
ABSTRACT
Introduction
Medulloblastoma (MB) is a heterogeneous tumor of the cerebellum that is divided into four main subgroups with distinct molecular and clinical features. Sonic Hedgehog MB (SHH-MB) is the most genetically understood and occurs predominantly in childhood. Current therapies consist of aggressive and non-targeted multimodal approaches that are often ineffective and cause long-term complications. These problems intensify the need to develop molecularly targeted therapies to improve outcome and reduce treatment-related morbidities. In this scenario, Hedgehog (HH) signaling, a developmental pathway whose deregulation is involved in the pathogenesis of several malignancies, has emerged as an attractive druggable pathway for SHH-MB therapy.
Areas covered
This review provides an overview of the advancements in the HH antagonist research field. We place an emphasis on Smoothened (SMO) and glioma-associated oncogene homolog (GLI) inhibitors and immunotherapy approaches that are validated in preclinical SHH-MB models and that have therapeutic potential for MB patients. Literature from Pubmed and data reported on ClinicalTrial.gov up to August 2020 were considered.
Expert opinion
Extensive-omics analysis has enhanced our knowledge and has transformed the way that MB is studied and managed. The clinical use of SMO antagonists has yet to be determined, however, future GLI inhibitors and multitargeting approaches are promising.
1. Introduction
The Hedgehog (HH) pathway was originally described in Drosophila as a regulator of embryonic patterning acting on cell fate determination and body−segment polarity. In mammals, HH signaling plays a crucial role in the development of tissues and organs, working as a morphogen, mitogen or differentiation factor [Citation1,Citation2]. In adult, the activity of the HH pathway is significantly reduced, except for tissue maintenance conditions [Citation3]; its aberrant reactivation is known to be related to the development of several human malignancies, i.e. basal cell carcinoma (BCC) and medulloblastoma (MB), thus representing a promising therapeutic target for cancer treatment [Citation4–8]. The main players of the HH pathway include the HH ligands Sonic (SHH), Indian (IHH) or Desert Hedgehog (DHH), the Patched transmembrane receptors (PTCH1 and 2), the G protein-coupled-like receptor Smoothened (SMO), and the glioma-associated oncogene (GLI) transcriptional factors (GLI1, GLI2 and GLI3). In a simplified model, the canonical activation of HH signaling is triggered by the binding of HH ligands to PTCH1. This event results in the releasing of its suppression on SMO. Then, SMO receptor translocates into the primary cilium, initiating a signaling cascade that culminates in the dissociation of GLIs from the negative regulator SUFU, and their subsequent nuclear localization. The activator forms of GLI factors promote the transcription of HH-target genes (i.e. CCND2, BMI1, MYCN and VEGF) hence regulating cell survival, invasion, and angiogenesis, as well as stem cell self-renewal, and epithelial–mesenchymal transition (EMT) [Citation9–13]. Notably, GLI1 and PTCH1 maintain their expression through an autoregulatory circuitry and contribute to a feedback loop that regulates the HH pathway. In addition to HH canonical signaling, non-canonical activation of GLI transcription factors can occur via SMO-independent signals, such as K-RAS, TGFβ, PI3K, PKC, TNF-α/mTOR/S6K1 and epigenetic regulators acting downstream of SMO [Citation14–18].
MB is the most common malignant brain tumor in childhood, and it comprises a heterogeneous group of embryonal tumors of the cerebellum [Citation19]. Historically classified on the basis of histopathology, the current availability of -omics data allows the molecular classification of MB in four consensus molecular subgroups: Wingless (WNT), Sonic-Hedgehog (SHH), Group 3 (G3) and Group 4 (G4). Each variant is characterized by peculiar transcriptional signatures, mutational spectra, epigenetic profiling, and clinical features [Citation20–22].
The SHH-activated MB subgroup is genetically the best understood, with the majority of patients harboring either germline or somatic mutations and copy-number alterations in critical genes of the SHH signaling pathway. These mutations frequently include loss-of-function or deletions in PTCH1 or SUFU, activating mutations in SMO and GLI1 or GLI2 amplifications [Citation23,Citation24]. In few cases, genes responsible for transcriptional regulation (MYCN) are recurrently amplified [Citation25]. All these genetic alterations lead to ligand-independent activation of the HH pathway, thus promoting tumorigenesis.
To date, a great deal of effort by pharmaceutical companies has been put into the development of SMO inhibitors [Citation26–29]. Clinical trials are being conducted to evaluate the efficacy of vismodegib and sonidegib in MB. These two drugs are known to be targeting the upstream receptor SMO and have already been approved by the Food and Drug Administration (FDA) for treating metastatic or locally advanced BCC [Citation30–32]. However, the responses to SMO inhibitors have been variable, likely due to SMO drug-resistance mutations. Moreover, genetic alterations downstream of SMO (i.e. amplifications or mutations of SUFU and GLI) or SMO-independent activation of GLI proteins, due to the crosstalk with other HH interacting pathways (i.e. PI3K/AKT/mTOR and RAS/RAF/MEK signaling) are causes of drug-resistance [Citation33]. Accordingly, several preclinical studies have tested interesting approaches to overcome these issues. Moreover, given the high heterogeneity of cancer cells, the combination of therapies targeting both key HH components and HH signaling regulators has become an innovative opportunity to enhance the treatment’s effectiveness [Citation34]. Recently, novel molecular players of the HH pathway (i.e. KCTD15 and ERAP1) have also been identified, thereby opening new perspectives for targeting HH-driven tumors [Citation35,Citation36].
In this review we discuss the limitations of SMO antagonists used in clinical practice, the promising results obtained in preclinical studies with inhibitors of GLI proteins, as well as the potential application of several immunotherapy strategies for the treatment of SHH-MB.
2. Medulloblastoma
2.1. SHH medulloblastoma (SHH-MB)
MB is a highly aggressive cerebellum malignancy, and one of the most frequent pediatric tumor, representing ~63% of childhood intracranial embryonal cancers [Citation37]. In 2016, the World Health Organization (WHO) classification of Central Nervous System Tumors described four genetically distinct variants of MB (WNT, SHH, G3 and G4) [Citation38], and among them the SHH-MB subgroup is the best characterized. This tumor is associated with aberrant activation of the HH pathway that causes the disruption of the developmental program of cerebellar neural progenitor cells (granule neuron precursors, GNPs), the cell of origin of SHH-MB group [Citation39]. In the cerebellum, GNPs migrate from the rhombic lip to the outer cerebellum surface (External Granule Layer, EGL), and subsequently differentiate and migrate toward the internal granule layer (IGL). Purkinje cell-derived SHH signaling keeps the GNPs undifferentiated, promoting cell expansion [Citation39]. Deregulation of HH signaling arrests cerebellar GNPs differentiation, resulting in the abnormal persistence of progenitor cells susceptible to malignant transformation [Citation39] .
Figure 1. Cells of origin of SHH-MB. (A) The figure depicts the cerebellum development at embryonic early stage. Under SHH stimuli secreted from Purkinje cells (orange), granule neuron precursors (GNPs, blue) migrate from the rhombic lip to the external granule layer (EGL), then undergo the subsequent differentiation and migration into the inner granule layer (IGL). (B) Schematic representation of SHH-induced GNPs development, and its deregulation in the tumorigenesis of MB
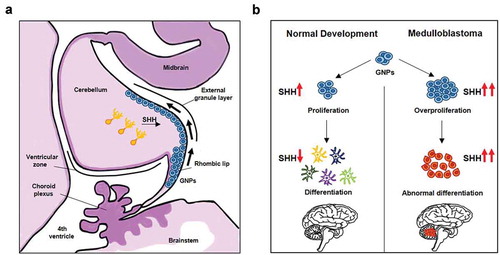
SHH-MB displays a bimodal age distribution and represents the most common molecular subgroup in both infants (< 5 years of age) and adults (> 17 years of age); only few cases have been diagnosed in childhood and adolescence. Demographically, SHH-MB is more common in males than in females (approximately 2:1) and represents the 30% of all MBs. The 5-years survival rate of SHH-MB patients is about 75%, a worse prognosis than WNT-MB, but more favorable than G3-MB patients [Citation22] .
Figure 2. Demographic, molecular and clinical features of SHH-MB and its four subtypes. SHH-MB accounts for 30% of MBs, has a 5-years survival rate of 75%, and occurs predominantly in male. Most SHH-MB patients are adult or infant. Values for histology variants (blue bars) and main gene alterations (orange bars) in SHH-MB are reported
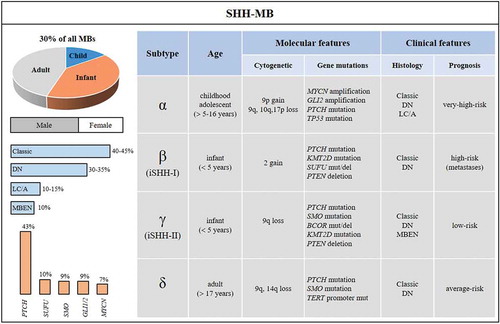
Given that all MBs and other pediatric brain tumors localize at posterior fossa, histopathological and molecular analysis are essential for the diagnosis of MB. Four different histological categories have been defined: classic, desmoplastic/nodular (DN), MB with extensive nodularity (MBEN), large cell/anaplastic (LC/A). In SHH-MBs subset all histological variants can be found. Classic and DN occur at similar frequencies (among 35–45% of tumors/each), MBEN and LC/A have been observed in ~10% and in ~15% of tumors, respectively. Standard diagnosis requires that the identification of morphology variants has to be integrated with genetically defined variants.
Molecular analysis reveals that SHH-MBs display specific chromosomal, genetic and epigenetic alterations whose understanding has allowed the improvement of disease risk assignment and has offered the possibility of using targeted and less toxic therapies against this devastating tumor.
Cytogenetically, SHH-MBs show frequent chromosomal aberrations, which include loss of chromosomes 9q, 10q, 14q and 17p, as well as gain of chromosomes 2 and 9p [Citation23]. Deletions of the long arms of chromosomes 9 and 10 or the short arm of chromosome 17 lead to the loss of heterozygosity of critical negative regulators of the HH pathway, such as PTCH, SUFU, TP53 and RENKCTD11 [Citation24,Citation40,Citation41].
Furthermore, somatic and germline mutations in key components of the HH pathway characterize this tumor. Germline mutations in PTCH and SUFU genes have been described in patients with Gorlin’s syndrome, a rare hereditary disease that predisposes to BCC and MB development [Citation42,Citation43]. Loss of function mutations or deletions in PTCH1 (43%) and SUFU (10%) have also been reported in sporadic MBs in the SHH subgroup. Alterations that lead to ligand-independent activation of HH signaling also include gain of function mutations in SMO (9%) [Citation44] and amplifications of SHH, GLI1 or GLI2 (9%) and MYCN (7%) [Citation24] .
Interestingly, these genetic events are highly age-dependent. Mutations in PTCH1 are found in all age groups, whereas SUFU alterations occur mostly in infants, and SMO mutations are found in adults [Citation24]. A subset of SHH-MB patients (between 3- and 16-years aged children) exhibits GLI2 and MYCN amplifications that are mutually exclusive with PTCH, but are frequently coincident with germline (Li-Fraumeni syndrome) or somatic mutations in TP53 (30%). These patients have a poor prognosis also due to the presence of chromosome shattering (chromothripsis) that can lead to an increased expression of SHH target genes favoring an aggressive tumor growth [Citation45]. Unlike infant tumors that exhibit PTCH or SUFU mutations, adult SHH-MB patients harbor recurrent alterations in both PTCH and SMO and rarely in IDH1 [Citation44,Citation46]. Of note, 98% of adult patients are characterized by mutations in telomerase reverse transcriptase (TERT) promoter, suggesting that alternative mechanisms of telomerase maintenance occur in younger SHH-MB patients [Citation24,Citation47].
Several SHH-MB mouse models have been generated and have confirmed GNPs as the cell origin of this MB entity. The main used models in preclinical study are mice harboring germline mutation in Ptch1 gene (Ptch±) [Citation48], or combined with TP53 deletion (Ptch±; p53-/-) [Citation49] or conditional mouse model leading to Ptch1 loss of heterozygosity [Citation50]. Other available MB mouse models are driven by constitutive activation of Smo, Sufu deletion or MycN overexpression [Citation51–53]. Interestingly, tumors comparison at transcriptional level between current SHH-MB mouse models and patients suggested that the available models are more molecularly similar to human adult SHH-MB [Citation54].
The complex heterogeneity of SHH-MB tumors, recently highlighted by DNA methylation and gene expression array datasets, led to the definition of four molecular SHH variants: SHHα, SHHβ, SHHγ and SHHδ [Citation55]. These subtypes reveal new biological and clinical patient clusters. SHHβ and SHHγ include infants of age ≤ 5 years old. SHHβ, also defined as iSHH-I subtype, is enriched with youngest patients harboring germline or somatic mutations in SUFU and chromosome 2 gain, whereas SHHγ, defined as iSHH-II subtype, exhibits activating SMO mutations and alterations of chromatin-modifying genes KMT2D and BCOR [Citation56]. SHHα and SHHδ include childhood/adolescent and adult patients, respectively. SHHα (which occurs in patients ≥ 5–16 years old) shows a preponderance of TP53 loss of function mutations, which frequently co-occur with GLI2 and/or MYCN amplification or chromothripsis, and it is associated with a worse prognosis than SHHδ (> 17 years old) .
The prognosis of SHH tumors is age-specific: infants belonging to SHHγ subtype have a good outcome compared to SHHβ MBs which are metastatic and have a bad prognosis. Older children and adolescents MB patients (SHHα) with TP53-mutant tumors are associated with poor survival compared to TP53-wild type (WT) ones, and are considered a very-high risk group [Citation23,Citation24,Citation57,Citation58]. Adult patients (SHHδ) rarely present TP53 alterations, but display a higher burden of genome-wide single-nucleotide variants (SNVs) and harbor mutations in PTCH and SMO with a frequency higher than 80% [Citation24]. For this reason, SHHδ subtype is an excellent candidate for molecular target therapies with SMO antagonists. Although the use of SMO inhibitors as therapeutic approach for HH-driven cancers has shown promising results in clinical trials, this strategy is not recommended for infants and children given the crucial role of HH signaling during development [Citation59].
2.2. Crosstalk between HH and other signaling pathways in MB
Evidence of crosstalk between HH and other signaling pathways have been described in several tumor types, thus adding more complexity to those mechanisms underlying the deregulation of this pathway in tumor development and progression [Citation60,Citation61]. The characterization of these intricate interplays is crucial for the development of combined SHH-MB therapies.
Phosphatidylinositol 3ʹ-Kinase/AKT/mTOR (PI3K/AKT/mTOR) cooperates with HH signaling to promote MB tumorigenesis, and it is associated with reduced expression of PTEN [Citation62,Citation63]. Activated mTOR/S6K1 pathway promotes GLI1 transcriptional activity and oncogenic function through GLI1 phosphorylation, with consequent release from its endogenous inhibitor SUFU [Citation18,Citation34]. The overexpression and activation of PI3K/mTOR signaling frequently occurs in SHH-MB resistant to SMO inhibitors and the crosstalk of HH with PI3K/mTOR pathway is associated with high-risk MB, belonging to SHH and G3 subgroups [Citation34,Citation64]. The concurrent combination of SMO antagonists with PI3K/mTOR inhibitors significantly delayed MB tumor growth, thus emerging as a viable therapeutic strategy to treat high-risk patients [Citation64].
Recent studies reported a crosstalk between the HH and RAS-RAF-MEK-ERK signaling pathways in MBin vitro models and identified the mitogen-activated kinase kinase 1 (MEKK1) as a therapeutic target [Citation65]. MEKK1 and MEKK2/3 exert an inhibitory effect on HH signaling through phosphorylation of GLI1. This event strongly reduces both GLI transcriptional activity and protein stability of GLI1. The exposure of MB cells to the MEKK1 activator Nocodazole inhibits GLI1 activity, resulting in the reduction of tumor cell proliferation and viability. Interestingly, MEKK2 and MEKK3, but not MEKK1, are activated in response to FGF signaling, a known potent inhibitor of the HH pathway in GNPs and MB cells [Citation65–67].
A crosstalk between HH and AMP-activated protein kinase (AMPK) signaling pathways in MB has also been well described. Activated AMPK phosphorylates GLI1 at three different sites (Ser102, Ser408 and Thr1074), reducing its stability and impairing the HH pathway activity in MB cells [Citation68]. Interestingly, a later study demonstrated that AMPK is a powerful inhibitor of GLI1 only in human MB cells, given that the consensus AMPK site Ser408 is conserved exclusively in primates [Citation69]. Furthermore, AMPK activation supports β-TrCP-mediated GLI1-ubiquitination and degradation, blocking GLI1 nuclear translocation, and promoting its interaction with β-TrCP [Citation70]. A further work showed that AMPK phosphorylates the zinc finger protein CNBP in response to HH activation. This event increases CNBP/SUFU association, thus leading to CNBP stabilization, ornithine decarboxylase (ODC) translation, and polyamine biosynthesis. Of note, the inhibition of this axis efficiently blocks HH-dependent proliferation of MB cells both in vitro and in vivo [Citation71].
Although in the last decade the knowledge of the intricate crosstalk between HH signaling and the other pathways in MB tumorigenesis has considerably increased, the understanding of these mechanisms should be improved; for example, the interplay between the HH and WNT pathways needs to be elucidated. It has been proven that the activation of Wnt/β-Catenin signaling can mediate the inhibition of the HH pathway and the proliferation of the SHH-dependent GNPs [Citation54]. In particular, Zinke and colleagues show that the stabilization of β-Catenin favors the degradation of GLI1, inhibiting MB cell proliferation [Citation72]. Noteworthy, these findings disagree with previous observation reported by Taylor and colleagues whose data suggest that loss of SUFU could lead to the activation of both pathways in GNPs, contributing to the pathogenesis of MB [Citation73].
At present, a wide number of enzymes identified as regulators of the HH pathway are considered as useful targets for inhibition of HH activity and MB growth. Among them, the phosphodiesterase 4D (PDE4D) has emerged as positive modulator of HH signaling, through the inhibition of PKA, which in turn promotes HH transduction [Citation74]. PDE4D interacts directly with Neuropilins (Nrp), previously identified as positive regulators of HH pathway [Citation75]. The Neuropilin ligand Semaphorin3 (Sema3) enhances this interaction, promoting PDE4D translocation to the cell membrane into close proximity to the site of cAMP production, thus favoring cAMP hydrolysis and the consequent inhibition of PKA. Targeting PDE4D to inhibit the Sema3-Nrp-PDE4D-PKA pathway blocks the growth of HH-related MBs that are resistant to SMO inhibitors [Citation74,Citation76].
This evidence underlines how the deep elucidation of the influence and the effects that other signaling pathways exert on HH signaling activation is fundamental for the development of more effective combinatorial therapies for MB treatments.
2.3. Targeting the HH pathway activity at upstream level: SMO antagonists in preclinical and clinical investigation for the treatment of MB
Deregulated HH signaling has been linked to a broad range of malignancies and has emerged as druggable pathway. The main strategy aimed to counteract its activity is focused on the inhibition at upstream level on SMO receptor .
Figure 3. SMO antagonists in SHH-MB. The figure highlights the compounds in preclinical and clinical studies impairing SMO receptor activity, and their action sites. Red stars indicate the SMO mutations involved in drug-resistance. Compounds entered in clinical trials for SHH-MB treatment are indicated in red. SHH: Sonic Hedgehog; PTCH: Patched receptor; SMO: Smoothened receptor; CRD: cysteine-rich domain; LD: linker domain; TMD: transmembrane domain; GLIs: glioma-associated oncogene transcriptional factors
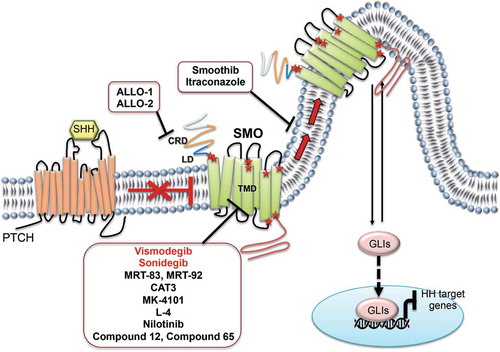
SMO is a G protein-coupled receptor type whose structure consists of an extracellular cysteine-rich domain (CRD), an extracellular linker domain (LD) and the seven-pass transmembrane domain (TMD) [Citation77]. SMO antagonists can be designed and classified accordingly to their ligand-binding sites in those that mostly bind to extracellular loops and those that deeply penetrate in the TMD cavity, which contains two subpockets (TM-1 and TM-2).
TMD portion features an orthosteric site along with a primary GPCR agonist-binding one, but to date no endogenous small molecules are known to bind it. However, the TMD cavity is slightly larger than the CRD one and provides multiple binding sites for several natural and synthetic ligands. The TMD’s plasticity can be attributed to gatekeeper role of the L325 residue in closing or opening TM-1 and TM-2. The mechanistic basis of antagonism mediated by small molecules could be explained by reinforcing the hydrogen bond network that stabilizes the inactive conformation of a GPCR. In particular, the D473, R400 and E518 residues play a pivotal role in stabilizing the inactive conformation state of SMO by hydrogen bonding. In the TM-1 subpocket, the inactive conformation is stabilized by an extra interaction, involving only a water molecule and the D473 amino acid, whose mutation is related with drug-resistance. Indeed, several acquired mutations in SMO have been identified in MB mouse models (D477G, L225R, N223D, S391N, D338N, G457S, E518K/A and W539L) as well as in MB and BCC patients (D473H, L221R, N219D, S387N, D384N, G453S, E518K/A and W535L) following treatment with SMO inhibitors [Citation78,Citation79].
2.3.1. Direct inhibitors of SMO receptor acting on the TMD domain
In the last decade, many antagonists whose binding sites reside into the TMD domain of SMO have been developed and their ability to interfere with the HH pathway activity has been demonstrated in different HH-dependent tumor models [Citation80–83].
The first SMO antagonist to be identified was cyclopamine [Citation84], a steroidal alkaloid that binds the TMD of SMO [Citation85]. Despite its inhibitory activity, this compound showed serious adverse effects, cytotoxicity, high chemical instability and poor aqueous solubility. These pitfalls prevented its further clinical investigation leading to the development of other small molecules with improved drug-like properties, potency, and bioavailability.
2.3.1.1. Vismodegib (GDC-0449)
Vismodegib, chemically described as 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide, binds the TMD of SMO (IC50 value of 21 nM in BODIPY-cyclopamine assay performed in HEK293T cells overexpressing SMO WT) [Citation86,Citation87]. Vismodegib treatment successfully induced tumor regression at 12.5 mg/Kg bis in die (BID) upon multiple dosing in a Ptch± derived MB allograft mouse model [Citation86]. Thanks to these promising results, the drug has been included in clinical trials for the treatment of MB. However, D473H SMO mutation was found in relapsed metastatic MB patient after three months of oral administration. This heterozygous G to C missense mutation in SMO determines a substitution that does not affect its ability to transduce HH signal, but made the receptor insensitive to the inhibitory effect of vismodegib by abrogating its physical interaction with the drug [Citation79,Citation88]. In 2012, vismodegib was the first in class selective SMO antagonist to become commercially available, following its approval by the FDA as a new treatment for locally advanced or metastatic BCC [Citation89]. Although different preclinical and clinical studies in BCC documented a significant initial efficacy of the treatment [Citation89–92], the onset of resistance events (unique SMO mutations, SUFU inactivation and GLI2 amplification) made the response to vismodegib effective in approximately 34% of advanced BCC after one year of treatment [Citation93,Citation94]. To date, vismodegib is adopted both as monotherapy and in combined therapies in eight ongoing clinical trials for the treatment of BCC, advanced gastric adenocarcinoma, advanced chondrosarcoma and metastatic pancreatic cancer. For the treatment of MB, vismodegib has been reported in four clinical trials (Table I). Among them, one trial (NCT01601184) has been conducted in combination with temozolomide in adult patients with recurrent or refractory SHH-MB. This trial terminated on May 2019 because the number of successes was not reached at the end of first stage of the phase II [Citation30,Citation95]. A phase II trial (NCT01878617) is now recruiting standard-risk or high-risk newly diagnosed MBs in skeletally mature SHH-activated patients to evaluate the feasibility and toxicity of oral vismodegib maintenance therapy after conventional chemotherapy (Table I).
Table 1. Clinical trials targeting SHH/GLI pathway for the treatment of newly diagnosed and relapsing/refractory MB patients
The biological complexity of SHH-MB highlights the difficulty to predict sensitivity or resistance to SMO inhibition with vismodegib. A comprehensive approach, including DNA methylation profiling, genome-wide copy number variations and DNA sequence analysis of key genes could provide a more detailed scenario of the course of disease as well as a more accurate understanding of the drug-resistance events occurring in individual patients [Citation96].
2.3.1.2. Sonidegib (NVP-LDE225, LDE-225)
Sonidegib belongs to a class of biphenyl carboxamides and has been identified as SMO antagonist able to bind its TMD domain. Sonidegib shows a strong inhibitory activity on both mSmo and hSMO (IC50 values of 1.3 and 2.5 nM in the fluorescent-labeled BODIPY-cyclopamine assay, respectively) [Citation97]. Daily oral administration of sonidegib in subcutaneous Ptch±; p53-/- MB allograft mouse model at doses of 5, 10, 20 mg/Kg inhibits tumor growth in a dose-dependent manner (33, 51 and 83% of tumor regression, respectively). Furthermore, this compound has shown the ability to cross the blood-brain barrier (BBB) inhibiting the orthotopic MB tumor growth [Citation97]. Sonidegib presents good safety profiles and in 2015 it has been approved by FDA for locally advanced BCC treatment [Citation97]. However, different mechanisms of resistance have been observed following the treatment with sonidegib (i.e. point mutations in SMO, GLI2 amplification, up-regulation of PI3K signaling) [Citation34]. The safety, efficacy and tolerability of sonidegib have been investigated as monotherapy or in combined therapies in human trials for the clinical management of several advanced solid tumors. For MB patients, the treatment with sonidegib has been reported in four clinical trials [Citation31,Citation32]. A phase I trial on patients with recurrent or refractory MB (NCT01125800) showed that oral once daily sonidegib administration (680 mg/m2 related to the Body Surface Area, BSA) was well tolerated in children and had antitumor activity in both pediatric and adult patients with relapsed SHH-MB (Table I) [Citation31]. The initial promising results from phase I have led to add a phase II part to the study. The aims of this phase II were to assess the efficacy of sonidegib in recurrent or progressive MB patients through radiographic response, analysis of GLI2 amplifications and SUFU mutations as outcomes of de novo resistance mechanisms, and through the evaluation of drug concentration in cerebrospinal fluid (CSF). Nevertheless, this phase II was closed prematurely due to the lack of sufficient patient material. Currently, a recruiting phase I trial (NCT03434262) is aimed to investigate subgroup-specific doublet combinations including the CDK4/6 inhibitor ribociclib and sonidegib for the treatment of SHH-activated patients (Table I).
2.3.1.3. MRT-83 and MRT-92
A novel class of SMO inhibitors based on acylthiourea, acylurea, and acylguanidine scaffolds has been recently developed [Citation98]. Among identified antagonists, the acylguanidine MRT-83 has a nanomolar antagonist efficiency toward SMO (IC50 value of 4.6 nM in BODIPY-cyclopamine binding assay performed in HEK-hSMO cells) [Citation99], and is able to block HH-mediated proliferation of GNPs [Citation100]. Mechanistically, MRT-83 abrogates SAG-induced trafficking of endogenous mSmo and hSMO to the primary cilium in C3H10T1/2 cells and NT2 testicular carcinoma cells, respectively. In vivo, stereotaxic MRT-83 injection into lateral ventricle of adult mice blocks Ptch gene transcription induced by SHH in the adjacent subventricular zone, demonstrating MRT-83-mediated HH signaling inhibition [Citation99].
The acylguanidine MRT-92, a derivative of MRT-83, exhibits a sub-nanomolar antagonistic activity against SMO (IC50 value of 8.4 nM in BODIPY-cyclopamine binding assay performed in HEK-hSMO cells) by blocking several overlapping sites of its TMD domain [Citation98]. Similar to MRT-83, MRT-92 blocks SAG-induced trafficking of SMO at the primary cilium and SAG-induced differentiation of C3H10T1/2 cells. Of note, this molecule maintains similar pharmacological characteristics when bound to vismodegib-resistant D473H SMO mutant [Citation98]. MRT-92 has also shown to inhibit tumor growth in vivo in melanoma mouse and colorectal cancer mouse models [Citation81,Citation82].
2.3.1.4. CAT3
Compound PF403 is a metabolite of the bioactive natural product 13a-(S)-deoxytylophorinine [Citation101] with strong inhibitory activity against HH pathway-hyperactivated MB cells (IC50 values of 0.013 nM values assayed by MTT assay in human MB DAOY cells). Mechanistic study revealed that PF403 directly binds SMO in a similar manner as vismodegib thereby inhibiting the receptor activity. Furthermore, PF403 promotes the interaction of SUFU and PKA with GLI1, thus reducing the nuclear translocation of the transcription factor [Citation27]. However, in vivo PF403 treatment was not effective. In order to improve the pharmacokinetic properties of PF403, its prodrug CAT3 (13a-(S)-3-pivaloyloxyl-6,7-dimethoxyphenanthro[9,10-b]-indolizidine) has been developed [Citation102]. CAT3 significantly suppresses DAOY orthotopic xenograft tumor growth with inhibition rate of 78.8% (dose of 12 mg/Kg), without showing toxicity [Citation102]. These results suggest that CAT3 might be a promising novel agent for the treatment of HH-driven MB.
2.3.1.5. MK-4101
MK-4101 (5-(3,3-difluorocyclobutyl)-3-[4-[4-methyl-5-[2-(trifluoromethyl)phenyl]-4 H-1,2,4-triazol-3-yl]bicyclo[2.2.2]oct-1-yl]-1,2,4-oxadiazole) was originally identified as an 11β-Hydroxy steroid dehydrogenase-1 inhibitor that caused embryonal toxicity and birth defects similarly to those elicited by mutations of the HH pathway [Citation103]. It was subsequently shown that MK-4101 inhibited HH signaling (IC50 value of 1.5 μmol/L in a luciferase reporter assay) through the binding to SMO receptor (IC50 value of 1.1 μmol/L assessed by a fluorescently labeled cyclopamine assay). Further, this compound showed efficacy also toward D477G Smo mutant [Citation103]. In vitro, MK-4101 inhibits the proliferation of MB cells derived from neonatally irradiated Ptch1± mice. In vivo, the efficacy of this molecule has been evaluated in allografts Ptch1± MB mouse model. MK-4101 impairs tumor growth (doses of 40 and 80 mg/Kg once a day) and induces tumor regression at the highest dose (80 mg/Kg twice a day), an effect that correlates with a dose-dependent downregulation of Gli1 mRNA levels. Of note, MK-4101 (dose of 80 mg/Kg BID for 35 days) completely eliminated MB, and administration prevents tumor relapse after three months from treatment termination. Finally, MK-4101 significantly improves survival of Ptch1± mice [Citation103].
2.3.1.6. Smoothib
Recently, the pyrazolo-imidazole smoothib has been identified as a SMO inhibitor targeting the heptahelical bundle of the receptor (IC50 value of 1.4 μM assessed by Gli-mediated luciferase expression in Shh-Light II cells). Interestingly, this compound is able to prevent SMO ciliary localization, to reduce the expression of HH target genes and to suppress Ptch+/− MB cells growth [Citation104].
2.3.1.7. L-4
L-4 featuring a dimethylpyridazine backbone has been recently identified as a potent, well-tolerated, orally active inhibitor of the HH pathway by directly targeting SMO in the same binding pocket of cyclopamine [Citation105]. This compound showed an IC50 value of 2.33 nM in the SHH-Light II assay. L-4 strongly inhibited the HH pathway in vitro, suppressing the proliferation of primary MB cells with nanomolar IC50 value similarly to vismodegib. Importantly, L-4 exhibited equivalent potency in reducing downstream HH targets expression induced by wild type SMO and D473H SMO mutant. Orally administration of L-4 provided remarkable dose-dependent antitumor effect in Ptch±;p53−/- MB allograft model without inducing loss of body weight side effect. Furthermore, L-4 revealed a good tolerance in acute toxicity test using ICR mice [Citation105].
2.3.1.8. Nilotinib
Nilotinib is an approved second generation protein tyrosine kinase inhibitor discovered as a potent SMO antagonist directly binding to its TMD domain [Citation106]. Nilotinib treatment reduces GLI1 protein levels in both SHH-MB PDX and human MB DAOY cells in vitro and tumor growth in subcutaneous MB mouse xenograft model [Citation106]. Nilotinib is an FDA-approved drug indicated for the treatment of chronic phase and accelerated phase Philadelphia chromosome-positive (Ph+) chronic myelogenous leukemia (CML) and has well-characterized pharmacokinetics and safety profile. Its newly discovered anti-HH activity makes Nilotinib an attractive therapeutic candidate against HH-dependent cancers, alone or in combination with surgery, radiotherapy and chemotherapy.
2.3.1.9. SMO antagonists derived from natural sources
In an effort to exploit natural products as profitable source of new SMO antagonists, a library of more than thousands of natural compounds and their derivatives has been screened in silico towards the crystallographic structure of SMO bound to cyclopamine affording the 2ʹ,4ʹ,5ʹ,3,4-pentamethoxychalcone (compound 12) as the most effective HH inhibitor [Citation107]. This compound has proven to be effective also on the drug-resistant D473H SMO variant, and showed antioncogenic activity in vitro and/or in vivo in HH-driven tumor cells (mouse ASZ001 BCC, human MB DAOY, human prostate carcinoma epithelial 22Rv1 cells, primary Ptch± MB cells) and inhibits MB stem-like cells self-renewal [Citation26].
A drug discovery program focused on the synthesis of a molecule incorporating the basic skeleton of the natural product artemisinin equipped with a SMO-targeting bullet led to the identification of the 2-(2,5-Dimethyl-5,6,7,8-tetrahydroquinolin-8-yl)-N-arylpropanamide compound 65 as the most potent HH inhibitor (IC50 value of 9.53 nM in a luciferase reporter assay). The molecule targets SMO receptor on the same binding site of cyclopamine. Compound 65 showed a good plasma exposure and an acceptable oral bioavailability, and antiproliferative effects in primary Ptch±; p53−/- MB cells both in vitro and in vivo [Citation108].
2.3.2. Direct inhibitors of SMO receptor acting on the CRD domain
2.3.2.1. ALLO-1 and ALLO-2
ALLO-1 and ALLO-2 have been identified as HH signaling inhibitors by high throughput screening of commercial compound libraries. These compounds act on both wild type and SMO mutants by binding the CRD domain of SMO, without any interaction with TMD (ALLO-1: IC50 values of 489 nM and 1.2 μM; ALLO-2: IC50 values of 132 nM and 440 nM against wild type and D477G Smo mutant in the TM3-Gli-Luc reporter cell line, respectively) [Citation109]. Both compounds inhibit the SHH-dependent proliferation of GNPs and Ptch1±; p53-/- MB cells in a dose-dependent manner [Citation109].
2.3.3. Alternative strategies to counteract SMO receptor activity
2.3.3.1. Itraconazole (ITZ)
Itraconazole is an FDA-approved drug for the treatment of fungal infection that has been proposed as a potent HH pathway inhibitor for its ability to prevent cilium translocation of SMO [Citation110]. This compound acts on SMO in a distinct binding site from cyclopamine, although its direct binding to SMO receptor has not been demonstrated yet [Citation110]. Interestingly, itraconazole is also active against drug-resistant SMO mutants [Citation111], and showed synergistic effects with other SMO antagonists, such as vismodegib and sonidegib [Citation110]. In vivo studies in a Ptch+/−; p53−/− MB allograft mouse model demonstrated that systemic administration of itraconazole suppresses tumor growth at a BID oral dosage of 75 or 100 mg/Kg after 18 days of treatment. Itraconazole has been clinically used for nearly 35 years as an antifungal agent and currently, thanks to its anticancer properties, it has entered in clinical trials for the treatment of many kinds of tumors [Citation112–115].
2.4. Targeting the HH pathway activity downstream of SMO: the development of GLI inhibitors
Glioma-associated oncogene homolog (GLI) transcription factors are the final effectors of the HH signaling pathway, and their activity is finely modulated by a balanced interplay among post-translational modifications and intersection with other pathways. Since GLI factors have a key role in embryogenesis and adult homeostasis, deregulation of their activity alters the feedback loop that controls HH response, leading to several pathological conditions and tumorigenesis. It is important to consider that every alteration linked to the activation of the HH pathway triggers the transcriptional activity of GLI1 effector, which in turn induces the expression of genes driving proliferation, stemness and survival. Furthermore, GLI1 promotes its own expression, thus representing an attractive target for the development of novel anticancer drugs. In the last years, a number of GLI1 inhibitors have been identified. These agents can be classified based on their mechanism of action as indirect (i.e. post-translational modifiers which impair GLI1 activity) and direct GLI1 inhibitors (small molecules blocking its transcriptional function) .
Figure 4. GLIs inhibitors in SHH-MB. The figure shows indirect (black) and direct (red) GLIs inhibitors. Negative regulators of GLIs activity are illustrated in blue boxes; positive regulators are illustrated in orange boxes. SHH: Sonic Hedgehog; PTCH: Patched receptor; SMO: Smoothened receptor; GLIs: glioma-associated oncogene transcriptional factors
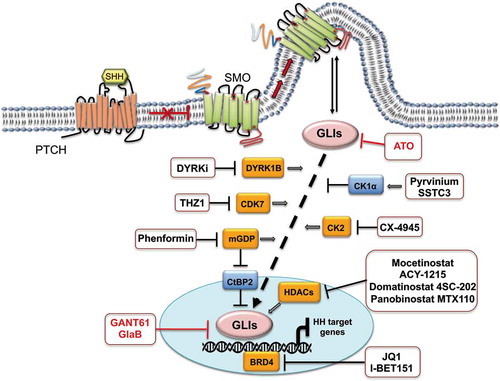
2.4.1. Modulation of GLI proteins function
2.4.1.1. BRD4 inhibitors
Epigenetic enzymes have emerged as therapeutic targets and critical regulators of HH transcriptional output [Citation116]. A new class of compounds targeting bromo and extra C-terminal (BET) bromodomain (BRD) proteins has been proposed as HH antagonists for their capability to affect GLI transcriptional activity. Members of the BET family proteins (BRD1–4) bind to histones acetylated lysines through their bromodomains, thus enhancing gene expression and regulating cellular processes such as cell cycle progression, chromatin compaction and chemoresistance [Citation117]. Among BET family members, BRD4 directly binds to GLI1 and GLI2 promoters, triggering the expression of HH target genes. Tang and collaborators identified the small molecule JQ1 as BRD4 inhibitor. JQ1 induces the downregulation of GLI1 target genes, and suppresses tumor growth in HH-dependent mouse models (BCC, MB and atypical teratoid rhabdoid tumor) resistant to SMO antagonists [Citation118]. Recently, Wang and colleagues demonstrate that encapsulation of JQ1 in apolipoprotein E nanoparticles (ApoE-NPs) significantly improves drug efficacy in orthotopic G3 MB bearing mice [Citation119], suggesting the potential use of this formulation for MB clinical management.
Another BRD4 inhibitor, I-BET151, has been discovered through a screening for inhibitors of epigenetic modulators that attenuate HH activity. This compound exerts an inhibitory effect on the HH pathway (IC50 value of 31 nM in Gli-luciferase reporter assay in SAG-activated Light II cells) and reduces HH signaling in Sufu-/- mouse embryonic fibroblasts, consistent with its mechanism of action downstream of SMO receptor. I-BET151 induces the dissociation of BRD4 from the proximal promoter region of GLI1 locus, thus confirming its BRD4-dependent HH antagonism. Of note, I-BET151 suppresses tumor growth in Ptch± MB allograft mouse model [Citation120].
2.4.1.2. CK1α agonist
The anthelmintic drug pyrvinium has been described for its ability to destabilize GLI proteins [Citation121,Citation122]. In particular, this compound works as allosteric activator of CK1α, a kinase that negatively regulates GLI transcription factors [Citation123]. In conditions of SMO-independent HH pathway hyperactivation, pyrvinium attenuates MB cells proliferation, both in vitro and in vivo, as consequence of GLI1 and PTCH1 down-regulation. Moreover, pyrvinium strongly suppresses HH signaling induced by the oncogenic SMO-M2 and the drug-resistant D473H variants [Citation122]. Although pyrvinium reduces the growth of SHH-MB, its poor ability to cross the BBB limits its efficacy.
Recently, SSTC3 has been described as a second-generation of CK1α activator. The compound inhibits HH signaling both in vitro and in vivo and possess improved pharmacokinetic and antioncogenic properties (crossing the BBB, attenuating the growth and metastases of SHH-MB mouse models and prolonging their survival) compared to pyrvinium [Citation124]. Most importantly, SSTC3 is effective against an orthotopically implanted SHH-MB PDX with a TRP53 mutation and MYCN amplification [Citation124].
2.4.1.3. CK2 inhibitor
The protein kinase CK2 emerged as a driver of phosphorylation events during the proliferative phase of GNPs growth. CK2 facilitates the HH pathway transduction regulating two steps: stabilizing GLI2 and enhancing its transcriptional activity [Citation125]. The highly specific CK2 inhibitor CX-4945 induces a dose-dependent reduction of GNPs proliferation in vitro, and of folia width during postnatal period. CX-4945 also decreases the viability of Ptch+/− mouse MB cell lines (MB21, MB53 and MB55; IC50 values of 2.5 to 5.3 μM) and human MB PDX cells (RCMB32, BT084, ICb-984 and ST01; IC50 values of 0.76 to 3.3 μM). In vivo, this compound blocks the growth of Ptch+/−;Tpr53−/− and Ptch+/−;Tpr53−/−;Smo D477G MB allografts mouse models resistant to currently available HH inhibitors, thereby extending the survival of tumor-bearing mice [Citation125]. CX-4945 is currently in a phase I/II trial (NCT03904862) in skeletally immature (phase I) and skeletally mature (phase II) SHH-recurrent or refractory MB patients .
2.4.1.4. CDKs inhibitors
The cyclin-dependent kinase 7 (CDK7) is a member of the cyclin-dependent kinase protein family involved in cell cycle regulation and transcription initiation or elongation. Through an unbiased screening of a collection of epigenetic or transcriptional targeted small-molecule compounds, the CDK7 inhibitor THZ1 has shown the higher inhibitory effect on cell viability in murine SHH-MB cell lines derived from Ptch1+/− mice [Citation126]. THZ1 induces down-regulation of GLI1 and GLI2 expression at both mRNA and protein levels in MB cell lines [Citation126]. THZ1, alone or in combination with BET inhibitors, effectively overcomes the resistance to SMO antagonists and inhibits HH-driven tumor growth both in vitro and in vivo. Since multiple CDK7-targeted drugs have recently entered phase I trial for tumor therapy, the evidence collected on THZ1 provide the preclinical rationale for enrolling CDK7 inhibitors in HH-dependent cancers treatment.
Besides CDK7, the CDK4/6/Cyclin D/RB pathway has been recently identified as a druggable target for all non-WNT MBs [Citation127]. Cyclin-dependent kinases 4 and 6 (CDK4/6) are involved in the regulation of cell cycle progression through the G1–S transition. In particular, they each form an active complex with Cyclin D, catalyzing the phosphorylation of the retinoblastoma (RB) protein. Hyperphosphorylation of RB favors the transcription of genes required for entry into S-phase and commitment to cell division. Palbociclib is a selective inhibitor of CDK4/6 that prevents RB hyperphosphorylation and promotes cell cycle arrest in the G1 phase [Citation128]. Palbociclib has been FDA-approved as part of a combination therapy for advanced breast cancer, and its efficacy has also been reported in a variety of RB-positive tumors including brain malignancies. Interestingly, palbociclib treatment (administered orally daily at 120 mg/Kg) induces tumor regression in SHH-PDX xenograft mouse models, showing an average reduction of 63% in tumor volume compared to vehicle-treated mice [Citation127]. These data encouraged the launch of a phase I clinical trial to test safety of palbociclib in patients with recurrent, progressive or refractory central nervous system tumors (NCT02255461) .
2.4.1.5. DYRK1B inhibitor
The dual-specificity tyrosine phosphorylation-regulated kinase (DYRK) family is involved in HH signaling regulation; among DYRK family members, DYRK1B has been identified as critical positive regulator of HH/GLI signaling downstream of SMO. DYRK1B inhibition induced by DYRKi impairs GLI1 expression in both vismodegib-sensitive and -resistant human MB DAOY cells (IC50 values of 1.16 μM and 1.04 μM, respectively) [Citation129].
2.4.1.6. HDACs inhibitors
Among epigenetic enzymes, histone deacetylases (HDACs) are strongly involved in the control of the HH pathway [Citation130,Citation131]. In particular, HDAC1 is up-regulated in MB and its mediated deacetylation of GLI1 and GLI2 promotes their transcriptional activity [Citation132,Citation133]. Therefore, HDAC1 inhibitors stand as effective drugs able to prevent the HH pathway activation through GLI1 and GLI2 hyperacetylation. The selective HDAC1/HDAC2 inhibitor mocetinostat has been recently described as potent HH inhibitor related to GLI1 K518-acetylation. Orally administration of mocetinostat increases the survival of SHH-MB mouse models and drastically impairs tumor growth by reducing the proliferation and increasing the apoptosis of tumor cells [Citation134].
HDAC6 is overexpressed in a murine model of SHH-MB, and its specific antagonist rocilinostat (ACY-1215) reduces tumor growth of primary MB99–1 MB cells (derived from the SmoA1 mouse model) as well as of MB99–1 allograft mouse models in vivo [Citation135].
Recently, the dual epigenetic inhibitor domatinostat 4SC-202 has also been tested in HH responsive human MB DAOY cells [Citation136,Citation137]. This is a very promising small molecule targeting class I HDACs 1/2/3 and the lysine-specific demethylase (LSD1) and has been evaluated in a phase I clinical trial in patients with advanced hematological malignancies, revealing an excellent safety profile. Several clinical studies aimed to evaluate 4SC-202 in combined therapies specifically in the immuno-oncology area are currently ongoing. Interestingly, treatment with increasing concentrations of 4SC-202 efficiently repressed SAG-induced GLI1 and Hedgehog interacting protein (HHIP) expression in DAOY cells (IC50 values of ~240 nM and ~140 nM, respectively) without affecting primary cilium formation [Citation137]. 4SC-202 treatment also reduced the proliferation rate of SAG-stimulated human MB DAOY cells. Importantly, mechanistic studies revealed that the repressive effect of 4SC-202 on HH/GLI1 signaling is attributed to class I HDACs inhibition, not involving LSD1 impairment.
Several chemical classes of HDACi are currently being tested in human clinical trials, and panobinostat is one of the synthetic inhibitors FDA-approved for cancer therapy [Citation138,Citation139]. Currently, a pilot phase I study (NCT04315064) is recruiting patients with histologically verified recurrent or progressed MB to assess the antitumor activity of simultaneous infusions of panobinostat MTX110 into the resection cavity. MTX110 is a gold nanoparticle-based formulation needed to solubilize the non-selective pan-deacetylase inhibitor panobinostat. This formulation can be directly injected into the brain, bypassing the BBB and delivering high concentrations of drug into the tumor while minimizing systemic toxicity [Citation140]. The first patient has been enrolled in March 2020, and the study has not started yet .
2.4.1.7. Biguanides
The biguanides metformin and phenformin are antidiabetic drugs associated with anticancer properties in preclinical and clinical settings [Citation141]. Several works sustained AMP-activated protein kinase (AMPK) as a key mediator of the direct anticancer properties of biguanides [Citation142], and AMPK-dependent inhibition of mTOR, which suppresses protein synthesis, cell growth, and viability [Citation143]. Recently, it has been demonstrated that clinically relevant doses (1–5 µM) of phenformin suppresses SHH-MB growth. Interestingly, this effect is mediated by the inhibition of mitochondrial glycerophosphate dehydrogenase (mGPD), a component of the glycerophosphate shuttle, without affecting complex I or AMPK activity [Citation144]. The inhibition of mGPD mimics phenformin action and increases redox state/NADH content. Elevated NADH levels promote the association between the corepressor CtBP2 and GLI1 [Citation144]. These findings show that phenformin suppresses SHH-MB growth via an interplay between metabolism and transcriptional repression.
2.4.2. Direct inhibition of GLIs transcriptional activity
2.4.2.1. GANTs
Besides HH inhibitors mentioned above, the most relevant contribution in the field of HH-driven tumor biology arises from the development of direct GLI inhibitors. GLI antagonists, or GANTs, have been discovered in 2007 by a cellular screening for small molecule inhibitors of GLI1-mediated transcription [Citation145]. This approach led to the identification of GANT58 and GANT61 as the first GLI antagonists (IC50 values of ≈ 5 μM for both compounds in Gli-luciferase reporter assays performed in SHH-Light II cells), with GANT61 more specific towards GLIs and more effective in reducing GLI1 and GLI2 DNA-binding ability. Although no records of clinical studies are available, GANT61 is the reference GLI1/GLI2 antagonist in many biological and drug design studies. Its mechanism of action has been controversial; GANT61 is highly unstable in physiological conditions, and quickly undergoes hydrolysis into the corresponding benzaldehyde and diamine derivative (GANT61-D). Accordingly, GANT61 can be considered as a prodrug able to release the biologically active form GANT61-D in physiological conditions, which might directly bind GLI1 in a groove between zinc finger-2 (ZF-2) and ZF-3, without interfering with the DNA-binding site [Citation146]. Nevertheless, recent studies elucidate the kinetics of GANT61 hydrolysis and show that GANT61-D might bind near ZF-1 and ZF-2 [Citation147]. GANT61 impairs GLI1- and GLI2-mediated transcription in vitro and in vivo, affecting GLI1/DNA interaction only in living cells, probably by inducing post-translational modifications of GLI1 [Citation145]. The effectiveness of this molecule has been demonstrated in several tumor types, including MB. GANT61 inhibits cell migration, invasion, and proliferation while enhances the apoptosis of human MB DAOY cells [Citation148]. A recent study reports that GANT61 is able to sensitize DAOY cells to particle radiation (i.e. protons and carbon ions), but not to conventional X-rays [Citation149] highlighting that the combination of GANT61 with particle radiation could offer a benefit for the treatment of specific cancer types.
2.4.2.2. Arsenic trioxide (ATO)
Arsenic trioxide antagonizes HH signaling both in vitro and in vivo directly interacting with GLI1. ATO inhibits GLI1 transcriptional activity (IC50 value of 2.7 μM assayed by Gli-luciferase reporter assay in HepG2 cells) without impairing the binding to DNA or modifying GLI1 cellular trafficking and stability [Citation150]. This molecule also inhibits GLI2 ciliary accumulation in short term, whereas enhances GLI2 degradation after long incubation time in MB cells [Citation151]. ATO inhibits Ptch±; p53-/- MB allografts tumor growth, and increases the survival of constitutively activated SMO transgenic mice [Citation150]. Recently, it has been reported that ATO promotes radiosensitivity in SHH-MB TP53 mutated cells reducing their clonogenic capacity [Citation152]. The effectiveness of ATO as anticancer therapeutic agent has been tested in several preclinical tumor models [Citation150,Citation153–157]. Data obtained from these studies sustain that ATO, used alone or in combination with other anticancer drugs, may represent a valuable therapeutic option to treat HH-dependent tumors, particularly those harboring drug-resistant SMO mutations [Citation111,Citation150,Citation151]. ATO is an FDA-approved drug for the treatment of acute myeloid leukemia (AML) patients used in combination with trans-retinoic acid therapy [Citation158,Citation159]. Moreover, it is currently in clinical trials ranging from phase I to phase IV for both solid tumors and hematological malignancies, as monotherapy or combined with chemo- and radiotherapy.
2.4.2.3. Glabrescione B (GlaB) and its derivatives
Natural compounds provide a significant contribution to the discovery of novel HH inhibitors [Citation160]. Isoflavones, a class of natural compounds particularly abundant in plants of the Leguminosae family, have high versatile scaffold and have long received attention due to their interesting biological activity and multiple benefits to human health [Citation161–163]. Recently, our research group has established a multi-disciplinary drug discovery program focused on the identification of natural products as direct GLI1 antagonists. Starting from the crystallographic structure of the GLI1-ZF domain in a complex with DNA [Citation164], we identified the strongest hot spots residues for GLI1/DNA interaction and GLI1 transcriptional functions [Citation165]. A subsequent virtual screening of a natural compounds library against these hot spots leads to the identification of Glabrescione B (GlaB), an isoflavone naturally occurring in Derris glabrescens, as a potent HH antagonist and direct inhibitor of GLI1. GlaB binds the ZF-4 and ZF-5 of GLI1 affecting its interaction with DNA and blocking its activity (IC50 value of 12 μM in GLI1-overexpressing HEK293T cells/firefly luciferase assay). The significant anticancer efficacy of GlaB has been demonstrated both in vitro and in vivo in HH-dependent MB and BCC model [Citation165]. These promising results have given relevance to the druggability of GLI1/DNA interaction in the treatment of HH-dependent tumors.
Exploiting the versatility of the isoflavone scaffold, Berardozzi and collaborators described that the insertion of a bulky substituent in meta or in para position of the isoflavone’s ring B enhances the specific affinity for GLI1 or SMO, respectively. These findings provided the first evidence of the synergistic effect induced by the combination of two HH inhibitors acting specifically at upstream or downstream level of HH signaling [Citation166]. In particular, the combined administration of different isoflavones behaving as SMO or GLI1 antagonists, showed synergistic HH inhibition in primary Ptch± MB cells at doses around 20-fold lower than individual compound doses [Citation166]. Based on these findings, a third generation of isoflavones able to target simultaneously SMO and GLI1 has been synthetized [Citation167]. In particular, the most promising multitarget compound 22 showed a strong inhibitory activity on HH signaling (IC50 of 0.79 μM in Gli-mediated luciferase expression in SHH-Light II cells), and HH-dependent tumor growth in human and murine MB cells at sub-micromolar concentration, inducing the reduction of GLI1 protein levels. An in vivo allograft model of MB shows the efficacy of intratumoral administration of compound 22 (5 mg/Kg), reducing HH-driven tumor growth by suppressing cell proliferation and promoting apoptosis [Citation167].
2.4.2.4. Cynanbungeigenin C and D
Cynanbungeigenin C (CBC) and D (CBD) have been isolated from Cynanchum bungei Decne plant and have emerged as GLI1 inhibitors although with an unclear mechanism. Both compounds are able to repress Gli1-luciferase reporter activity (IC50 values of 2.9 and 3.7 μM, respectively), and to inhibit HH signaling in cells expressing D473H and W535L drug-resistant SMO mutants. Moreover, CBC and CBD suppress cell proliferation in Ptch1±;p53-/- MB models both in vitro and in in vivo (dose of 50 mg/Kg by i.p. injection in allograft nu/nu mouse model). Of note, pharmacokinetic studies demonstrated the capability of these compounds to cross the BBB [Citation168]. CBC and CBD stand as potential lead compounds in the treatment of MB and other HH-dependent malignancies.
2.4.2.5. Other GLI1 inhibitors
Following a common feature pharmacophore generation approach by known GLI1 inhibitors, a virtual screening protocol of commercially available databases recently led to identification of three different chemical scaffolds as GLI1 modulators [Citation169]. The diprenylxanthone α-mangostine SST0673, the thiophene derivative SST0682 and the pyrazolo[1,5-a]pyrimidine analogous SST0704 were able to inhibit the HH pathway activity by affecting GLI1 protein levels, and to impair proliferation of both human melanoma A375 (IC50 values of 2.7, 12 and 2.2 μM, respectively) and MB DAOY (IC50 values of 1.9, 0.9 and 2.3 μM, respectively) cells. Several derivatives of hit compounds have been synthesized and extensive SAR has been established, providing a good starting point for further steps in the development of GLI1 negative modulators.
2.5. Immunotherapy in MB
Immunotherapy represents an attractive therapeutic approach in several solid tumors and has recently been investigated for the treatment of central nervous system (CNS) malignancies [Citation170–173]. In particular, in MB, immunotherapy has emerged as a valuable strategy to limit the side effects caused by radiation and chemotherapy, due to its potential ability to target tumor cells while preserving the surrounding normal brain tissue. A deep understanding of the connections between the immune system and MB is fundamental to design effective and specific immunologic-based approaches.
Preclinical models of MB molecular subtypes in mice have shown higher percentages of dendritic cells, infiltrating lymphocytes, myeloid-derived suppressor cells and tumor-associated macrophages in murine SHH subgroup compared to G3 MB [Citation174].
A recent study on the microenvironment populations involving 763 human MBs belonging to the four molecular subgroups (70 WNT, 223 SHH, 144 G3 and 326 G4) has shown a higher distinctive pattern of microenvironmental cells in SHH subgroup compared to the others [Citation175]. In particular, SHH-MB displays a stronger signature of T cells, fibroblasts and macrophages together with a lower numbers of neutrophils compared to the other MB subgroups others [Citation175,Citation176]. Moreover, greater expression of inflammation-related genes (CD14, PTX3, CD4, CD163, CSF1R and TGFB2) is observed in tumors of the SHH subgroup in comparison to those of the G3 and G4 [Citation176].
Regardless of the subtype, MBs show low levels of cytotoxic lymphocytes and endothelial cells, as well as impaired antigen presentation due to the down-regulated expression of MHC-I components (i.e. LMP2, LMP7, calnexin and b2-microglobulin) [Citation175–178]. These findings have suggested that the effectiveness of immunotherapy in MB depends on the immunologic differences in MB molecular subgroups. At present, several clinical trials using immunotherapy in MB are ongoing and most of them are at early stages [Citation179]. The main immune-based strategies that are currently considered for treatment of this malignancy and here reviewed include: natural killer (NK) cells, CAR-T therapy, immune checkpoint inhibitors (ICIs), oncolytic viruses.
2.5.1. Natural Killer
Natural Killer (NK) cells have emerged as a promising immune-strategy for hematological malignancies and solid tumors thanks to their capacity to lyse directly specific ligand-targeted cancer cells [Citation180–182]. In the treatment of MB, one of the advantages of using NK cells is that this tumor shows the down-regulation of MHC-I that renders malignant cells more susceptible to NK cells-mediated lytic activity [Citation183]. In addition, MB cells express ligands for triggering NK cells receptors and are sensitive to NK-mediated cytotoxicity in vitro [Citation184,Citation185]. In particular, human DAOY MB cells express high levels of NKG2D, a NK-activating receptor that plays a major role in the killing of this tumor cell line [Citation184]. Recently, in vivo studies demonstrated that the intratumor injection of activated NK cells suppresses the tumor growth of DAOY cells implanted into the cerebella of NSG mice [Citation183]. A phase I trial is currently ongoing (NCT02271711) for the study of autologous NK cells delivered via the fourth ventricle catheter after surgery in patients with recurrent MB .
Table 2. Ongoing clinical trials for the treatment of MB patients with immunotherapy
2.5.2. Chimeric antigen receptor T cell therapy
The use of chimeric antigen receptor (CAR) T cell therapy has shown efficacy especially in hematologic cancers, while the main challenge for their applicability in solid cancers is the identification of specific tumor-associated antigens enriched in tumors, but not expressed in normal tissues [Citation186,Citation187]. Receptor tyrosine-protein kinase ERBB2 (HER2) is a known immunotherapy target that is overexpressed in several adult and pediatric tumors, including 40% of MB, while is not detected in normal brain [Citation188–191]. To this regard, human HER2-CAR T cells, containing the CD3zeta and 4–1BB inducible co-stimulator receptor (HER2-BBz-CAR T), showed, at low doses, strong antitumor activity against MB cell lines both in vitro and in vivo orthotopic models [Citation192]. Autologous CD4+ and CD8+ T cells transduced with lentiviral particles expressing HER2 antigen receptor and EGFRt (a truncated form of the human epidermal growth factor receptor) are currently under investigation in a phase I trial (NCT03500991) for the treatment of recurrent/refractory MB patients HER2-positive . Recently, Donovan and colleagues identified high expression of EPHA2, HER2 and interleukin 13 receptor α2 (IL-13Rα2) in G3 MBs and ependymomas, but not in the normal developing brain. They demonstrated the efficacy of locoregional CSF delivery of EPHA2 monovalent, HER2 monovalent and EPHA2–HER2–IL-13Rα2 trivalent CAR T cell therapy in xenograft mouse models of primary, metastatic and recurrent G3 MBs and posterior fossa group A (PFA) ependymomas. These findings provide a rationale for clinical trials of these approaches in patients [Citation193].
2.5.3. Immune checkpoint inhibitors
One of the most successful immunotherapy approaches in cancer is the use of immune checkpoint inhibitors (ICIs), especially those acting against the checkpoint protein PD-1 or its partner protein PD-L1 [Citation194–196]. Several recent studies have evaluated PD-L1 expression in MB with controversial results, demonstrating that SHH cell lines showed both constitutive and inducible expression of PD-L1, while G3 and G4 MB cells had only inducible expression [Citation197]. Currently, two clinical trials are investigating the effectiveness of ICIs in MB. The aim of the phase I trial NCT02359565 is to assess the side effects and best dose of the monoclonal antibody MK-3475 (Pembrolizumab; Anti-PD-1) in children with recurrent/progressed/refractory brain tumors, including MB . The effectiveness of nivolumab, a human immunoglobulin G4 monoclonal antibody that binds to the PD-1, is under evaluation in a phase II trial (NCT03173950) for the treatment of adult patients with rare CNS malignancies including MB . Furthermore, nivolumab with and without ipilimumab, another checkpoint inhibitor that targets the Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4), is under investigation in a phase II trial of patients with high-grade CNS malignancies including MB (NCT03130959) .
2.5.4. Oncolytic viruses
Different types of oncolytic viruses (OVs) are being investigated as anticancer-therapy for pediatric brain tumors with satisfactory results in preclinical models of MB [Citation198,Citation199]. Orthotopic murine models of SHH-dependent MB treated with intratumoral administration of myxoma virus or with the double-stranded RNA reovirus have shown a significant prolonged survival [Citation200,Citation201]. The rodent parvovirus H-1 (H-1PV) and the adenovirus Ad5 Delta-24 revealed lytic effects and reduction of viability in vitro in several MB cell lines, included human DAOY [Citation202,Citation203]. So far, in vivo studies evaluating the effect of these viruses in MB animal models have not been performed. At present, a phase I clinical trial (NCT03911388) is ongoing to determine the safety of inoculating the oncolytic variants of the Herpes Simplex Virus G207 into recurrent brain tumors including MB [Citation204,Citation205]. Moreover, WNT and G3 MB groups have shown high levels of poliovirus receptor CD155, and a rhinovirus recombinant form of polio (PVSRIPO) is currently under phase I testing in patients with brain tumors including MB (NCT03043391) [Citation206].
The increasing number of clinical trials using several immunotherapy strategies for the treatment of CNS tumors, underlines how the deep characterization of the microenvironmental phenotypes is fundamental in these malignancies for the development of more specific and effective immune-based opportunities. In particular, in the context of MB, in which the molecular classification seems to be related also to subgroup-specific immune response and different strategies of immune escape, the study of molecular mechanisms related to immune system machinery is a critical point for the identification of novel druggable targets. Recently, accordant to this scenario, it has emerged the role of ERAP1, a known key regulator of innate and adaptive antitumor immune responses, as positive player of the HH signaling pathway. Indeed, ERAP1 is able to promote βTrCP degradation through the binding with the deubiquitylase enzyme USP47, resulting in GLI transcription factors modulation and enhancement of the HH activity. Pharmacological inhibition of ERAP1 drastically reduced SHH-MB growth in orthotopic and Patients derived xenograft (PDX) mouse models [Citation36]. These findings open the way for targeting ERAP1 in SHH-driven MBs in order to suppress tumor growth both by blocking cell proliferation and making tumor cells more susceptible to immune system.
3. Conclusion
Although MB is one of the most common malignancies of the CNS, a definitive cure is elusive. Several therapeutic strategies for SHH-MB are under evaluation in preclinical and clinical studies. Significant progress has been made in the development of HH inhibitors that target the SMO receptor. However, only few of these inhibitors have begun clinical trials, and those that have been clinically approved (vismodegib, sonidegib and glasdegib) do not include treatment for MB. Indeed, numerous limitations and drug-resistance issues have hindered the efficacy and safety of these drugs when translated to humans. Furthermore, the limited enrollment of pediatric SHH-MB patients precludes a robust conclusion on treatment assessments. Targeting the downstream transcription factors GLI, or other players involved in HH activity modulation, is an emerging approach with great potential. The extensive molecular investigations conducted recently have clarified and highlighted the deep heterogeneity of MB; its advanced molecular subclassification will offer new perspectives on therapy.
4. Expert opinion
The HH pathway is considered an attractive therapeutic target for various solid and hematologic tumors, especially for MB. However, among the FDA-approved HH inhibitors, only vismodegib and sonidegib have been investigated and are ongoing into clinical phase I/II for recurrent MB. Results from these studies highlight that only a group of patients would benefit from their clinical development. The variable response to SMO inhibitors is strikingly linked to specific HH pathway gene mutations that differ according to patient age at diagnosis and molecular subtypes [Citation55,Citation56]. Indeed, tumors with PTCH1 mutations were sensitive to SMO inhibitors, whereas no beneficial response was observed in patients with SUFU alterations or TP53 mutations in concomitance with MYCN and GLI2 amplifications. Moreover, the severe skeletal defects observed in young patients under treatment with SMO antagonists have narrowed their administration only in adults [Citation59,Citation207]. Children treated with vismodegib in early phase clinical studies developed widespread growth plate fusions that also continue after cessation of therapy. This adverse effect is one of the main reasons why the interest in inhibiting the HH pathway in MB has been reduced. Clinical development of SMO antagonists has also been restricted due to low selectivity on cancer stem cells, the emergence of drug-resistance and the downstream SMO pathway activation. Mechanisms of resistance to SMO inhibition involve the activation of alternative oncogenic pathways that directly impinge GLI activities (i.e. PI3K-mTOR, BRD4 and PDE4D signaling [Citation34,Citation76,Citation118,Citation208,Citation209], leading to a positive clinical response only for some patients.
The development of GLI1 and GLI2 inhibitors represents a concrete opportunity to overcome the pitfalls of the existing therapeutic approaches to treat SHH-MB. The efficacy of GANT61 and ATO observed in preclinical and clinical settings, strongly supports the translation of GLI inhibitors in clinical practice. However, their limited potency and BBB permeability restrain their use in MB. Future efforts should be focalized in the design and development of next generation of GLI inhibitors, more specific and with optimal druglike and pharmacokinetic properties. In this regard, natural products could offer a valuable alternative given their potential as a unique source of remedies and medicines since ancient times, and their interesting activity as HH inhibitors. It is expected that in the next years, the development of these agents will reach the clinical phase, also thanks to the interest by pharmaceutical companies in continuing investing in HH inhibitors. Given the crucial role of GLI proteins in tumor onset, progression, metastases, and CSCs maintenance, hopes for GLI inhibitors stem from the fact that these drugs would not only be beneficial to treat primary but also secondary tumors. Nevertheless, it is important to keep in mind the common failure of the single agent-based therapies in cancer. A single-cell RNA-seq performed to analyze cellular diversity in MB show that even in tumors with a single pathway-activating mutation, diverse mechanisms drive tumor growth. This diversity confers early resistance to vismodegib, demonstrating the need to target multiple pathways simultaneously [Citation210].
Therefore, a huge clinical impact is expected by multitargeting approaches. Hitting the HH pathway both at upstream and downstream level and/or alternative routes leading to HH activation represents a valuable attempt for a better clinical practice [Citation14,Citation166,Citation167]. Design and optimization of small molecules able to target simultaneously SMO and GLI [Citation167] represent a promising strategy.
With the aim to design more effective biological therapies, many other aspects should be taken under consideration, such as the tumor microenvironment, the role of immune system and the bypassing of the BBB. For this purpose, the use of nanoparticles or polymeric micelles carrying small molecules would result in effective treatment of MB and would avoid the use of organic solvents or pharmaceutical excipients, which may elicit toxic effects.
The difficulties encountered in the use of HH inhibitors in MB treatment should not discourage the research in this direction. Multi-omics analysis highlighted a substantial biological MB heterogeneity, but also confirmed the high percentage of patients harboring mutations in key components of HH signaling in SHH-MB (about 43% of PTCH mutations), indicating that SMO receptor, as well as GLI factors, have to still be considered as targets for MB therapy. Interestingly, a recent review on the current available phase I and II clinical data of vismodegib and sonidegib reported that both drugs were well tolerated and demonstrated antitumor activity in SHH-MB over than non-SHH-MB subgroups [Citation211]. This report highlights the need to identify SHH-MB patients with mutation upstream of PTCH that respond to vismodegib and sonidegib and stratify SHH-MB patients for treatment.
Tremendous progress has been made; however, considerable efforts and preclinical evaluation are still required. Further extensive -omics analysis on larger patient cohorts will allow the identification of novel driver and/or cooperating genes capable of promoting tumorigenesis, thus giving the opportunity to unveil novel potential therapeutic targets for the development of tailored MB treatments to increase cure rate and to improve quality of life of MB patients.
Article highlights
Medulloblastoma (MB) is the most common pediatric brain tumor for which a definitive cure is elusive. Sonic Hedgehog medulloblastoma subgroup (SHH-MB) is the most characterized molecular variant and represents ~30% of all MBs.
Hedgehog (HH) signaling is a therapeutic target in SHH-MB subgroup because its aberrant activation is involved in the tumorigenesis.
Vismodegib and sonidegib are the only SMO antagonists to enter clinical trials for SHH-MB but resulted in several side effects and SMO drug-resistance mutations.
The inhibition of the HH pathway at the downstream level acting on GLI1 could represent a valid therapeutic option to overcome the limitations of SMO antagonists.
Multitargeted therapies and immunotherapy strategies are promising platforms for the treatment of SHH-MB.
Extensive -omics analysis on larger patient cohorts will allow the identification of novel driver and/or cooperating genes capable of promoting tumorigenesis, thus providing the opportunity to unveil new potential therapeutic targets for the development of tailored MB treatments.
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001 Dec;15(23):3059–3087.
- Ruiz I Altaba A, Sánchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer. 2002 May;2(5):361–372.
- Petrova R, Joyner AL. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development. 2014 Sep;141(18):3445–3457.
- Skoda AM, Simovic D, Karin V, et al. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn J Basic Med Sci. 2018 Feb;18(1):8–20. DOI:10.17305/bjbms.2018.2756.
- Teglund S, Toftgård R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 2010 Apr;1805(2):181–208.
- Ingallina C, Costa PM, Ghirga F, et al. Polymeric glabrescione B nanocapsules for passive targeting of Hedgehog-dependent tumor therapy in vitro. Nanomedicine (Lond). 2017 Apr;12(7):711–728. DOI:10.2217/nnm-2016-0388.
- D’Alessandro G, Quaglio D, Monaco L, et al. H-NMR metabolomics reveals the Glabrescione B exacerbation of glycolytic metabolism beside the cell growth inhibitory effect in glioma. Cell Commun Signal. 2019 08; 17(1): 108. Doi:10.1186/s12964-019-0421-8.
- Girardi D, Barrichello A, Fernandes G, et al. Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives. Cells. 2019 02;8:2. DOI:10.3390/cells8020153
- Kogerman P, Grimm T, Kogerman L, et al. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999 Sep;1(5):312–319. DOI:10.1038/13031.
- Infante P, Faedda R, Bernardi F, et al. Itch/β-arrestin2-dependent non-proteolytic ubiquitylation of SuFu controls Hedgehog signalling and medulloblastoma tumorigenesis. Nat Commun. 2018 03; 9(1): 976. Doi:10.1038/s41467-018-03339-0.
- Lee RT, Zhao Z, Ingham PW. Hedgehog signalling. Development. 2016 Feb;143(3):367–372.
- Zhang J, Tian XJ, Xing J. Signal Transduction Pathways of EMT Induced by TGF-β, SHH, and WNT and Their Crosstalks. J Clin Med.. 2016 Mar;5(4):4. DOI:10.3390/jcm5040041
- Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013 Jul;14(7):416–429.
- Pietrobono S, Gagliardi S, Stecca B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front Genet. 2019;10:556.
- Di Magno L, Coni S, Di Marcotullio L, et al. Digging a hole under Hedgehog: downstream inhibition as an emerging anticancer strategy. Biochim Biophys Acta. 2015 Aug;1856(1):62–72.
- Wang Y, Jin G, Li Q, et al. Hedgehog signaling non-canonical activated by pro-inflammatory cytokines in pancreatic ductal adenocarcinoma. J Cancer. 2016;7(14):2067–2076. DOI:10.7150/jca.15786.
- Colavito SA, Zou MR, Yan Q, et al. Significance of glioma-associated oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway. Breast Cancer Res. 2014 Sep;16(5):444. DOI:10.1186/s13058-014-0444-4.
- Wang Y, Ding Q, Yen CJ, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 2012 Mar;21(3):374–387. DOI:10.1016/j.ccr.2011.12.028.
- Northcott PA, Robinson GW, Kratz CP, et al. Medulloblastoma. Nat Rev Dis Primers. 2019 02;5(1):11.
- Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011 Apr;29(11):1408–1414. DOI:10.1200/JCO.2009.27.4324.
- Ramaswamy V, Taylor MD. Medulloblastoma: from myth to molecular. J Clin Oncol. 2017 Jul;35(21):2355–2363.
- Hovestadt V, Ayrault O, Swartling FJ, et al. Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat Rev Cancer. 2020 Jan;20(1):42–56. DOI:10.1038/s41568-019-0223-8.
- Northcott PA, Buchhalter I, Morrissy AS, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017 07;547(7663):311–317. .
- Kool M, Jones DT, Jäger N, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014 Mar;25(3):393–405. DOI:10.1016/j.ccr.2014.02.004.
- Roussel MF, Robinson GW. Role of MYC in medulloblastoma. Cold Spring Harbor Perspect Med. 2013 Nov;3(11):11.
- Infante P, Alfonsi R, Ingallina C, et al. Inhibition of Hedgehog-dependent tumors and cancer stem cells by a newly identified naturally occurring chemotype. Cell Death Dis. 2016 09; 7(9): e2376. Doi:10.1038/cddis.2016.195.
- Ghirga F, Mori M, Infante P. Current trends in Hedgehog signaling pathway inhibition by small molecules. Bioorg Med Chem Lett. 2018 10;28(19):3131–3140.
- Palermo R, Ghirga F, Piccioni MG, et al. Natural products inspired modulators of cancer stem cells-specific signaling pathways notch and hedgehog. Curr Pharm Des. 2018;24(36):4251–4269. DOI:10.2174/1381612825666190111124822.
- Quaglio D, Infante P, Di Marcotullio L, et al. Hedgehog signaling pathway inhibitors: an updated patent review (2015-present). Expert Opin Ther Pat. 2020 Apr;30(4):235–250. DOI:10.1080/13543776.2020.1730327.
- Gajjar A, Stewart CF, Ellison DW, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res. 2013 Nov;19(22):6305–6312. DOI:10.1158/1078-0432.CCR-13-1425.
- Kieran MW, Chisholm J, Casanova M, et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. 2017 Oct;19(11):1542–1552.
- Minami H, Ando Y, Ma BB, et al. Phase I, multicenter, open-label, dose-escalation study of sonidegib in Asian patients with advanced solid tumors. Cancer Sci. 2016 Oct;107(10):1477–1483. DOI:10.1111/cas.13022.
- Liu X, Ding C, Tan W, et al. Medulloblastoma: Molecular understanding, treatment evolution, and new developments. Pharmacol Ther. 2020 Jun;210:107516.
- Buonamici S, Williams J, Morrissey M, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010 Sep;2(51):51ra70. DOI:10.1126/scitranslmed.3001599.
- Spiombi E, Angrisani A, Fonte S, et al. KCTD15 inhibits the Hedgehog pathway in Medulloblastoma cells by increasing protein levels of the oncosuppressor KCASH2. Oncogenesis. 2019 Nov;8(11):64. DOI:10.1038/s41389-019-0175-6.
- Bufalieri F, Infante P, Bernardi F, et al. ERAP1 promotes Hedgehog-dependent tumorigenesis by controlling USP47-mediated degradation of βTrCP. Nat Commun. 2019 Jul;10(1):3304. DOI:10.1038/s41467-019-11093-0.
- Ostrom QT, Gittleman H, Truitt G, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 2018 10; 20(suppl_4): iv1–iv86. Doi:10.1093/neuonc/noy131.
- Louis DN, Perry A, Reifenberger G, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016 06; 131(6): 803–820. Doi:10.1007/s00401-016-1545-1.
- Ruiz I Altaba A, Palma V, Dahmane N. Hedgehog-gli signalling and the growth of the brain. Nat Rev Neurosci. 2002 Jan;3(1):24–33.
- Northcott PA, Rutka JT, Taylor MD. Genomics of medulloblastoma: from Giemsa-banding to next-generation sequencing in 20 years. Neurosurg Focus. 2010 Jan;28(1):E6.
- Di Marcotullio L, Ferretti E, De Smaele E, et al. REN(KCTD11) is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma. Proc Natl Acad Sci U S A. 2004 Jul;101(29):10833–10838. DOI:10.1073/pnas.0400690101.
- Wolter M, Reifenberger J, Sommer C, et al. Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1997 Jul;57(13):2581–2585.
- Taylor MD, Liu L, Raffel C, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002 Jul;31(3):306–310. DOI:10.1038/ng916.
- Lam CW, Xie J, To KF, et al. A frequent activated smoothened mutation in sporadic basal cell carcinomas. Oncogene. 1999 Jan;18(3):833–836. DOI:10.1038/sj.onc.1202360.
- Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012 Jan;148(1–2):59–71. DOI:10.1016/j.cell.2011.12.013.
- Snuderl M, Triscott J, Northcott PA, et al. Deep sequencing identifies IDH1 R132S mutation in adult medulloblastoma. J Clin Oncol. 2015 Feb;33(6):e27–31. DOI:10.1200/JCO.2013.49.4864.
- Remke M, Ramaswamy V, Peacock J, et al. TERT promoter mutations are highly recurrent in SHH subgroup medulloblastoma. Acta Neuropathol. 2013 Dec;126(6):917–929. DOI:10.1007/s00401-013-1198-2.
- Goodrich LV, Milenković L, Higgins KM, et al. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997 Aug;277(5329):1109–1113. DOI:10.1126/science.277.5329.1109.
- Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001 Jan;61(2):513–516.
- Yang ZJ, Ellis T, Markant SL, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008 Aug;14(2):135–145. DOI:10.1016/j.ccr.2008.07.003.
- Hatton BA, Villavicencio EH, Tsuchiya KD, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008 Mar;68(6):1768–1776. DOI:10.1158/0008-5472.CAN-07-5092.
- Schüller U, Heine VM, Mao J, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008 Aug;14(2):123–134. DOI:10.1016/j.ccr.2008.07.005.
- Swartling FJ, Grimmer MR, Hackett CS, et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010 May;24(10):1059–1072. DOI:10.1101/gad.1907510.
- Pöschl J, Bartels M, Ohli J, et al. Wnt/β-catenin signaling inhibits the Shh pathway and impairs tumor growth in Shh-dependent medulloblastoma. Acta Neuropathol. 2014 Apr;127(4):605–607. DOI:10.1007/s00401-014-1258-2.
- Cavalli FMG, Remke M, Rampasek L, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017 06;31(6):737–754.e6. DOI:10.1016/j.ccell.2017.05.005 .
- Robinson GW, Rudneva VA, Buchhalter I, et al. Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol. 2018 06; 19(6): 768–784. Doi:10.1016/S1470-2045(18)30204-3.
- Ramaswamy V, Remke M, Bouffet E, et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 2016 06; 131(6): 821–831. Doi:10.1007/s00401-016-1569-6.
- Schwalbe EC, Lindsey JC, Nakjang S, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol. 2017 07; 18(7): 958–971. Doi:10.1016/S1470-2045(17)30243-7.
- Robinson GW, Kaste SC, Chemaitilly W, et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget. 2017 Sep;8(41):69295–69302.
- Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev. 2014 Jul;40(6):750–759.
- Pandolfi S, Stecca B. Cooperative integration between HEDGEHOG-GLI signalling and other oncogenic pathways: implications for cancer therapy. Expert Rev Mol Med.. 2015 Feb 17:e5. doi:10.1017/erm.2015.3.
- Hartmann W, Digon-Söntgerath B, Koch A, et al. Phosphatidylinositol 3ʹ-kinase/AKT signaling is activated in medulloblastoma cell proliferation and is associated with reduced expression of PTEN. Clin Cancer Res. 2006 May;12(10):3019–3027. DOI:10.1158/1078-0432.CCR-05-2187.
- Castellino RC, Barwick BG, Schniederjan M, et al. Heterozygosity for Pten promotes tumorigenesis in a mouse model of medulloblastoma. PLoS One. 2010 May;5(5):e10849. DOI:10.1371/journal.pone.0010849.
- Chaturvedi NK, Kling MJ, Coulter DW, et al. Improved therapy for medulloblastoma: targeting hedgehog and PI3K-mTOR signaling pathways in combination with chemotherapy. Oncotarget. 2018 Mar;9(24):16619–16633. DOI:10.18632/oncotarget.24618.
- Antonucci L, Di Magno L, D’Amico D, et al. Mitogen-activated kinase kinase kinase 1 inhibits hedgehog signaling and medulloblastoma growth through GLI1 phosphorylation. Int J Oncol. 2019 Feb;54(2):505–514.
- Lu J, Liu L, Zheng M, et al. MEKK2 and MEKK3 suppress Hedgehog pathway-dependent medulloblastoma by inhibiting GLI1 function. Oncogene. 2018 07; 37(28): 3864–3878. Doi:10.1038/s41388-018-0249-5.
- Fogarty MP, Emmenegger BA, Grasfeder LL, et al. Fibroblast growth factor blocks Sonic hedgehog signaling in neuronal precursors and tumor cells. Proc Natl Acad Sci USA. 2007 Feb;104(8):2973–2978. DOI:10.1073/pnas.0605770104.
- Li YH, Luo J, Mosley YY, et al. AMP-activated protein kinase directly phosphorylates and destabilizes hedgehog pathway transcription factor GLI1 in medulloblastoma. Cell Rep. 2015 Jul;12(4):599–609. DOI:10.1016/j.celrep.2015.06.054.
- Di Magno L, Basile A, Coni S, et al. The energy sensor AMPK regulates Hedgehog signaling in human cells through a unique Gli1 metabolic checkpoint. Oncotarget. 2016 Feb;7(8):9538–9549. DOI:10.18632/oncotarget.7070.
- Zhang R, Huang SY, Ka-Wai Li K, et al. Dual degradation signals destruct GLI1: AMPK inhibits GLI1 through β-TrCP-mediated proteasome degradation. Oncotarget. 2017 Jul;8(30):49869–49881. DOI:10.18632/oncotarget.17769.
- D’Amico D, Antonucci L, Di Magno L, et al. Non-canonical Hedgehog/AMPK-mediated control of polyamine metabolism supports neuronal and medulloblastoma cell growth. Dev Cell. 2015 Oct;35(1):21–35. DOI:10.1016/j.devcel.2015.09.008.
- Zinke J, Schneider FT, Harter PN, et al. β-Catenin-Gli1 interaction regulates proliferation and tumor growth in medulloblastoma. Mol Cancer. 2015;14:17.
- Taylor MD, Zhang X, Liu L, et al. Failure of a medulloblastoma-derived mutant of SUFU to suppress WNT signaling. Oncogene. 2004 Jun;23(26):4577–4583. DOI:10.1038/sj.onc.1207605.
- Ge X, Milenkovic L, Suyama K, et al. Phosphodiesterase 4D acts downstream of Neuropilin to control Hedgehog signal transduction and the growth of medulloblastoma. eLife. 2015;4:e07068. DOI:10.7554/eLife.07068
- Hillman RT, Feng BY, Ni J, et al. Neuropilins are positive regulators of Hedgehog signal transduction. Genes Dev. 2011 Nov;25(22):2333–2346. DOI:10.1101/gad.173054.111.
- Williams CH, Hempel JE, Hao J, et al. An in vivo chemical genetic screen identifies phosphodiesterase 4 as a pharmacological target for hedgehog signaling inhibition. Cell Rep. 2015 Apr;11(1):43–50. DOI:10.1016/j.celrep.2015.03.001.
- Schulte G, Bryja V. The Frizzled family of unconventional G-protein-coupled receptors. Trends Pharmacol Sci. 2007 Oct;28(10):518–525. DOI:10.1016/j.tips.2007.09.001
- Kieran MW. Targeted treatment for sonic hedgehog-dependent medulloblastoma. Neuro Oncol. 2014 Aug;16(8):1037–1047.
- Yauch RL, Dijkgraaf GJ, Alicke B, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009 Oct;326(5952):572–574. DOI:10.1126/science.1179386.
- Kumar V, Chaudhary AK, Dong Y, et al. Design, synthesis and biological evaluation of novel hedgehog inhibitors for treating pancreatic cancer. Sci Rep. 2017 05; 7(1): 1665. Doi:10.1038/s41598-017-01942-7.
- Pietrobono S, Santini R, Gagliardi S, et al. Targeted inhibition of Hedgehog-GLI signaling by novel acylguanidine derivatives inhibits melanoma cell growth by inducing replication stress and mitotic catastrophe. Cell Death Dis. 2018 02; 9(2): 142. Doi:10.1038/s41419-017-0142-0.
- Vesci L, Milazzo FM, Stasi MA, et al. Hedgehog pathway inhibitors of the acylthiourea and acylguanidine class show antitumor activity on colon cancer in vitro and in vivo. Eur J Med Chem.. 2018 Sep;157:368–379. DOI:10.1016/j.ejmech.2018.07.053
- Williams JA, Guicherit OM, Zaharian BI, et al. Identification of a small molecule inhibitor of the hedgehog signaling pathway: effects on basal cell carcinoma-like lesions. Proc Natl Acad Sci USA. 2003 Apr;100(8):4616–4621. DOI:10.1073/pnas.0732813100.
- Cooper MK, Porter JA, Young KE, et al. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998 Jun;280(5369):1603–1607. DOI:10.1126/science.280.5369.1603.
- Taipale J, Chen JK, Cooper MK, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000 Aug;406(6799):1005–1009. DOI:10.1038/35023008.
- Robarge KD, Brunton SA, Castanedo GM, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett. 2009 Oct;19(19):5576–5581. DOI:10.1016/j.bmcl.2009.08.049.
- Ishii T, Shimizu Y, Nakashima K, et al. Inhibition mechanism exploration of investigational drug TAK-441 as inhibitor against Vismodegib-resistant Smoothened mutant. Eur J Pharmacol. 2014 Jan;723:305–313.
- Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009 Sep;361(12):1173–1178. DOI:10.1056/NEJMoa0902903.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012 Jun;366(23):2171–2179. DOI:10.1056/NEJMoa1113713.
- Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009 Sep;361(12):1164–1172. DOI:10.1056/NEJMoa0905360.
- LoRusso PM, Rudin CM, Reddy JC, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011 Apr;17(8):2502–2511. DOI:10.1158/1078-0432.CCR-10-2745.
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med. 2012 Jun;366(23):2180–2188. DOI:10.1056/NEJMoa1113538.
- Danhof R, Lewis K, Brown M. Small molecule inhibitors of the hedgehog pathway in the treatment of basal cell carcinoma of the skin. Am J Clin Dermatol. 2018 Apr;19(2):195–207.
- Basset-Seguin N, Hauschild A, Grob JJ, et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): a pre-planned interim analysis of an international, open-label trial. Lancet Oncol. 2015 Jun;16(6):729–736. DOI:10.1016/S1470-2045(15)70198-1.
- Robinson GW, Orr BA, Wu G, et al. Vismodegib Exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol. 2015 Aug;33(24):2646–2654.
- Lou E, Nelson AC, Kool M. Differential response of SHH-expressing adult medulloblastomas to the sonic hedgehog inhibitor vismodegib: whole-genome analysis. Cancer Biol Ther. 2019;20(11):1398–1402.
- Pan S, Wu X, Jiang J, et al. Discovery of NVP-LDE225, a potent and selective smoothened antagonist. ACS Med Chem Lett. 2010 Jun;1(3):130–134. DOI:10.1021/ml1000307.
- Hoch L, Faure H, Roudaut H, et al. MRT-92 inhibits Hedgehog signaling by blocking overlapping binding sites in the transmembrane domain of the Smoothened receptor. FASEB J. 2015 May;29(5):1817–1829. DOI:10.1096/fj.14-267849.
- Roudaut H, Traiffort E, Gorojankina T, et al. Identification and mechanism of action of the acylguanidine MRT-83, a novel potent Smoothened antagonist. Mol Pharmacol. 2011 Mar;79(3):453–460. DOI:10.1124/mol.110.069708.
- Solinas A, Faure H, Roudaut H, et al. Acylthiourea, acylurea, and acylguanidine derivatives with potent hedgehog inhibiting activity. J Med Chem. 2012 Feb;55(4):1559–1571. DOI:10.1021/jm2013369.
- Lv H, Ren J, Ma S, et al. Synthesis, biological evaluation and mechanism studies of deoxytylophorinine and its derivatives as potential anticancer agents. PLoS One. 2012;7(1):e30342. DOI:10.1371/journal.pone.0030342.
- Chen J, Lv H, Hu J, et al. CAT3, a novel agent for medulloblastoma and glioblastoma treatment, inhibits tumor growth by disrupting the Hedgehog signaling pathway. Cancer Lett. 2016 10; 381(2): 391–403. Doi:10.1016/j.canlet.2016.07.030.
- Filocamo G, Brunetti M, Colaceci F, et al. MK-4101, a potent inhibitor of the hedgehog pathway, is highly active against medulloblastoma and basal cell carcinoma. Mol Cancer Ther. 2016 06; 15(6): 1177–1189. Doi:10.1158/1535-7163.MCT-15-0371.
- Kremer L, Schultz-Fademrecht C, Baumann M, et al. Discovery of a novel inhibitor of the hedgehog signaling pathway through cell-based compound discovery and target prediction. Angew Chem Int Ed Engl. 2017 10; 56(42): 13021–13025. Doi:10.1002/anie.201707394.
- Zhu M, Wang H, Wang C, et al. L-4, a Well-Tolerated and Orally Active Inhibitor of Hedgehog Pathway, Exhibited Potent Anti-tumor Effects Against Medulloblastoma. Front Pharmacol. 2019;10:89.
- Chahal KK, Li J, Kufareva I, et al. Nilotinib, an approved leukemia drug, inhibits smoothened signaling in Hedgehog-dependent medulloblastoma. PLoS One. 2019;14(9):e0214901. DOI:10.1371/journal.pone.0214901.
- Weierstall U, James D, Wang C, et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat Commun. 2014;5(1):3309. DOI:10.1038/ncomms4309.
- Liu G, Xue D, Yang J, et al. Design, synthesis, and pharmacological evaluation of 2-(2,5-Dimethyl-5,6,7,8-tetrahydroquinolin-8-yl)-N-aryl propanamides as novel smoothened (smo) antagonists. J Med Chem. 2016 12; 59(24): 11050–11068. Doi:10.1021/acs.jmedchem.6b01247.
- Tao H, Jin Q, Koo DI, et al. Small molecule antagonists in distinct binding modes inhibit drug-resistant mutant of smoothened. Chem Biol. 2011 Apr;18(4):432–437. DOI:10.1016/j.chembiol.2011.01.018.
- Kim J, Tang JY, Gong R, et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010 Apr;17(4):388–399. DOI:10.1016/j.ccr.2010.02.027.
- Kim J, Aftab BT, Tang JY, et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013 Jan;23(1):23–34. DOI:10.1016/j.ccr.2012.11.017.
- Antonarakis ES, Heath EI, Smith DC, et al. Repurposing itraconazole as a treatment for advanced prostate cancer: a noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer. Oncologist. 2013;18(2):163–173. DOI:10.1634/theoncologist.2012-314.
- Lee M, Hong H, Kim W, et al. Itraconazole as a noncastrating treatment for biochemically recurrent prostate cancer: A phase 2 study. Clin Genitourin Cancer. 2019 Feb;17(1):e92–e96. DOI:10.1016/j.clgc.2018.09.013.
- Dirix L, Swaisland H, Verheul HM, et al. Effect of Itraconazole and rifampin on the pharmacokinetics of olaparib in patients with advanced solid tumors: results of two phase I open-label studies. Clin Ther. 2016 Oct;38(10):2286–2299. DOI:10.1016/j.clinthera.2016.08.010.
- Rudin CM, Brahmer JR, Juergens RA, et al. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J Thorac Oncol. 2013 May;8(5):619–623. DOI:10.1097/JTO.0b013e31828c3950.
- Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010 Dec;468(7327):1067–1073. DOI:10.1038/nature09504.
- Yang Z, Yik JH, Chen R, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005 Aug;19(4):535–545. DOI:10.1016/j.molcel.2005.06.029.
- Tang Y, Gholamin S, Schubert S, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014 Jul;20(7):732–740. DOI:10.1038/nm.3613.
- Wang Q, Kumar V, Lin F, et al. ApoE mimetic peptide targeted nanoparticles carrying a BRD4 inhibitor for treating Medulloblastoma in mice. JControlled Release. 2020;323:463–474.
- Long J, Li B, Rodriguez-Blanco J, et al. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J Biol Chem. 2014 Dec;289(51):35494–35502. DOI:10.1074/jbc.M114.595348.
- Li B, Flaveny CA, Giambelli C, et al. Repurposing the FDA-approved pinworm drug pyrvinium as a novel chemotherapeutic agent for intestinal polyposis. PLoS One. 2014;9(7):e101969. DOI:10.1371/journal.pone.0101969.
- Li B, Fei DL, Flaveny CA, et al. Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer Res. 2014 Sep;74(17):4811–4821. DOI:10.1158/0008-5472.CAN-14-0317.
- Ruiz I Altaba A, Mas C, Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007 Sep;17(9):438–447.
- Rodriguez-Blanco J, Li B, Long J, et al. A CK1α activator penetrates the brain and shows efficacy against drug-resistant metastatic medulloblastoma. Clin Cancer Res. 2019 02; 25(4): 1379–1388. Doi:10.1158/1078-0432.CCR-18-1319.
- Purzner T, Purzner J, Buckstaff T, et al. Developmental phosphoproteomics identifies the kinase CK2 as a driver of Hedgehog signaling and a therapeutic target in medulloblastoma. Sci Signal. 2018 09; 11(547): 547. DOI:10.1126/scisignal.aau5147.
- Liu F, Jiang W, Sui Y, et al. CDK7 inhibition suppresses aberrant hedgehog pathway and overcomes resistance to smoothened antagonists. Proc Natl Acad Sci U S A. 2019 06; 116(26): 12986–12995. Doi:10.1073/pnas.1815780116.
- Cook Sangar ML, Genovesi LA, Nakamoto MW, et al. Inhibition of CDK4/6 by palbociclib significantly extends survival in medulloblastoma patient-derived xenograft mouse models. Clin Cancer Res. 2017 Oct;23(19):5802–5813. DOI:10.1158/1078-0432.CCR-16-2943.
- Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–1438.
- Gruber W, Hutzinger M, Elmer DP, et al. DYRK1B as therapeutic target in Hedgehog/GLI-dependent cancer cells with Smoothened inhibitor resistance. Oncotarget. 2016 Feb;7(6):7134–7148. DOI:10.18632/oncotarget.6910.
- Canettieri G, Di Marcotullio L, Coni S, et al. Turning off the switch in medulloblastoma: the inhibitory acetylation of an oncogene. Cell Cycle. 2010 Jun;9(11):2047–2048. DOI:10.4161/cc.9.11.11860.
- De Smaele E, Di Marcotullio L, Moretti M, et al. Identification and characterization of KCASH2 and KCASH3, 2 novel Cullin3 adaptors suppressing histone deacetylase and Hedgehog activity in medulloblastoma. Neoplasia. 2011 Apr;13(4):374–385. DOI:10.1593/neo.101630.
- Canettieri G, Di Marcotullio L, Greco A, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010 Feb;12(2):132–142. DOI:10.1038/ncb2013.
- Coni S, Antonucci L, D’Amico D, et al. Gli2 acetylation at lysine 757 regulates hedgehog-dependent transcriptional output by preventing its promoter occupancy. PLoS One. 2013;8(6):e65718. DOI:10.1371/journal.pone.0065718.
- Coni S, Mancuso AB, Di Magno L, et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci Rep. 2017 03; 7(1): 44079. DOI:10.1038/srep44079.
- Dhanyamraju PK, Holz PS, Finkernagel F, et al. Histone deacetylase 6 represents a novel drug target in the oncogenic Hedgehog signaling pathway. Mol Cancer Ther. 2015 Mar;14(3):727–739. DOI:10.1158/1535-7163.MCT-14-0481.
- Messerli SM, Hoffman MM, Gnimpieba EZ, et al. 4SC-202 as a potential treatment for the pediatric brain tumor medulloblastoma. Brain Sci. 2017 Nov;7(12):11. DOI:10.3390/brainsci7110147.
- Gruber W, Peer E, Elmer DP, et al. Targeting class I histone deacetylases by the novel small molecule inhibitor 4SC-202 blocks oncogenic hedgehog-GLI signaling and overcomes smoothened inhibitor resistance. Int J Cancer. 2018 03; 142(5): 968–975. Doi:10.1002/ijc.31117.
- Lea MA, Rasheed M, Randolph VM, et al. Induction of histone acetylation and inhibition of growth of mouse erythroleukemia cells by S-allylmercaptocysteine. Nutr Cancer. 2002;43(1):90–102. DOI:10.1207/S15327914NC431_11.
- Ververis K, Hiong A, Karagiannis TC, et al. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics. 2013;7:47–60.
- Singleton WGB, Bienemann AS, Woolley M, et al. The distribution, clearance, and brainstem toxicity of panobinostat administered by convection-enhanced delivery. J Neurosurg Pediatr. 2018 09; 22(3): 288–296. Doi:10.3171/2018.2.PEDS17663.
- Pollak M. Potential applications for biguanides in oncology. J Clin Invest. 2013 Sep;123(9):3693–3700.
- Moreno D, Knecht E, Viollet B, et al. A769662, a novel activator of AMP-activated protein kinase, inhibits non-proteolytic components of the 26S proteasome by an AMPK-independent mechanism. FEBS Lett. 2008 Jul;582(17):2650–2654. DOI:10.1016/j.febslet.2008.06.044.
- Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf).. 2009 May;196(1):65–80. DOI:10.1111/j.1748-1716.2009.01972.x
- Di Magno L, Manni S, Di Pastena F, et al. Phenformin inhibits hedgehog-dependent tumor growth through a complex i-independent redox/corepressor module. Cell Rep. 2020 Feb;30(6):1735–1752.e7. DOI:10.1016/j.celrep.2020.01.024.
- Lauth M, Bergström A, Shimokawa T, et al. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci USA. 2007 May;104(20):8455–8460. DOI:10.1073/pnas.0609699104.
- Agyeman A, Jha BK, Mazumdar T, et al. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget. 2014 Jun;5(12):4492–4503. DOI:10.18632/oncotarget.2046.
- Calcaterra A, Iovine V, Botta B, et al. Chemical, computational and functional insights into the chemical stability of the Hedgehog pathway inhibitor GANT61. J Enzyme Inhib Med Chem. 2018 Dec;33(1):349–358. DOI:10.1080/14756366.2017.1419221.
- Lin Z, Li S, Sheng H, et al. Suppression of GLI sensitizes medulloblastoma cells to mitochondria-mediated apoptosis. J Cancer Res Clin Oncol.. 2016 Dec;142(12):2469–2478. DOI:10.1007/s00432-016-2241-1.
- Konings K, Vandevoorde C, Belmans N, et al. The combination of particle irradiation with the hedgehog inhibitor GANT61 differently modulates the radiosensitivity and migration of cancer cells compared to X-Ray irradiation. Front Oncol. 2019;9:391.
- Beauchamp EM, Ringer L, Bulut G, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J Clin Invest. 2011 Jan;121(1):148–160. DOI:10.1172/JCI42874.
- Kim J, Lee JJ, Gardner D, et al. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci U S A. 2010 Jul;107(30):13432–13437. DOI:10.1073/pnas.1006822107.
- Dos Santos Klinger PH, Delsin LEA, Cruzeiro GAV, et al. Arsenic Trioxide exerts cytotoxic and radiosensitizing effects in pediatric Medulloblastoma cell lines of SHH Subgroup. Sci Rep. 2020 Apr;10(1):6836. DOI:10.1038/s41598-020-63808-9.
- Di Magno L, Manzi D, D’Amico D, et al. Druggable glycolytic requirement for Hedgehog-dependent neuronal and medulloblastoma growth. Cell Cycle. 2014;13(21):3404–3413. DOI:10.4161/15384101.2014.952973.
- Chen GQ, Zhu J, Shi XG, et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood. 1996 Aug;88(3):1052–1061. DOI:10.1182/blood.V88.3.1052.1052.
- Li X, Ding X, Adrian TE. Arsenic trioxide causes redistribution of cell cycle, caspase activation, and GADD expression in human colonic, breast, and pancreatic cancer cells. Cancer Invest. 2004;22(3):389–400.
- Wang C, Li B, Zhang H, et al. Effect of arsenic trioxide on uveal melanoma cell proliferation in vitro. Ophthalmic Res. 2007;39(6):302–307. DOI:10.1159/000109985.
- Zhao S, Tsuchida T, Kawakami K, et al. Effect of As2O3 on cell cycle progression and cyclins D1 and B1 expression in two glioblastoma cell lines differing in p53 status. Int J Oncol. 2002 Jul;21(1):49–55.
- Valenzuela M, Glorieux C, Stockis J, et al. Retinoic acid synergizes ATO-mediated cytotoxicity by precluding Nrf2 activity in AML cells. Br J Cancer. 2014 Aug;111(5):874–882. DOI:10.1038/bjc.2014.380.
- Raffoux E, Rousselot P, Poupon J, et al. Combined treatment with arsenic trioxide and all-trans-retinoic acid in patients with relapsed acute promyelocytic leukemia. J Clin Oncol. 2003 Jun;21(12):2326–2334. DOI:10.1200/JCO.2003.01.149.
- Petricci E, Manetti F. Targeting the hedgehog signaling pathway with small molecules from natural sources. Curr Med Chem. 2015;22(35):4058–4090.
- Zaheer K, Humayoun Akhtar M. An updated review of dietary isoflavones: Nutrition, processing, bioavailability and impacts on human health. Crit Rev Food Sci Nutr. 2017 Apr;57(6):1280–1293. DOI:10.1080/10408398.2014.989958
- Hussain H, Green IR. A patent review of the therapeutic potential of isoflavones (2012-2016). Expert Opin Ther Pat. 2017 Oct;27(10):1135–1146.
- Ko KP. Isoflavones: chemistry, analysis, functions and effects on health and cancer. Asian Pac J Cancer Prev. 2014;15(17):7001–7010.
- Pavletich NP, Pabo CO. Crystal structure of a five-finger GLI-DNA complex: new perspectives on zinc fingers. Science. 1993 Sep;261(5129):1701–1707.
- Infante P, Mori M, Alfonsi R, et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015 Jan;34(2):200–217.
- Berardozzi S, Bernardi F, Infante P, et al. Synergistic inhibition of the Hedgehog pathway by newly designed Smo and Gli antagonists bearing the isoflavone scaffold. Eur J Med Chem. 2018 Aug;156:554–562. DOI:10.1016/j.ejmech.2018.07.017
- Lospinoso Severini L, Quaglio D, Basili I, et al. A Smo/Gli multitarget hedgehog pathway inhibitor impairs tumor growth. Cancers (Basel). 2019 Oct;11(10):10. DOI:10.3390/cancers11101518.
- Li XY, Zhou LF, Gao LJ, et al. Cynanbungeigenin C and D, a pair of novel epimers from Cynanchum bungei, suppress hedgehog pathway-dependent medulloblastoma by blocking signaling at the level of Gli. Cancer Lett. 2018 04;420:195–207. DOI:10.1016/j.canlet.2018.02.005
- Manetti F, Stecca B, Santini R, et al. Pharmacophore-based virtual screening for identification of negative modulators of GLI1 as potential anticancer agents. ACS Med Chem Lett. 2020 May;11(5):832–838. DOI:10.1021/acsmedchemlett.9b00639.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015 Jun;372(26):2521–2532. DOI:10.1056/NEJMoa1503093.
- Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016 11;375(19):1823–1833.
- Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015 Nov;373(19):1803–1813. DOI:10.1056/NEJMoa1510665.
- Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017 03;31(3):326–341. DOI:10.1016/j.ccell.2017.02.009.
- Pham CD, Flores C, Yang C, et al. Differential Immune microenvironments and response to immune checkpoint blockade among molecular subtypes of murine medulloblastoma. Clin Cancer Res. 2016 Feb;22(3):582–595. DOI:10.1158/1078-0432.CCR-15-0713.
- Bockmayr M, Mohme M, Klauschen F, et al. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology. 2018;7(9):e1462430. DOI:10.1080/2162402X.2018.1462430.
- Margol AS, Robison NJ, Gnanachandran J, et al. Tumor-associated macrophages in SHH subgroup of medulloblastomas. Clin Cancer Res. 2015 Mar;21(6):1457–1465. DOI:10.1158/1078-0432.CCR-14-1144.
- Majd N, Penas-Prado M. Updates on Management of Adult Medulloblastoma. Curr Treat Options Oncol.. 2019 Jun;20(8):64. DOI:10.1007/s11864-019-0663-0
- Raffaghello L, Nozza P, Morandi F, et al. Expression and functional analysis of human leukocyte antigen class I antigen-processing machinery in medulloblastoma. Cancer Res. 2007 Jun;67(11):5471–5478. DOI:10.1158/0008-5472.CAN-06-4735.
- Kabir TF, Kunos CA, Villano JL, et al. Immunotherapy for medulloblastoma: current perspectives. Immuno Targets Ther. 2020;9:57. DOI:10.2147/ITT.S198162.
- Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011 Jan;331(6013):44–49. DOI:10.1126/science.1198687.
- Seidel UJ, Schlegel P, Lang P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front Immunol. 2013;4:76.
- Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. 2020 Mar;19(3):200–218.
- Kennis BA, Michel KA, Brugmann WB, et al. Correction to: Monitoring of intracerebellarly-administered natural killer cells with fluorine-19 MRI. J Neurooncol. 2019 May;142(3):409. DOI:10.1007/s11060-019-03162-7.
- Castriconi R, Dondero A, Negri F, et al. Both CD133+ and CD133- medulloblastoma cell lines express ligands for triggering NK receptors and are susceptible to NK-mediated cytotoxicity. Eur J Immunol. 2007 Nov;37(11):3190–3196. DOI:10.1002/eji.200737546.
- Fernández L, Portugal R, Valentín J, et al. In vitro natural killer cell immunotherapy for medulloblastoma. Front Oncol. 2013;3:94.
- Mohanty R, Chowdhury CR, Arega S, et al. CAR T cell therapy: A new era for cancer treatment (Review). Oncol Rep. 2019 Dec;42(6):2183–2195.
- Gauthier J, Yakoub-Agha I. Chimeric antigen-receptor T-cell therapy for hematological malignancies and solid tumors: Clinical data to date, current limitations and perspectives. Curr Res Transl Med. 2017 09;65(3):93–102. DOI:10.1016/j.retram.2017.08.003.
- Orentas RJ, Lee DW, Mackall C. Immunotherapy targets in pediatric cancer. Front Oncol. 2012;2:3.
- Fousek K, Ahmed N. The evolution of T-cell therapies for solid malignancies. Clin Cancer Res. 2015 Aug;21(15):3384–3392.
- Ahmed N, Salsman VS, Kew Y, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010 Jan;16(2):474–485. DOI:10.1158/1078-0432.CCR-09-1322.
- Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015 Jan;347(6220):1260419. DOI:10.1126/science.1260419.
- Nellan A, Rota C, Majzner R, et al. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J Immunother Cancer. 2018 04; 6(1): 30. Doi:10.1186/s40425-018-0340-z.
- Donovan LK, Delaidelli A, Joseph SK, et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat Med. 2020;26(5):720–731. DOI:10.1038/s41591-020-0827-2.
- Sharpe AH. Introduction to checkpoint inhibitors and cancer immunotherapy. Immunol Rev. 2017 03;276(1):5–8. DOI:10.1111/imr.12531.
- Topalian SL, Taube JM, Anders RA, et al. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016 05; 16(5): 275–287. Doi:10.1038/nrc.2016.36.
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015 Apr;348(6230):56–61.
- Martin AM, Nirschl CJ, Polanczyk MJ, et al. PD-L1 expression in medulloblastoma: an evaluation by subgroup. Oncotarget. 2018 Apr;9(27):19177–19191.
- Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015 Sep;14(9):642–662.
- Varela-Guruceaga M, Tejada-Solís S, García-Moure M, et al. Oncolytic viruses as therapeutic tools for pediatric brain tumors. Cancers (Basel). 2018 Jul;10(7):226. DOI:10.3390/cancers10070226.
- Yang WQ, Senger D, Muzik H, et al. Reovirus prolongs survival and reduces the frequency of spinal and leptomeningeal metastases from medulloblastoma. Cancer Res. 2003 Jun;63(12):3162–3172.
- Lun XQ, Zhou H, Alain T, et al. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007 Sep;67(18):8818–8827. DOI:10.1158/0008-5472.CAN-07-1214.
- Lacroix J, Schlund F, Leuchs B, et al. Oncolytic effects of parvovirus H-1 in medulloblastoma are associated with repression of master regulators of early neurogenesis. Int J Cancer. 2014 Feb;134(3):703–716. DOI:10.1002/ijc.28386.
- Stolarek R, Gomez-Manzano C, Jiang H, et al. Robust infectivity and replication of Delta-24 adenovirus induce cell death in human medulloblastoma. Cancer Gene Ther. 2004 Nov;11(11):713–720. DOI:10.1038/sj.cgt.7700731.
- Friedman GK, Moore BP, Nan L, et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro Oncol. 2016 Feb;18(2):227–235.
- Studebaker AW, Hutzen BJ, Pierson CR, et al. Oncolytic herpes virus rrp450 shows efficacy in orthotopic xenograft group 3/4 medulloblastomas and atypical teratoid/rhabdoid tumors. Mol Ther Oncolytics. 2017 Sep;6:22–30.
- Thompson EM, Brown M, Dobrikova E, et al. Poliovirus receptor (CD155) expression in pediatric brain tumors mediates oncolysis of medulloblastoma and pleomorphic xanthoastrocytoma. J Neuropathol Exp Neurol. 2018;77(8):696–702. DOI:10.1093/jnen/nly045.
- Kimura H, Ng JM, Curran T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell. 2008 Mar;13(3):249–260.
- Wu F, Zhang Y, Sun B, et al. Hedgehog signaling: From basic biology to cancer therapy. Cell Chem Biol. 2017 Mar;24(3):252–280. DOI:10.1016/j.chembiol.2017.02.010.
- Atwood SX, Li M, Lee A, et al. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature. 2013 Feb;494(7438):484–488. DOI:10.1038/nature11889.
- Ocasio J, Babcock B, Malawsky D, et al. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat Commun. 2019 12; 10(1): 5829. Doi:10.1038/s41467-019-13657-6.
- Li Y, Song Q, Day BW. Phase I and phase II sonidegib and vismodegib clinical trials for the treatment of paediatric and adult MB patients: a systemic review and meta-analysis. Acta Neuropathol Commun. 2019 07;7(1):123. DOI:10.1186/s40478-019-0773-8.