
ABSTRACT
Introduction: Translesion synthesis (TLS) is a DNA damage tolerance (DDT) mechanism that employs error-prone polymerases to bypass replication blocking DNA lesions, contributing to a gain in mutagenesis and chemo-resistance. However, recent findings illustrate an emerging role for TLS in replication gap suppression (RGS), distinct from its role in post-replication gap filling. Here, TLS protects cells from replication stress (RS)-induced toxic single-stranded DNA (ssDNA) gaps that accumulate in the wake of active replication. Intriguingly, TLS-mediated RGS is specifically observed in several cancer cell lines and contributes to their survival. Thus, targeting TLS has the potential to uniquely eradicate tumors without harming non-cancer tissues.
Areas Covered: This review provides an innovative perspective on the role of TLS beyond its canonical function of lesion bypass or post-replicative gap filling. We provide a comprehensive analysis that underscores the emerging role of TLS as a cancer adaptation necessary to overcome the replication stress response (RSR), an anti-cancer barrier.
Expert Opinion: TLS RGS is critical for tumorigenesis and is a new hallmark of cancer. Although the exact mechanism and extent of TLS dependency in cancer is still emerging, TLS inhibitors have shown promise as an anti-cancer therapy in selectively targeting this unique cancer vulnerability.
1. Introduction
1.1. Replication stress response: an anti-cancer barrier
DNA replication is fundamental to the propagation of all life forms and its integrity is critical for heredity and genome stability [Citation1,Citation2]. Accordingly, dysregulated replication invariably generates DNA mutations and/or chromosomal instability. Moreover, defects in replication proficiency are associated with premature aging and cancer [Citation3,Citation4]. To maintain genome integrity, DNA replication employs a dynamic process that readily responds to a range of DNA perturbations, generally described as replication stress (RS) [Citation5]. RS derives from endogenous metabolic byproducts such as aldehydes, oxygen and nitrogen free radicals that modify DNA bases, or from environmental sources such as ultraviolet (UV) light or chemotherapies that modify DNA structure with adducts or crosslinks which, if left unrepaired, leads to DNA damage [Citation5]. DNA replication is also challenged by insufficient building blocks, which occurs by depletion of nucleotides, for example,when cells are treated with the ribonucleotide reductase inhibitor, hydroxyurea (HU) or when cells are prematurely driven into S phase [Citation6,Citation7]. Additional sources of stress include DNA secondary structures that impede DNA replication, such as G-quadruplexes (G4), common fragile sites (CFS) or loop formations occurring in highly repetitive sequences such as microsatellites [Citation8–12].
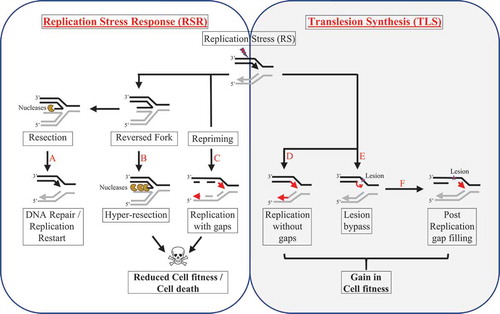
These diverse sources of stress activate the replication stress response (RSR) that alters replication fork dynamics to either temporarily stall or fully restrict replication through activation of checkpoint responses that signal a global arrest [Citation5]. Replication fork slowing is often associated with fork reversal that results in the formation of intermediary DNA structures called reversed forks. The reversed fork structure (also sometimes referred to as the ‘chicken foot’ structure) is thought to facilitate DNA repair by physically distancing the replication machinery from the stress invoking DNA lesion [Citation13–15]. Along with access to enhance repair processing, the RSR coordinates global replication which will either pause, terminate, or eventually restart following sufficient DNA repair. If mechanisms to re-initiate the preexisting replication fork fail, replication recovery can be achieved by the firing of new origins or by favoring replication by re-priming downstream from the site of stress [Citation16]. Alternatively, under severe stress, cell death responses can be triggered thereby eliminating cells with under replicated or grossly unstable genomes [Citation17–22].
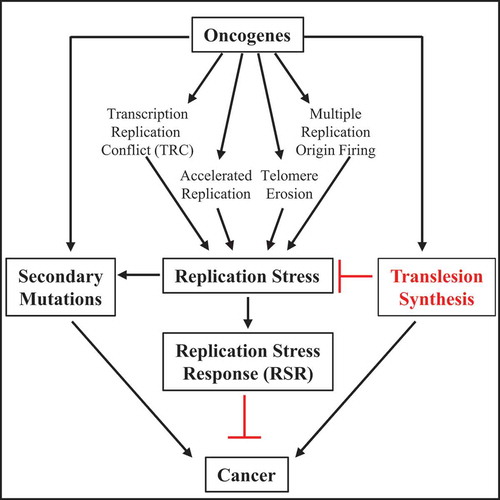
Despite the fact that RSR has been proposed to be an Achilles’ heel, cancer still emerges [Citation23]. For example, and counterintuitively, oncogenes that drive cancer development induce RS, and accordingly activate the RSR that induces cell cycle arrest, senescence, and/or cell death, thereby preventing accumulating mutations and effectively counteracting cancer initiation. As such, the RSR is considered a critical anti-cancer barrier [Citation5,Citation24–28] (). Oncogene expression induces RS via a range of cellular responses. For example, some oncogenes slow replication whereas others accelerate replication and induce the firing of multiple replication origins [Citation24–26]. Additionally, oncogenes are associated with reduced nucleotide pools, the erosion of telomeres, as well as the induction of reactive oxygen species and transcription-replication conflicts (TRC) [Citation24]. Thus, it has largely been a mystery as to how cells with oncogene activation ultimately overcome the RSR and promote cancer. Mitigation of the oncogene-induced RSR could explain secondary mutations and other adaptations occurring during cancer progression. This is underscored by the high incidence of mutations in the tumor suppressor gene, p53, that functions in checkpoint activation and apoptosis [Citation29]. However, p53 loss can interfere with the restart of DNA replication [Citation30] and potentially elevate RS induced by oncogenes, raising the idea that other events are critical to suppress stress and confer cancer development.
Figure 1. Schematic illustration to depict replication fork dynamics upon RS
1.2. TLS a cancer adaptation
Conventionally, RS deriving from DNA lesions are thought to be countered by lesion tolerance mechanisms such as translesion synthesis (TLS). Lower-fidelity TLS polymerases facilitate polymerization across from a DNA lesion in cases where the high-fidelity DNA replicative polymerases stall and cannot replicate past the lesion [Citation31]. One critical point of regulation that engages TLS is the mono-ubiquitination of lysine 164 (K164) of proliferating cell nuclear antigen (PCNA) that is mediated by the RAD18/RAD6 ubiquitin ligases, which initiates the switch from replicative to TLS polymerases. This ubiquitination serves as a platform to recruit TLS polymerases (POL η, POL ι, POL κ, REV1, POL ζ [also known as REV3/REV7], POL θ, and POL ν) that mediate lesion bypass either in the course of DNA replication or in a process of post-replication gap filling [Citation32–38]. By interacting with one or more TLS polymerases, PCNA functions as a ‘tool belt’ to coordinate TLS polymerases in a concerted response that initiates with a TLS polymerase such as POL η or POL κ that inserts a nucleotide at the site of the lesion [Citation39–43]. Extension past the lesion is mediated by a distinct TLS polymerase such as POL ζ followed by a final switch to the replicative polymerases [Citation44,Citation45]. Alternatively, TLS can be engaged independently of PCNA ubiquitination (PCNA Ub) via a REV1 scaffold domain ‘bridge’ that interacts with several TLS polymerases [Citation46,Citation47]. PRIMPOL, a DNA primase and TLS polymerase, can also operate independently of PCNA to restart stalled forks or re-prime replication ahead of a lesion [Citation48–50]. These well-orchestrated TLS polymerase switching events and their regulating mechanisms are reviewed at length in the following articles [Citation51–54].
Cells confronting replication stress also benefit from TLS activation. Aside from the bypass of DNA lesions that block the replicative polymerases, TLS also facilitates replication during stress that does not necessarily damage DNA [Citation55]. Demonstrating their essentiality for replication during stress, POL κ protects against fork degradation and maintains DNA replication during HU-mediated RS [Citation56]. Moreover, human and mouse cells deficient in distinct TLS polymerases have reduced fork progression and fork stalling when challenged with UV [Citation57–59]. In addition to altering fork dynamics, TLS has been shown to counteract replication gaps that are generated from distinct forms of RS. In yeast cells, TLS suppresses ssDNA gaps when replication forks are under stress due to methyl methane-sulfonate (MMS) or UV [Citation14,Citation60,Citation61]. Recent analysis indicates that these stress-induced gaps form in the wake of DNA replication forks at regions distinct from DNA double-strand breaks (DSBs) or stalled forks [Citation62–66]. Thus, TLS impacts a range of RS-related outcomes aside from bypassing DNA lesions ().
Hinting that replication gaps in the wake of replication forks are a fundamental problem for human cells that must be suppressed for cancer to evolve, it was recently found that gaps correlate with a reduction in replication proficiency and cell fitness [Citation67]. By engineering the activation of TLS, it was further illustrated that replication and cell fitness were elevated and notably gaps were also suppressed [Citation67]. Indeed, many RS-inducing agents generate gaps in human cells as noted in response to UV as early as 1976 [Citation68,Citation69]. In addition to UV, gaps form in response to HU, cisplatin, or inhibitors of ataxia telangiectasia and Rad3-related (ATR), WEE1 or poly(ADP-ribose) polymerases (PARP) as well as following oncogene expression [Citation67,Citation70,Citation71]. The toxicity of these agents is however reduced in pro-TLS cells as gaps are also suppressed [Citation67]. Correspondingly, POL η protects against MYC-induced replication arrest and cell death by rescuing fork stalling [Citation72] and RAD18 – mediated activation of POL κ prevents cell-cycle arrest and death induced by Cyclin E expression [Citation73,Citation74]. Interestingly, a range of genotoxic agents have been shown to increase TLS activity [Citation75–77], suggesting that TLS in human cells is analogous to the SOS response in bacteria that activates TLS in response to stress to restore cell homeostasis [Citation78–80].
Another mechanism by which cells adapt to oncogene-induced RS is by overexpressing checkpoint mediator proteins CLASPIN and TIMELESS [Citation81]. In fact, yeast homologues of CLASPIN and TIMELESS, mediator of replication checkpoint 1 (Mrc1) and Topoisomerase 1 interacting factor (Tof1) also have been shown to alleviate RS [Citation82]. As CLASPIN and TIMELESS contribute to PCNA-Ub, conceivably their countering of oncogene-induced stress is linked to TLS activation as well [Citation83–85].
Consistent with TLS being a cancer adaptation necessary to avert the initial oncogene-induced RS, comparative examination of tumor and adjacent normal tissue samples demonstrates upregulation of TLS polymerases as a widespread phenomenon in cancer [Citation86]. Notably, POL η is overexpressed in head and neck squamous cell carcinoma (HNSCC) [Citation87], POL θ expression is associated with poor patient survival in breast cancer [Citation88], and POL κ is overexpressed in glioblastoma and lung cancer patients [Citation89–91]. Additionally, POL ι expression is tightly correlated with cancer incidence in esophageal squamous cell carcinoma [Citation92–95]. Further evidence for elevated TLS activity comes from whole-genome sequencing in cancers in which TLS specific mutational signatures are consistent with the upregulation of POL η and POL ε. In particular, cancers such as leukemia and lymphoma display the classical POL η mutational signature 9 [Citation96–99]. Colorectal and uterine cancers have signature 10 attributed to POL ε [Citation100]. Collectively, cancer evolutions could be mediated by TLS that mitigates stress, bypasses lesions, and other replication impediments, and enhances mutability ().
Figure 2. The role of oncogene-induced RS in the development of cancer
1.3. TLS-induced mutagenesis vs RGS in cancer development
Although TLS-induced mutations and lesion bypass cannot be overlooked as providing a selective advantage for tumorigenesis and/or chemoresistance, TLS-induced RGS should also be considered. First, RGS mitigates replication gaps that cause replication catastrophe, a situation in which ssDNA becomes exposed to nucleases when ssDNA exceeds protection by the ssDNA binding protein, RPA [Citation101]. Conceivably, oncogene-induced senescence is also linked to replication catastrophe so that oncogenic transformation requires either RGS by TLS or higher RPA levels. In contrast, oncogenes are not expected to induce DNA lesions that would require a bypass to overcome stress. Likewise, TLS-induced mutations are not likely to counteract the rapid induction of senescence following oncogene expression. Second, it is worth considering that TLS also operates to suppress mutagenesis in some cellular contexts. This thinking is based on the fact that ssDNA gaps often act as a key initiator for APOBEC proteins/enzymes induced high mutational signature clusters [Citation102–105]. Thus, oncogene-induced gaps may not only induce senescence but also grossly elevate mutation loads via APOBEC activity that exceed a viability threshold. Here, TLS could overcome this gap-driven restriction point. Of note, catalytic-inactive REV1 mutants exhibit increased loss of heterozygosity (LOH), consistent with the function of TLS polymerases promoting genomic stability [Citation106]. Likewise, gaps are able to initiate sister chromosomal rearrangements that, when unchecked, limit cell viability [Citation107]. Moreover, by countering ssDNA gaps, TLS could limit the formation of secondary structures that form at CFS. Accordingly, POL η is recruited to CFS during normal replication and RS to prevent under-replication during mitosis and to maintain genomic stability [Citation108]. Given these points, RGS by TLS to gain fitness could be equal to, or possibly exceed, the role of TLS-induced mutagenesis to develop cancer, the challenge will be to separate these simultaneous phenomena.
A clear and important caveat in considering the targeting of TLS is that its loss also causes cancer. This likely stems from the fact that TLS can operate in an error-free manner such as in the bypass of UV lesions by POL η. Indeed, POL η deficiency results in Xeroderma pigmentosum variant (XPV), a hereditary condition with high susceptibility to skin cancer [Citation109]. Consistent with a TLS deficiency contributing to tumorigenesis, a down-regulation of DNA polymerases κ, η, ι, and ζ is observed in lung, stomach, and colorectal cancers [Citation110] and various mouse models of disrupted TLS polymerase activity increase the incidence of lymphomas, squamous cell carcinomas, and mammary gland tumorigenesis [Citation111,Citation112]. Notably, polθ−/ – and polη−/- mice display a greater susceptibility to UV-induced skin cancer [Citation59]. It is also worth considering that gaps due to loss of TLS may lead to unchecked inflammation that is complicit in cancer. This phenomenon is observed in Rev1 deficient mice, which when crossed to skin-cancer prone mice, exhibit inflammatory hyperplasia and an acceleration of skin cancer [Citation113]. Of note, pol κ deficient mice have increased inflammation-mediated mutagenesis, and pol ζ deficient cells exhibit chromosomal breaks and increased sensitivity to nitric oxide, a mediator of inflammatory responses [Citation114,Citation115]. Thus, TLS can potentially alleviate inflammation-mediated DNA lesions and help mitigate stress responses [Citation116–118].
1.4. Role of TLS in chemoresistance
In addition to the role of TLS in tumor initiation, clinical evidence points to a relationship between TLS and the development of chemoresistance. Chemoresistance correlates with increased POL η in head neck squamous cell carcinoma and increased POL κ in glioblastomas [Citation87,Citation89,Citation91]. Furthermore, RAD18 expression is elevated in glioblastoma stem cells that develop cisplatin resistance [Citation119]. Additionally, REV1 and POL ζ have critical roles in acquired chemoresistance in prostate cancer xenograft models [Citation120]. Furthermore, targeting TLS limits chemoresistance. In particular, depletion of TLS polymerases, Rev1 or Rev3L re-sensitizes chemo-resistant cancer models to cisplatin [Citation121,Citation122]. More recently, inactivation of REV7 in testicular germ cell tumors was shown to restore chemosensitivity in both in vitro and in vivo cancer models [Citation123].
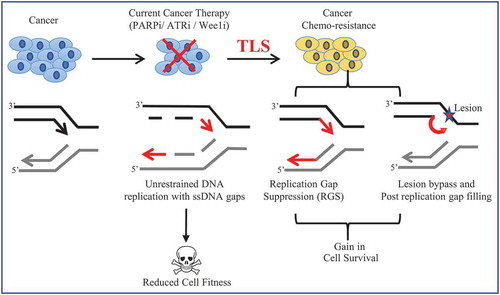
Although lesion-bypass is conventionally considered a major driver of chemoresistance, the emergence of new functions for TLS in fork integrity and fork restart suggests other mechanisms also contribute to TLS-mediated chemoresistance [Citation56,Citation57,Citation77,Citation124,Citation125]. Our findings also indicate that TLS-RGS is relevant and likely overlaps with the function of TLS in fork protection (FP). Notably, TLS activation suppresses fork stalling as well as fork degradation following sustained RS [Citation67]. As such, TLS activation elevates FP in BRCA2-deficient cells that have a hyper-fork degradation phenotype [Citation67,Citation126]. Similarly, loss of the chromatin remodeler, CHD4 confers cisplatin resistance in BRCA2-mutant cancer cells, activates TLS [Citation127], and restores FP. Indeed, FP as a mechanism of cisplatin resistance in BRCA2-deficient cancer has gained attention with a series of papers showing a range of genetic means to restore FP [Citation126,Citation128–131]. Interestingly, aside from restored FP, loss of CHD4 along with EZH2 or FEN1 restores RGS to BRCA2-mutant cells [Citation71]. In contrast, loss of genes such as MRE11 or SMARCAL1 that restore FP, do not lead to RGS, nor confer cisplatin resistance. Moreover, low expression of these genes does not predict poor survival for BRCA2-mutant patients as found for low expression of CHD4, EZH2, or FEN1 [Citation71]. Together, these findings indicate that restored FP alone through inhibition of fork reversal and/or nuclease degradation is not sufficient to confer chemotherapy resistance. Rather, RGS is central to chemoresistance because it not only limits fork reversal and degradation but also enables the continuation of replication during stress (). In this context, it is important to note that the BRCA-RAD51 pathway also promotes RGS and when defective, TLS-RGS may be particularly important to maintain cell viability and resistance to therapy. Thus, TLS-RGS would be an effective target to kill or chemo-sensitize BRCA-deficient cancer.
Figure 3. Schematic illustration to depict TLS-induced chemo-resistance in cancer
1.5. Targeting TLS as an anti-cancer approach
Based on the ability of TLS polymerases to counteract anti-cancer drugs, they have been an attractive target for the development of cancer therapies. Small molecule inhibitors targeting TLS polymerases have been met with varying success [Citation132]. For example, the TLS inhibitor (TLSi) (JH-RE-06) targets the interaction between REV1 and the REV7 subunit of POL ζ. It inhibited mutagenic TLS and enhanced cisplatin-induced toxicity in cultured human and mouse cell lines. Further, when combined with cisplatin treatment, JH-RE-06 synergistically suppressed the growth of xenograft human melanomas in mice by inducing a senescent phenotype, recapitulating findings observed in Rev7-deficient models of chemo-resistant lung cancers [Citation133–135]. Similarly, small molecule inhibitors that specifically target the C-terminal domain of TLS polymerase REV1 (REV1-CT) scaffolding protein (compounds 4 and 5) inhibit the interaction between REV1 and TLS polymerases to block TLS activation [Citation136]. These compounds effectively increased both the sensitivity and mutagenesis following cisplatin treatment [Citation136].
Given the finding that stress from a range of sources including oncogene expression induced gaps that are countered by TLS, it was considered that a rewiring of TLS could emerge as cancer develops. If so, inhibition of TLS without additional chemotherapy agents could limit the replication and proliferation of cancer cells by promoting the persistence and toxicity of ssDNA gaps and thereby result in loss of viability. Consistent with this idea, the REV1-CT inhibitor compound 4 [Citation136] uniquely reduced the colony-forming potential of TLS-dependent cancer cells and induced widespread ssDNA gaps [Citation67]. The compound also was effective as a single agent to suppress the proliferation of ovarian patient-derived ascites [Citation67].
As described above, gaps are induced following treatment with inhibitors of ATR, or WEE1. However, gaps were less evident in cancer cells rewired to engage in TLS or lines engineered to favor TLS. When TLSi were co-incubated with ATR or WEE1 inhibitors, these pro-TLS lines exhibited wide-spread gap induction, reduced proliferation, and loss of viability. These findings indicate that co-targeting of TLS will elevate ssDNA gap incidences and enhance the therapeutic success of these inhibitors. Efficacy and selectivity of TLS inhibitors await in vivo studies to determine the therapeutic potential of both mono and co-administered regimens.
In addition to inhibitors that directly target the TLS polymerases, another promising strategy is to identify compounds that alter regulators of TLS. Validating the strategy of inhibiting TLS regulators, a small molecule inhibitor of RAD6 reduced PCNA-Ub, enhanced sensitivity to cisplatin in cancer cells, and diminished tumor growth in xenografts, which was further attenuated upon treatment with RAD6 inhibitor and cisplatin [Citation137]. Conversely, negative regulators of TLS such as the deubiquitinase of PCNA, USP1/UAF1 could be modulated to limit TLS further. Another strategy to inhibit TLS are compounds that indirectly impair Ub-PCNA, as observed with AKT inhibitors that impair PCNA-Ub and TLS activity [Citation138]. Notably, AKT inhibitors induce synthetic lethality in homologous recombination-deficient cells upon RS, in a mechanism dependent on diminished TLS as verified by the synthetic lethality also conferred by loss of PCNA-Ub or RAD18 expression under similar conditions [Citation138]. Additionally, an inactivating PCNA mutation (PCNA K164R) that leads to loss of DDT can effectively sensitive tumors to platinum therapy [Citation139]. These findings suggest that there will be yet several unexpected routes and great potential to limit TLS in cancer alone or in combination with RS-inducing agents.
2. Conclusion
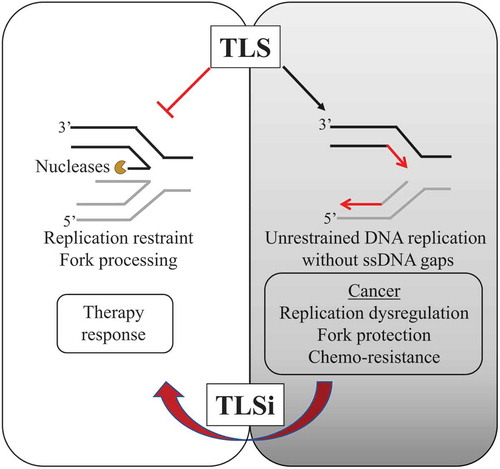
While most TLS-centric reviews provide a comprehensive analysis of multiple functions of TLS polymerases [Citation46,Citation75,Citation140,Citation141], the newly identified role for TLS in countering RS and suppressing replication gaps that arise behind the replication fork, is yet to be discussed [Citation67]. Here, we review the function of TLS to blunt the RS-associated anti-cancer barrier and also to suppress ssDNA gap formation, and how these roles function as an important cancer adaptation during cancer evolution. Accordingly, the enhanced dependence on TLS observed in cancer cells and diminished survival that occurs upon TLS inhibition prompts us to propose that TLS be considered as a new non-oncogenic addiction and a hallmark of cancer. Therefore, the nuanced understanding of TLS functions underscores the importance of re-analyzing how TLS impacts cancer growth and chemoresistance, and how could be further exploited as a cancer vulnerability. Ideally, these recent insights into TLS function in cancer development open a pandora’s box with untapped potential for improved anti-cancer therapies ().
Figure 4. Model proposing TLS as a new evolving target for cancer therapy
3. Expert opinion
Although a role for TLS in cancer has been described for over a decade [Citation109,Citation142,Citation143], its exact function in tumorigenesis remains unresolved given the multiple disparate consequences of altered TLS. A role for TLS in lesion bypass and post-replication gap filling is well characterized and conventionally thought to contribute to chemoresistance. Here we highlight a less well-appreciated role for TLS in countering RS by suppressing toxic replication gaps that arise behind the replication fork. We propose that similar to other TLS functions, TLS-RGS mediates chemoresistance. However, we also contend that TLS-RGS provides a foothold for cancer. In particular, RGS opposes oncogene-mediated senescence, genomic instability, and cell death. This understanding of an additional yet distinct TLS function underscores the importance of reconsidering how TLS contributes to cancer growth and chemoresistance. Toward this goal, we review evidence supporting a dependence on TLS in cancer. Although the mechanisms of TLS rewiring in cancer have yet to be fully elucidated, it is likely achieved not only by increasing the expression of TLS polymerases but also by changes in positive and negative regulators [Citation144–146]. We further propose that the reliance of different cancer cells on TLS indicates that it becomes an addiction, and therefore required to overcome metabolic, replication, or other stresses that arise during tumorigenesis [Citation147,Citation148].
Cancer therapy has evolved considerably over the last few decades with the development of selective and targeted therapies. Traditional approaches to cancer therapy leveraged the failure of cancer cells to repair damaged DNA or to elicit checkpoint responses, but unfortunately, often these strategies prove futile as cells adapt and no longer respond to these interventions. On the contrary, therapies specifically targeting proteins contributing to cell addictions have been successful therapeutic strategies [Citation149–151]. Thus, targeting essentialities in cancer, such as TLS could provide a durable response to chemotherapy [Citation67]. In accordance, to exploit this cancer vulnerability, we emphasize the need to investigate existing and newly synthesized small molecule TLSi [Citation132,Citation136]. TLSi exhibit great promise for widespread cancer therapeutic as either a monotherapy or combination therapy [Citation67,Citation133]. Indeed, TLSi have already demonstrated great potential to preferentially kill cancer cells in combination with traditional platinum-based cancer therapies [Citation67].
Additionally, rational combination strategies that combine TLSi with small molecule inhibitors specifically designed to target the DNA damage sensors and expose ssDNA gaps, such as inhibitors of ataxia telangiectasia-mutated (ATM), ATR, CHK1, WEE1, and PARP1 will be another important therapeutic avenue to investigate. Indeed, combined inhibition of TLS and ATR or WEE1 resulted in increased toxicity in cancer cells [Citation67]. Finally, given the increased reliance of cancer cells on TLS, cancer cells are preferentially sensitive to TLSi, thereby reducing bystander effects on noncancer cells. Finally, with this newly defined role for TLS during replication in RGS and its seemingly widespread prevalence in cancer development, we postulate that TLS rewiring is a necessary non-oncogene addiction required for cancer development which now provides a new framework for the development of novel and highly specific anti-cancer therapies.
Article highlights
A newly defined role for TLS at the replication fork in countering replication stress (RS) by facilitating replication gap suppression (RGS), distinct from its role in canonical lesion bypass and post-replication gap filing.
Replication-associated ssDNA gaps are toxic consequences of replication stress, induced by oncogene expression or other agents that determine cell fitness and therapy response.
TLS-mediated RGS is a key phenomenon to overcome the oncogene-induced anti-cancer barrier and to promote tumorigenesis.
TLS RGS is a wide-spread cancer adaptation and plays an important role in resistance to cancer therapies.
Small molecule inhibitors of TLS have promise as monotherapies or in combination with other drugs designed to expose ssDNA gaps to target the dependency of cancer cells on RGS.
Replication gaps are a cancer vulnerability, suggesting that TLS-associated RGS is a hallmark of cancer.
This box summarizes key points contained in the article.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Acknowledgments
We thank Dr. Kyle Hadden along with the members of the Cantor laboratory for helpful discussions.
Additional information
Funding
References
- Hejna JA, Moses RE. In Schaechter M (Ed.) Encyclopedia of Microbiology (Third. Oxford: Academic Press. 2009. 113–122.
- Samson RY, Bell SD. The Enzymes. Kaguni LS, Oliveira MT (Eds), Academic Press. 2016;Vol. 39:169–190.
- Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–1485.
- Mazouzi A, Velimezi G, Loizou JI. DNA replication stress: causes, resolution and disease. Exp Cell Res. 2014;329(1):85–93.
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2–9.
- Poli J, et al. dNTP pools determine fork progression and origin usage under replication stress. Embo J. 2012;31(4):883–894.
- Bester AC, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446.
- Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6(10):729–742.
- Kim JC, Mirkin SM. The balancing act of DNA repeat expansions. Curr Opin Genet Dev. 2013;23(3):280–288.
- Bochman ML, Paeschke K, Zakian VA. DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet. 2012;13(11):770–780.
- Pomerantz RT, O’Donnell M. What happens when replication and transcription complexes collide? Cell Cycle. 2010;9(13):2537–2543.
- Helmrich A, Ballarino M, Nudler E, et al. Transcription-replication encounters, consequences and genomic instability. Nat Struct Mol Biol. 2013;20(4):412–418.
- Neelsen KJ, Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol. 2015;16(4):207–220.
- Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297(5581):599–602.
- Atkinson J, McGlynn P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009;37(11):3475–3492.
- Berti M, Vindigni A. Replication stress: getting back on track. Nat Struct Mol Biol. 2016;67(2):103–109.
- Branzei D, Foiani M. The checkpoint response to replication stress. DNA Repair (Amst). 2009;178(9):1038–1046.
- Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11(3):208–219.
- Jackson SP, Bartek J, The DN. A-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078.
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28(5):739–745.
- Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297(5581):547–551.
- Bakkenist CJ, Kastan MB. Initiating cellular stress responses. Cell. 2004;118(1):9–17.
- Da-Rè C, Halazonetis TD. DNA replication stress as an achilles’ heel of cancer. Oncotarget. 2015;6(1):1–2.
- Kotsantis P, Petermann E, Boulton SJ. Mechanisms of oncogene-induced replication stress: jigsaw falling into place. Cancer Discov. 2018;8(5):537–555.
- Macheret M, Halazonetis TD. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature. 2018;555(7694):112–116.
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced dna damage model for cancer development. Science. 2008;319(5868):1352–1355.
- Bartkova J, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444(7119):633–637.
- Primo LMF, Teixeira LK. DNA replication stress: oncogenes in the spotlight. Genet Mol Biol. 2019;10(1 suppl 1):e20190138–e20190138.
- Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6:a026104–a026104.
- Roy S, et al. p53 orchestrates DNA replication restart homeostasis by suppressing mutagenic RAD52 and POLtheta pathways. Elife. 2018;43. DOI:10.7554/eLife.31723.
- Ghosal G, Chen J. DNA damage tolerance: a double-edged sword guarding the genome.. Transl Cancer Res. 2018;7(3):107–129.
- Arana ME, Seki M, Wood RD, et al. Low-fidelity DNA synthesis by human DNA polymerase theta. Nucleic Acids Res. 2008;36(11):3847–3856.
- Watanabe K, et al. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. Embo J. 2004;23(19):3886–3896.
- Guo C, et al. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol Cell. 2006;23(2):265–271.
- Kannouche PL, Lehmann AR. Ubiquitination of PCNA and the polymerase switch in human cells. Cell Cycle. 2004;3(8):1011–1013.
- Vaisman A, Woodgate R. Translesion DNA polymerases in eukaryotes: what makes them tick? Crit Rev Biochem Mol Biol. 2017;52(3):274–303.
- Moon AF, et al. The X family portrait: structural insights into biological functions of X family polymerases. DNA Repair (Amst). 2007;6(12):1709–1725.
- Blanca G, et al. Human DNA polymerases λ and β show different efficiencies of translesion DNA synthesis past abasic sites and alternative mechanisms for frameshift generation. Biochemistry. 2004;43(36):11605–11615.
- Masutani C, et al. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. Embo J. 1999;43(12):3491–3501.
- Masutani C, Kusumoto R, Iwai S, et al. Mechanisms of accurate translesion synthesis by human DNA polymerase eta. Embo J. 2000;19:3100–3109.
- Zhao Y, et al. Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proc Natl Acad Sci U S A. 2012;109(19):7269–7274.
- Zhang Y, et al. Error-free and error-prone lesion bypass by human DNA polymerase kappa in vitro. Nucleic Acids Res. 2000;28(21):4138–4146.
- Huang X, et al. Effects of base sequence context on translesion synthesis past a bulky (+)-trans-anti-B[a]P-N2-dG lesion catalyzed by the Y-family polymerase pol kappa. Biochemistry. 2003;42(8):2456–2466.
- Livneh Z, Ziv O, Shachar S. Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis. Cell Cycle. 2010;9(4):729–735.
- Johnson RE, Washington MT, Haracska L, et al. Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature. 2000;406(6799):1015–1019.
- Powers KT, Washington MT. Eukaryotic translesion synthesis: choosing the right tool for the job. DNA Repair (Amst). 2018;71:127–134.
- Boehm EM, Spies M, Washington MT. PCNA tool belts and polymerase bridges form during translesion synthesis. Nucleic Acids Res. 2016;44(17):8250–8260.
- Martinez-Jimenez MI, et al. Alternative solutions and new scenarios for translesion DNA synthesis by human PrimPol. DNA Repair (Amst). 2015;29:127–138.
- Torregrosa-Munumer R, et al. PrimPol is required for replication reinitiation after mtDNA damage. Proc Natl Acad Sci U S A. 2017;114:11398–11403.
- Makarova AV, Boldinova EO, Belousova EA, et al. In vitro lesion bypass by human PrimPol. DNA Repair (Amst). 2018;70:18–24.
- Choe KN, Moldovan GL. Forging Ahead through Darkness: PCNA, still the principal conductor at the replication fork. Mol Cell. 2017;65(3):380–392.
- Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol. 2012;13(3):141–152.
- Chang DJ, Cimprich KA. DNA damage tolerance: when it’s OK to make mistakes. Nat Chem Biol. 2009;5(2):82–90.
- Yamanaka K, Chatterjee N, Hemann MT, et al. Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet. 2017;13(8):e1006842.
- Northam MR, Garg P, Baitin DM, et al. A novel function of DNA polymerase zeta regulated by PCNA. Embo J. 2006;25(18):4316–4325.
- Tonzi P, Yin Y, Lee CWT, et al. Translesion polymerase kappa-dependent DNA synthesis underlies replication fork recovery. Elife. 2018;7. DOI:10.7554/eLife.41426
- Quinet A, et al. Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Res. 2016;44(12):5717–5731.
- Quinet A, et al. Gap-filling and bypass at the replication fork are both active mechanisms for tolerance of low-dose ultraviolet-induced DNA damage in the human genome. DNA Repair (Amst). 2014;14:27–38.
- Yoon JH, et al. Error-prone replication through UV lesions by DNA polymerase theta protects against skin cancers. Cell. 2019;176:1295–1309 e1215.
- Prado F. Homologous recombination: to fork and beyond. Genes (Basel). 2018;9(12). DOI:10.3390/genes9120603
- Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell. 2006;21(1):15–27.
- Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell. 2008;30(4):519–529.
- Temviriyanukul P, et al. Temporally distinct translesion synthesis pathways for ultraviolet light-induced photoproducts in the mammalian genome. DNA Repair (Amst). 2012;11(6):550–558.
- Jansen JG, et al. Separate domains of Rev1 mediate two modes of DNA damage bypass in mammalian cells. Mol Cell Biol. 2009;29(11):3113–3123.
- Hedglin M, Benkovic SJ. Eukaryotic translesion DNA synthesis on the leading and lagging strands: unique detours around the same obstacle. Chem Rev. 2017;117(12):7857–7877.
- Wong RP, Garcia-Rodriguez N, Zilio N, et al. Processing of DNA polymerase-blocking lesions during genome replication is spatially and temporally segregated from replication forks. Mol Cell. 2020;77(1):3–16 e14.
- Nayak S, et al. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci Adv. 2020;6(24):eaaz7808.
- Meneghini R. Gaps in DNA synthesized by ultraviolet light-irradiated WI38 human cells. Biochim Biophys Acta Nucleic Acids Protein Synth. 1976;425(4):419–427.
- Meneghini R, Cordeiro-Stone M, Schumacher RI. Size and frequency of gaps in newly synthesized DNA of xeroderma pigmentosum human cells irradiated with ultraviolet light. Biophys J. 1981;33(1):81–92.
- Cong K, et al. PARPi synthetic lethality derives from replication-associated single-stranded DNA gaps. bioRxiv. 2019;781989.
- Panzarino NJ, et al. Replication gaps underlie BRCA-deficiency and therapy response. Cancer Res. canres.1602.2020. 2020;canres.1602.2020. DOI:10.1158/0008-5472.CAN-20-1602.
- Kurashima K, et al. Polη, a Y-family translesion synthesis polymerase, promotes cellular tolerance of Myc-induced replication stress. J Cell Sci. 2018;131(12):jcs212183.
- Yang Y, et al. Diverse roles of RAD18 and Y-family DNA polymerases in tumorigenesis. Cell Cycle. 2018;17(7):833–843.
- Yang Y, et al. DNA repair factor RAD18 and DNA polymerase Polkappa confer tolerance of oncogenic DNA replication stress. J Cell Biol. 2017;216(10):3097–3115.
- Zafar MK, Eoff RL. Translesion DNA synthesis in cancer: molecular mechanisms and therapeutic opportunities. Chem Res Toxicol. 2017;30(11):1942–1955.
- Tomicic MT, et al. Translesion polymerase η is upregulated by cancer therapeutics and confers anticancer drug resistance. Cancer Res. 2014;74(19):5585–5596.
- Lerner LK, et al. Predominant role of DNA polymerase eta and p53-dependent translesion synthesis in the survival of ultraviolet-irradiated human cells. Nucleic Acids Res. 2017;45(3):1270–1280.
- Weigle JJ. Induction of mutations in a bacterial virus. Proc Natl Acad Sci U S A. 1953;39(7):628–636.
- Witkin EM. The radiation sensitivity of Escherichia coli B: a hypothesis relating filament formation and prophage induction. Proc Natl Acad Sci U S A. 1967;57(5):1275–1279.
- Patel M, Jiang Q, Woodgate R, et al. A new model for SOS-induced mutagenesis: how RecA protein activates DNA polymerase V. Crit Rev Biochem Mol Biol. 2010;45(3):171–184.
- Bianco JN, et al. Overexpression of Claspin and timeless protects cancer cells from replication stress in a checkpoint-independent manner. Nat Commun. 2019;10(1):910.
- Katou Y, et al. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature. 2003;424(6952):1078–1083.
- Yang XH, Zou L. Dual functions of DNA replication forks in checkpoint signaling and PCNA ubiquitination. Cell Cycle. 2009;8(2):191–194.
- Yang XH, Shiotani B, Classon M, et al. Chk1 and Claspin potentiate PCNA ubiquitination. Genes Dev. 2008;22(9):1147–1152.
- Unsal-Kacmaz K, et al. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol. 2007;27(8):3131–3142.
- Albertella MR, Lau A, O’Connor MJ. The overexpression of specialized DNA polymerases in cancer. DNA Repair (Amst). 2005;4(5):583–593.
- Zhou W, et al. Expression of DNA translesion synthesis polymerase eta in head and neck squamous cell cancer predicts resistance to gemcitabine and cisplatin-based chemotherapy. PLoS One. 2013;8(12):e83978.
- Lemee F, et al. DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc Natl Acad Sci U S A. 2010;107(30):13390–13395.
- Bostian AC, et al. Kynurenine signaling increases dna polymerase Kappa expression and promotes genomic instability in glioblastoma cells. Chem Res Toxicol. 2016;29(1):101–108.
- O-Wang, et al. DNA polymerase kappa, implicated in spontaneous and DNA damage-induced mutagenesis, is overexpressed in lung cancer.. Cancer Res. 2018;17(14):5366–5369.
- Peng C, et al. The error-prone dna polymerase κ promotes Temozolomide resistance in glioblastoma through Rad17-dependent activation of ATR-Chk1 signaling. Cancer Res. 2016;76(8):2340–2353.
- Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231(1):11–30.
- Ohshima H, Yermilov V, Yoshie Y, et al. Advances in DNA Damage and Repair: oxygen Radical Effects, Cellular Protection, and Biological Consequences. Dizdaroglu M, Karakaya AE, Eds. Boston, MA: Springer US; 1999. p. 329–339.
- McIntyre J, Woodgate R. Regulation of translesion DNA synthesis: posttranslational modification of lysine residues in key proteins. DNA Repair (Amst). 2015;29:166–179.
- McIntyre J. Polymerase iota - an odd sibling among Y family polymerases. DNA Repair (Amst). 2020;86:102753.
- Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421.
- Puente XS, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–105.
- Puente XS, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519–524.
- Agishev TT, et al. Determination of oxygen perfusion in the area of radiation-induced fibrosis of the skin in patients with breast cancer and its role in pathogenesis of late radiation injury. Exp Oncol. 2018;40:235–238.
- Alexandrov LB, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101.
- Ercilla A, et al. Physiological tolerance to ssDNA enables strand uncoupling during DNA replication. Cell Rep. 2020;30(7):2416–2429 e2417.
- Seplyarskiy VB, et al. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016;26(2):174–182.
- Roberts SA, et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell. 2012;46(4):424–435.
- Taylor BJ, et al. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife. 2013;2:e00534.
- Chen J, Miller BF, Furano AV. Repair of naturally occurring mismatches can induce mutations in flanking DNA. Elife. 2014;3:e02001.
- Fujikawa Y, et al. Involvement of Rev1 in alkylating agent-induced loss of heterozygosity in oryzias latipes. Genes Cells. 2020;25(2):124–138.
- Nagaraju G, Scully R. Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair (Amst). 2007;13(7):1018–1031.
- Bergoglio V, et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J Cell Biol. 2013;46(3):395–408.
- Masutani C, et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399(6737):700–704.
- Pan Q, Fang Y, Xu Y, et al. Down-regulation of DNA polymerases kappa, eta, iota, and zeta in human lung, stomach, and colorectal cancers. Cancer Lett. 2005;217(2):139–147.
- Wittschieben JP, et al. Loss of DNA polymerase zeta enhances spontaneous tumorigenesis. Cancer Res. 2010;25(7):2770–2778.
- Lange SS, et al. Dual role for mammalian DNA polymerase zeta in maintaining genome stability and proliferative responses. Proc Natl Acad Sci U S A. 2013;110(8):E687–696.
- Tsaalbi-Shtylik A, et al. Error-prone translesion replication of damaged DNA suppresses skin carcinogenesis by controlling inflammatory hyperplasia. Proc Natl Acad Sci U S A. 2009;25(51):21836–21841.
- Wu X, et al. Critical roles for polymerase ζ in cellular tolerance to Nitric Oxide–Induced DNA damage. Cancer Res. 2006;66(2):748–754.
- Hakura A, Sui H, Sonoda J, et al. DNA polymerase kappa counteracts inflammation-induced mutagenesis in multiple organs of mice. Environ Mol Mutagen. 2019;60(4):320–330.
- Yoon J-H, Johnson RE, Prakash L, et al. Genetic evidence for reconfiguration of DNA polymerase θ active site for error-free translesion synthesis in human cells. J Biol Chem. 2020;26(18):5918–5927.
- Yasui M, et al. Translesion synthesis past 2ʹ-deoxyxanthosine, a nitric oxide-derived DNA adduct, by mammalian DNA polymerases. J Mol Biol. 2004;344(3):665–674.
- Yasui M, et al. Miscoding properties of 2ʹ-deoxyinosine, a nitric oxide-derived DNA Adduct, during translesion synthesis catalyzed by human DNA polymerases. J Mol Biol. 2008;46(4):1015–1023.
- Kermi C, et al. RAD18 Is a maternal limiting factor silencing the UV-dependent dna damage checkpoint in xenopus embryos. Dev Cell. 2015;34(3):364–372.
- Xu X, et al. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc Natl Acad Sci U S A. 2013;399(46):18638–18643.
- Xie K, Doles J, Hemann MT, et al. Error-prone translesion synthesis mediates acquired chemoresistance. Proc Natl Acad Sci U S A. 2010;107(48):20792–20797.
- Doles J, et al. Suppression of Rev3, the catalytic subunit of Pol{zeta}, sensitizes drug-resistant lung tumors to chemotherapy. Proc Natl Acad Sci U S A. 2010;107(48):20786–20791.
- Sakurai Y, et al. Inactivation of REV7 enhances chemosensitivity and overcomes acquired chemoresistance in testicular germ cell tumors. Cancer Lett. 2020;489:100–110.
- Yang Y, et al. FANCD2 and REV1 cooperate in the protection of nascent DNA strands in response to replication stress. Nucleic Acids Res. 2015;43(17):8325–8339.
- Sokol AM, Cruet-Hennequart S, Pasero P, et al. DNA polymerase η modulates replication fork progression and DNA damage responses in platinum-treated human cells. Sci Rep. 2013;3(1):3277.
- Schlacher K, et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145(4):529–542.
- Guillemette S, et al. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 2015;29(5):489–494.
- Mijic S, et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun. 2017;8(1):859.
- Ray Chaudhuri A, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535(7612):382–387.
- Kolinjivadi AM, et al. Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017;591(8):1083–1100.
- Kolinjivadi AM, et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell. 2017;67(5):867–881 e867.
- Korzhnev DM, Hadden MK. Targeting the translesion synthesis pathway for the development of anti-cancer chemotherapeutics. J Med Chem. 2016;59(20):9321–9336.
- Wojtaszek JL, et al. A small molecule targeting mutagenic translesion synthesis improves chemotherapy. Cell. 2019;178(1):152–159 e111 .
- Chatterjee N, et al. REV1 inhibitor JH-RE-06 enhances tumor cell response to chemotherapy by triggering senescence hallmarks. Proc Natl Acad Sci U S A. 2020;117(46):28918–28921.
- Vassel FM, Bian K, Walker GC, et al. Rev7 loss alters cisplatin response and increases drug efficacy in chemotherapy-resistant lung cancer. Proc Natl Acad Sci U S A. 2020;117(46):28922–28924.
- Sail V, et al. Identification of small molecule translesion synthesis inhibitors that target the Rev1-CT/RIR protein-protein interaction. ACS Chem Biol. 2017;106(7):1903–1912.
- Sanders MA, Haynes B, Nangia-Makker P, et al. Pharmacological targeting of RAD6 enzyme-mediated translesion synthesis overcomes resistance to platinum-based drugs. J Biol Chem. 2017;292(25):10347–10363.
- Villafanez F, et al. AKT inhibition impairs PCNA ubiquitylation and triggers synthetic lethality in homologous recombination-deficient cells submitted to replication stress. Oncogene. 2019;38(22):4310–4324.
- Buoninfante OA, et al. Precision cancer therapy: profiting from tumor specific defects in the DNA damage tolerance system. Oncotarget. 2018;9(27):18832–18843.
- Ma X, Tang TS, Guo C. Regulation of translesion DNA synthesis in mammalian cells. Environ Mol Mutagen. 2020;295(7):680–692.
- Pilzecker B, Jacobs H. Mutating for good: DNA damage responses during somatic hypermutation. Front Immunol. 2019;34:438.
- Loeb LA, Monnat RJ Jr. DNA polymerases and human disease. Nat Rev Genet. 2008;9(8):594–604.
- Watson NB, Mukhopadhyay S, McGregor WG. Translesion DNA replication proteins as molecular targets for cancer prevention. Cancer Lett. 2006;107(1):13–22.
- Haynes B, Gajan A, Nangia-Makker P, et al. RAD6B is a major mediator of triple negative breast cancer cisplatin resistance: regulation of translesion synthesis/Fanconi anemia crosstalk and BRCA1 independence. Biochim Biophys Acta Mol Basis Dis. 2020;1866(1):165561.
- Gao Y, Tateishi S, Vaziri C. Pathological trans-lesion synthesis in cancer. Cell Cycle. 2016;15(22):3005–3006.
- Lessel D, et al. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet. 2014;46:1239–1244.
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–837.
- Nagel R, Semenova EA, Berns A. Drugging the addict: non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016;17(11):1516–1531.
- Gad H, et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature. 2014;508(7495):215–221.
- Luo J, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–848.
- Li M, et al. Non-oncogene addiction to SIRT3 plays a critical role in lymphomagenesis. Cancer Cell. 2019;35(6):916–931 e919.