specificity of most targeting agents and the pleiotrophic functions of activin A still pose considerable challenges to the task of defining the clinical scenarios in which cancer patients will receive the highest benefit with the least side effects.
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Additional information
Funding
References
- Gregory SJ, Kaiser UB. Regulation of gonadotropins by inhibin and activin. Semin Reprod Med. 2004;22:253–267.
- Namwanje M, Brown CW. Activins and inhibins: roles in development, physiology, and disease. Cold Spring Harb Perspect Biol. 2016;8. DOI:10.1101/cshperspect.a021881
- Lewis KA, Gray PC, Blount AL, et al. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature. 2000;404:411–414..
- Wang X, Fischer G, Hyvonen M. Structure and activation of pro-activin A. Nat Commun. 2016;7:12052.
- Weiss A, Attisano L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol. 2013;2:47–63.
- Michael IP, Saghafinia S, Tichet M, et al. ALK7 signaling manifests a homeostatic tissue barrier that is abrogated during tumorigenesis and metastasis. Dev Cell. 2019;49:409–24 e6.
- Tsuchida K, Nakatani M, Yamakawa N, et al. Activin isoforms signal through type I receptor serine/threonine kinase ALK7. Mol Cell Endocrinol. 2004;220:59–65.
- Bertolino P, Holmberg R, Reissmann E, et al. Activin B receptor ALK7 is a negative regulator of pancreatic beta-cell function. Proc Natl Acad Sci U S A. 2008;105:7246–7251.
- Pituello F, Yamada G, Gruss P. Activin A inhibits Pax-6 expression and perturbs cell differentiation in the developing spinal cord in vitro. Proc Natl Acad Sci U S A. 1995;92:6952–6956.
- Attardi B, Miklos J. Rapid stimulatory effect of activin-A on messenger RNA encoding the follicle-stimulating hormone beta-subunit in rat pituitary cell cultures. Mol Endocrinol. 1990;4:721–726.
- Kaneda H, Arao T, Matsumoto K, et al. Activin A inhibits vascular endothelial cell growth and suppresses tumour angiogenesis in gastric cancer. Br J Cancer. 2011;105:1210–1217.
- Burdette JE, Jeruss JS, Kurley SJ, et al. Activin A mediates growth inhibition and cell cycle arrest through Smads in human breast cancer cells. Cancer Res. 2005;65:7968–7975.
- Bartholin L, Maguer-Satta V, Hayette S, et al. Transcription activation of FLRG and follistatin by activin A, through Smad proteins, participates in a negative feedback loop to modulate activin A function. Oncogene. 2002;21:2227–2235.
- Ishisaki A, Yamato K, Nakao A, et al. Smad7 is an activin-inducible inhibitor of activin-induced growth arrest and apoptosis in mouse B cells. J Biol Chem. 1998;273:24293–24296.
- Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982..
- Hata A, Chen YG. TGF-beta signaling from receptors to smads. Cold Spring Harb Perspect Biol. 2016;8. DOI:10.1101/cshperspect.a022061
- Miller DSJ, Schmierer B, Hill CS. TGF-β family ligands exhibit distinct signalling dynamics that are driven by receptor localisation. J Cell Sci. 2018;6(14):jcs234039.
- de Guise C, Lacerte A, Rafiei S, et al. Activin inhibits the human Pit-1 gene promoter through the p38 kinase pathway in a Smad-independent manner. Endocrinology. 2006;147:4351–4362.
- Dean M, Davis DA, Burdette JE. Activin A stimulates migration of the fallopian tube epithelium, an origin of high-grade serous ovarian cancer, through non-canonical signaling. Cancer Lett. 2017;391:114–124.
- Tamminen JA, Yin M, Ronty M, et al. Overexpression of activin-A and -B in malignant mesothelioma - attenuated Smad3 signaling responses and ERK activation promote cell migration and invasive growth. Exp Cell Res. 2015;332:102–115.
- Shao L, Frigon NL Jr., Sehy DW, et al. Regulation of production of activin A in human marrow stromal cells and monocytes. Exp Hematol. 1992;20:1235–1242.
- Shao LE, Frigon NL Jr., Yu A, et al. Contrasting effects of inflammatory cytokines and glucocorticoids on the production of activin A in human marrow stromal cells and their implications. Cytokine. 1998;10:227–235.
- Breit S, Ashman K, Wilting J, et al. The N-myc oncogene in human neuroblastoma cells: down-regulation of an angiogenesis inhibitor identified as activin A. Cancer Res. 2000;60:4596–4601.
- Makanji Y, Zhu J, Mishra R, et al. Inhibin at 90: from discovery to clinical application, a historical review. Endocr Rev. 2014;35:747–794.
- Mellor SL, Ball EM, O’Connor AE, et al. Activin betaC-subunit heterodimers provide a new mechanism of regulating activin levels in the prostate. Endocrinology. 2003;144:4410–4419.
- Nakamura T, Takio K, Eto Y, et al. Activin-binding protein from rat ovary is follistatin. Science. 1990;247:836–838..
- Sugino K, Kurosawa N, Nakamura T, et al. Molecular heterogeneity of follistatin, an activin-binding protein. Higher affinity of the carboxyl-terminal truncated forms for heparan sulfate proteoglycans on the ovarian granulosa cell. J Biol Chem. 1993;268:15579–15587.
- Glister C, Regan SL, Samir M, et al. Gremlin, Noggin, Chordin and follistatin differentially modulate BMP induced suppression of androgen secretion by bovine ovarian theca cells. J Mol Endocrinol. 2019;62:15–25.
- Amthor H, Nicholas G, McKinnell I, et al. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev Biol. 2004;270:19–30.
- Tsuchida K, Arai KY, Kuramoto Y, et al. Identification and characterization of a novel follistatin-like protein as a binding protein for the TGF-beta family. J Biol Chem. 2000;275:40788–40796.
- Martello G, Zacchigna L, Inui M, et al. MicroRNA control of Nodal signalling. Nature. 2007;449:183–188..
- Li D, Duan M, Feng Y, et al. MiR-146a modulates macrophage polarization in systemic juvenile idiopathic arthritis by targeting INHBA. Mol Immunol. 2016;77:205–212.
- Seder CW, Hartojo W, Lin L, et al. Upregulated INHBA expression may promote cell proliferation and is associated with poor survival in lung adenocarcinoma. Neoplasia. 2009;11:388–396.
- Seder CW, Hartojo W, Lin L, et al. INHBA overexpression promotes cell proliferation and may be epigenetically regulated in esophageal adenocarcinoma. J Thorac Oncol. 2009;4:455–462.
- Hoda MA, Munzker J, Ghanim B, et al. Suppression of activin A signals inhibits growth of malignant pleural mesothelioma cells. Br J Cancer. 2012;107:1978–1986.
- Razanajaona D, Joguet S, Ay A-S, et al. Silencing of FLRG, an antagonist of activin, inhibits human breast tumor cell growth. Cancer Res. 2007;67(15):7223–7229.
- Grusch M, Drucker C, Peter-Vorosmarty B, et al. Deregulation of the activin/follistatin system in hepatocarcinogenesis. J Hepatol. 2006;45:673–680.
- Jung B, Doctolero RT, Tajima A, et al. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology. 2004;126:654–659.
- Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435–457.
- Schwall RH, Robbins K, Jardieu P, et al. Activin induces cell death in hepatocytes in vivo and in vitro. Hepatology. 1993;18:347–356..
- Zauberman A, Oren M, Zipori D. Involvement of p21(WAF1/Cip1), CDK4 and Rb in activin A mediated signaling leading to hepatoma cell growth inhibition. Oncogene. 1997;15:1705–1711.
- Vejda S, Erlach N, Peter B, et al. Expression of activins C and E induces apoptosis in human and rat hepatoma cells. Carcinogenesis. 2003;24:1801–1809.
- Dalkin AC, Gilrain JT, Bradshaw D, et al. Activin inhibition of prostate cancer cell growth: selective actions on androgen-responsive LNCaP cells. Endocrinology. 1996;137:5230–5235.
- Jung BH, Beck SE, Cabral J, et al. Activin type 2 receptor restoration in MSI-H colon cancer suppresses growth and enhances migration with activin. Gastroenterology. 2007;132:633–644.
- Jiang L, Si T, Yu M, et al. The tumor suppressive role of inhibin betaA in diffuse large B-cell lymphoma. Leuk Lymphoma. 2018;59:1202–1212.
- Panopoulou E, Murphy C, Rasmussen H, et al. Activin A suppresses neuroblastoma xenograft tumor growth via antimitotic and antiangiogenic mechanisms. Cancer Res. 2005;65:1877–1886.
- Ohga E, Matsuse T, Teramoto S, et al. Effects of activin A on proliferation and differentiation of human lung fibroblasts. Biochem Biophys Res Commun. 1996;228:391–396.
- Gotzmann J, Mikula M, Eger A, et al. Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat Res. 2004;566:9–20.
- Dignass AU, Jung S, Harder-d’Heureuse J, et al. Functional relevance of activin A in the intestinal epithelium. Scand J Gastroenterol. 2002;37:936–943.
- Nomura M, Tanaka K, Wang L, et al. Activin type IB receptor signaling in prostate cancer cells promotes lymph node metastasis in a xenograft model. Biochem Biophys Res Commun. 2013;430:340–346.
- Kalli M, Mpekris F, Wong CK, et al. Activin A signaling regulates IL13Ralpha2 expression to promote breast cancer metastasis. Front Oncol. 2019;9:32.
- Chang KP, Kao HK, Liang Y, et al. Overexpression of activin A in oral squamous cell carcinoma: association with poor prognosis and tumor progression. Ann Surg Oncol. 2010;17:1945–1956.
- Yi Y, Cheng J-C, Klausen C, et al. Activin A promotes ovarian cancer cell migration by suppressing E-cadherin expression. Exp Cell Res. 2019;382(2):111471.
- Bashir M, Damineni S, Mukherjee G, et al. Activin-A signaling promotes epithelial–mesenchymal transition, invasion, and metastatic growth of breast cancer. NPJ Breast Cancer. 2015;1(1):15007.
- Loomans HA, Andl CD. Intertwining of Activin A and TGFbeta Signaling: dual Roles in Cancer Progression and Cancer Cell Invasion. Cancers (Basel). 2014;7:70–91.
- Hyuga S, Kawasaki N, Hashimoto O, et al. Possible role of hepatocyte growth factor/scatter factor and activin A produced by the target organ in liver metastasis. Cancer Lett. 2000;153(1–2):137–143.
- Portale F, Cricri G, Bresolin S, et al. ActivinA: a new leukemia-promoting factor conferring migratory advantage to B-cell precursor-acute lymphoblastic leukemic cells. Haematologica. 2019;104:533–545.
- Ferrara N, Hillan KJ, Gerber HP, et al. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400.
- Kozian DH, Ziche M, Augustin HG. The activin-binding protein follistatin regulates autocrine endothelial cell activity and induces angiogenesis. Lab Invest. 1997;76:267–276.
- Maeshima K, Maeshima A, Hayashi Y, et al. Crucial role of activin a in tubulogenesis of endothelial cells induced by vascular endothelial growth factor. Endocrinology. 2004;145:3739–3745.
- Krneta J, Kroll J, Alves F, et al. Dissociation of angiogenesis and tumorigenesis in follistatin- and activin-expressing tumors. Cancer Res. 2006;66:5686–5695..
- Heinz M, Niederleithner HL, Puujalka E, et al. Activin A is anti-lymphangiogenic in a melanoma mouse model. J Invest Dermatol. 2015;135:212–221.
- Phillips DJ, de Kretser DM, Hedger MP. Activin and related proteins in inflammation: not just interested bystanders. Cytokine Growth Factor Rev. 2009;20:153–164.
- Morianos I, Papadopoulou G, Semitekolou M, et al. Activin-A in the regulation of immunity in health and disease. J Autoimmun. 2019;104:102314.
- Antsiferova M, Huber M, Meyer M, et al. Activin enhances skin tumourigenesis and malignant progression by inducing a pro-tumourigenic immune cell response. Nat Commun. 2011;2:576..
- Antsiferova M, Piwko-Czuchra A, Cangkrama M, et al. Activin promotes skin carcinogenesis by attraction and reprogramming of macrophages. EMBO Mol Med. 2017;9:27–45.
- Donovan P, Dubey OA, Kallioinen S, et al. Paracrine Activin-A signaling promotes melanoma growth and metastasis through immune evasion. J Invest Dermatol. 2017;137:2578–2587.
- Ni X, Tao J, Barbi J, et al. YAP is essential for treg-mediated suppression of antitumor immunity. Cancer Discov. 2018;8:1026–1043.
- Sainz B Jr., Alcala S, Garcia E, et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut. 2015;64:1921–1935.
- Xiao L, Yuan X, Sharkis SJ. Activin A maintains self-renewal and regulates fibroblast growth factor, Wnt, and bone morphogenic protein pathways in human embryonic stem cells. Stem Cells. 2006;24:1476–1486..
- Jiang J, Au M, Lu K, et al. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells. 2007;25:1940–1953.
- Cai J, Zhao Y, Liu Y, et al. Directed differentiation of human embryonic stem cells into functional hepatic cells. Hepatology. 2007;45:1229–1239.
- Lonardo E, Hermann PC, Mueller MT, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 2011;9:433–446.
- Liu R, Wang JH, Xu C, et al. Activin pathway enhances colorectal cancer stem cell self-renew and tumor progression. Biochem Biophys Res Commun. 2016;479:715–720.
- Wamsley JJ, Kumar M, Allison DF, et al. Activin Upregulation by NF-κB Is Required to Maintain Mesenchymal Features of Cancer Stem–like Cells in Non–Small Cell Lung Cancer. Cancer Res. 2015;75(2):426–435.
- Rodrigues CFD, Serrano E, Patricio MI, et al. Stroma-derived IL-6, G-CSF and Activin-A mediated dedifferentiation of lung carcinoma cells into cancer stem cells. Sci Rep. 2018;8:11573.
- Ohno Y, Shingyoku S, Miyake S, et al. Differential regulation of the sphere formation and maintenance of cancer-initiating cells of malignant mesothelioma via CD44 and ALK4 signaling pathways. Oncogene. 2018;37:6357–6367.
- Cioffi M, Trabulo SM, Sanchez-Ripoll Y, et al. The miR-17-92 cluster counteracts quiescence and chemoresistance in a distinct subpopulation of pancreatic cancer stem cells. Gut. 2015;64:1936–1948.
- Marini KD, Croucher DR, McCloy RA, et al. Inhibition of activin signaling in lung adenocarcinoma increases the therapeutic index of platinum chemotherapy. Sci Transl Med2018;10. DOI:10.1126/scitranslmed.aat3504.
- Paajanen J, Ilonen I, Lauri H, et al. Elevated circulating Activin A levels in patients with malignant pleural mesothelioma are related to cancer cachexia and reduced response to platinum-based chemotherapy. Clin Lung Cancer. 2020;21:e142–e150.
- Tanaka T, Toujima S, Umesaki N. Growth-inhibitory signals by activin A do not affect anticancer drug-sensitivity and acquired multi-drug-resistance in human ovarian endometrioid adenocarcinoma OVK-18 cells. Oncol Rep. 2004;11:667–671.
- Huang YW, Lee WH, Tsai YH, et al. Activin A induction of erythroid differentiation sensitizes K562 chronic myeloid leukemia cells to a subtoxic concentration of imatinib. Am J Physiol Cell Physiol. 2014;306:C37–44.
- Matzuk MM, Finegold MJ, Mather JP, et al. Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc Natl Acad Sci U S A. 1994;91:8817–8821.
- Coerver KA, Woodruff TK, Finegold MJ, et al. Activin signaling through activin receptor type II causes the cachexia-like symptoms in inhibin-deficient mice. Mol Endocrinol. 1996;10:534–543.
- Zhou X, Wang JL, Lu J, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531–543..
- Togashi Y, Kogita A, Sakamoto H, et al. Activin signal promotes cancer progression and is involved in cachexia in a subset of pancreatic cancer. Cancer Lett. 2015;356:819–827.
- Chen JL, Walton KL, Winbanks CE, et al. Elevated expression of activins promotes muscle wasting and cachexia. Faseb J. 2014;28:1711–1723.
- Winbanks CE, Murphy KT, Bernardo BC, et al. Smad7 gene delivery prevents muscle wasting associated with cancer cachexia in mice. Sci Transl Med. 2016;8:348ra98.
- Loumaye A, de Barsy M, Nachit M, et al. Role of Activin A and myostatin in human cancer cachexia. J Clin Endocrinol Metab. 2015;100:2030–2038.
- Hatakeyama S, Summermatter S, Jourdain M, et al. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti-cancer treatments. Skelet Muscle. 2016;6:26.
- Barreto R, Kitase Y, Matsumoto T, et al. ACVR2B/Fc counteracts chemotherapy-induced loss of muscle and bone mass. Sci Rep. 2017;7:14470.
- Vallet S, Mukherjee S, Vaghela N, et al. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proc Natl Acad Sci U S A. 2010;107:5124–5129.
- Carrancio S, Markovics J, Wong P, et al. An activin receptor IIA ligand trap promotes erythropoiesis resulting in a rapid induction of red blood cells and haemoglobin. Br J Haematol. 2014;165:870–882.
- Raftopoulos H, Laadem A, Hesketh PJ, et al. Sotatercept (ACE-011) for the treatment of chemotherapy-induced anemia in patients with metastatic breast cancer or advanced or metastatic solid tumors treated with platinum-based chemotherapeutic regimens: results from two phase 2 studies. Support Care Cancer. 2016;24:1517–1525.
- Fields SZ, Parshad S, Anne M, et al. Activin receptor antagonists for cancer-related anemia and bone disease. Expert Opin Investig Drugs. 2013;22:87–101.
- Pirisi M, Fabris C, Luisi S, et al. Evaluation of circulating activin-A as a serum marker of hepatocellular carcinoma. Cancer Detect Prev. 2000;24:150–155.
- Reis FM, Cobellis L, Tameirao LC, et al. Serum and tissue expression of activin a in postmenopausal women with breast cancer. J Clin Endocrinol Metab. 2002;87(5):2277–2282.
- Hoda MA, Rozsas A, Lang E, et al. High circulating activin A level is associated with tumor progression and predicts poor prognosis in lung adenocarcinoma. Oncotarget. 2016;7:13388–13399.
- Hoda MA, Dong Y, Rozsas A, et al. Circulating activin A is a novel prognostic biomarker in malignant pleural mesothelioma - A multi-institutional study. Eur J Cancer. 2016;63:64–73.
- Kim SH, Kim JW, Hwang IG, et al. Serum biomarkers for predicting overall survival and early mortality in older patients with metastatic solid tumors. J Geriatr Oncol. 2019;10:749–756.
- Leto G, Incorvaia L, Badalamenti G, et al. Activin A circulating levels in patients with bone metastasis from breast or prostate cancer. Clin Exp Metastasis. 2006;23:117–122.
- Loumaye A, de Barsy M, Nachit M, et al. Circulating Activin A predicts survival in cancer patients. J Cachexia Sarcopenia Muscle. 2017;8:768–777.
- Schuster C, Akslen LA, Stokowy T, et al. Predictive value of angiogenic proteins in patients with metastatic melanoma treated with bevacizumab monotherapy. J Pathol Clin Res. 2019;5:53–62.
- Okano M, Yamamoto H, Ohkuma H, et al. Significance of INHBA expression in human colorectal cancer. Oncol Rep. 2013;30:2903–2908.
- Oshima T, Yoshihara K, Aoyama T, et al. Relation of INHBA gene expression to outcomes in gastric cancer after curative surgery. Anticancer Res. 2014;34:2303–2309.
- Kelner N, Rodrigues PC, Bufalino A, et al. Activin A immunoexpression as predictor of occult lymph node metastasis and overall survival in oral tongue squamous cell carcinoma. Head Neck. 2015;37(4):479–486.
- Mylonas I. Inhibin-betaA subunit immunolabeling as a prognostic factor in endometrioid adenocarcinomas: a matter of evaluation? Arch Gynecol Obstet. 2011;284:467–476.
- Lee HY, Li CC, Huang CN, et al. INHBA overexpression indicates poor prognosis in urothelial carcinoma of urinary bladder and upper tract. J Surg Oncol. 2015;111:414–422.
- Si T, Lu Y, Li F, et al. High expression of INHBA is an adverse prognostic factor for de novo acute myeloid leukemia. Leuk Lymphoma. 2018;59:114–120.
- Vanhoutte F, Liang S, Ruddy M, et al. Pharmacokinetics and pharmacodynamics of garetosmab (Anti-Activin A): results from a first-in-human Phase 1 study. J Clin Pharmacol. 2020 Jun 18. online ahead of print. DOI:10.1002/jcph.1638
- Lach-Trifilieff E, Minetti GC, Sheppard K, et al. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol. 2014;34:606–618.
- Rooks D, Praestgaard J, Hariry S, et al. Treatment of sarcopenia with Bimagrumab: results from a Phase II, randomized, controlled, proof-of-concept study. J Am Geriatr Soc. 2017;65:1988–1995.
- Polkey MI, Praestgaard J, Berwick A, et al. Activin Type II receptor blockade for treatment of muscle depletion in chronic obstructive pulmonary disease. a randomized trial. Am J Respir Crit Care Med. 2019;199:313–320.
- Hanna MG, Badrising UA, Benveniste O, et al. Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): a randomised, double-blind, placebo-controlled phase 2b trial. Lancet Neurol. 2019;18:834–844.
- Motta I, Scaramellini N, Cappellini MD. Investigational drugs in phase I and phase II clinical trials for thalassemia. Expert Opin Investig Drugs. 2017;26:793–802.
- Verma A, Suragani RNVS, Aluri S, et al. Biological basis for efficacy of activin receptor ligand traps in myelodysplastic syndromes. J Clin Invest. 2020;130(2):582–589.
- Abdulkadyrov KM, Salogub GN, Khuazheva NK, et al. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br J Haematol. 2014;165:814–823.
- Tao JJ, Cangemi NA, Makker V, et al. First-in-Human Phase I study of the Activin A inhibitor, STM 434, in patients with granulosa cell ovarian cancer and other advanced solid tumors. Clin Cancer Res. 2019;25:5458–5465.
- Welt CK, Lambert-Messerlian G, Zheng W, et al. Presence of activin, inhibin, and follistatin in epithelial ovarian carcinoma. J Clin Endocrinol Metab. 1997;82:3720–3727.
- Pearsall RS, Davies MV, Cannell M, et al. Follistatin-based ligand trap ACE-083 induces localized hypertrophy of skeletal muscle with functional improvement in models of neuromuscular disease. Sci Rep. 2019;9:11392.
- Mendell JR, Sahenk Z, Al-Zaidy S, et al. Follistatin gene therapy for sporadic inclusion body myositis improves functional outcomes. Mol Ther. 2017;25:870–879.
- Forrester HB, de Kretser DM, Leong T, et al. Follistatin attenuates radiation-induced fibrosis in a murine model. PLoS One. 2017;12:e0173788.
- Herbertz S, Sawyer JS, Stauber AJ, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. 2015;9:4479–4499..
- Melisi D, Garcia-Carbonero R, Macarulla T, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer. 2018;119:1208–1214.
- van Zandwijk N, Pavlakis N, Kao SC, et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: a first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017;18:1386–1396.
- Krug M, Hilgeroth A. Recent advances in the development of multi-kinase inhibitors. Mini Rev Med Chem. 2008;8:1312–1327.
- Tournier I, Marlin R, Walton K, et al. Germline mutations of inhibins in early-onset ovarian epithelial tumors. Hum Mutat. 2014;35:294–297.
- Cunha SI, Pietras K. ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood. 2011;117:6999–7006.
- Holmgaard RB, Schaer DA, Li Y, et al. Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer. 2018;6:47.
- Chen W, Ten Dijke P. Immunoregulation by members of the TGFbeta superfamily. Nat Rev Immunol. 2016;16:723–740.