
ABSTRACT
Introduction: Disturbances in myocardial lipid metabolism are increasingly being recognized as drivers of the development and progression of heart disease. Therefore, there is a need for treatments that can directly target lipid metabolic defects in heart failure. The membrane-associated glycoprotein CD36 plays a pivotal role in governing myocardial lipid metabolism by mediating lipid signaling and facilitating the cellular uptake of long-chain fatty acids. Emerging evidence suggests that CD36 is a prominent target in the treatment of heart failure.
Areas covered: This article provides an overview of the key role of CD36 for proper contractile functioning of a healthy heart, its implications in the development of cardiac disease (ischemia/reperfusion, cardiac hypertrophy, and diabetic cardiomyopathy), and its application as a target to normalize cardiac metabolism as part of so-called metabolic modulation therapy.
Expert opinion: CD36 appears a promising and effective therapeutic target in the treatment of heart failure. Natural compounds and chemical agents known to alter the amount or subcellular distribution of CD36 or inhibit its functioning, should be evaluated for their potency to correct cardiac metabolism and cure heart disease.
1. Introduction
In recent years, it has been established that chronic changes in cardiac energy metabolism are an important cause of impaired cardiac contractile performance [Citation1]. Such metabolic changes may either directly underlie cardiac disease or aggravate the progression of cardiac disease elicited by other factors. For instance, lipid overload due to Western lifestyle or obesity elicits cardiac insulin resistance and contractile dysfunction often eventually leading to diabetic cardiomyopathy [Citation2]. Furthermore, it has been found that interventions aimed at normalizing cardiac substrate metabolism are an effective therapeutic approach for cardiac diseases [Citation3,Citation4].
Of special interest is cardiac lipid metabolism because long-chain fatty acids comprise the main source of myocardial energy provision [Citation5]. Studies performed in the last two decades have shown that the transmembrane glycoprotein CD36, which facilitates long-chain fatty acid uptake into cardiac myocytes, is primarily responsible for the proper regulation of myocardial lipid metabolism. Thus, in the healthy heart the presence and activity of CD36 in the sarcolemma appears to be closely adjusted to the cellular need for fatty acids, avoiding both too low uptake rates (risk of energy deficit) and too high uptake rates (risk of lipotoxicity) [Citation6,Citation7]. In line with the dynamic nature of cardiac energy metabolism, whereby changes in demand may occur almost instantaneously, the sarcolemmal presence and activity of CD36 can be adapted on a short timescale. As a result, CD36 serves as a dynamic gatekeeper of myocardial lipid metabolism.
In this perspective, we will briefly review the physiological role of CD36 in the regulation of myocardial lipid metabolism and of lipid signaling, and its consequently pivotal role in the development of cardiac disease. Thereafter, we will discuss exciting evidence indicating the effective application of CD36 as suitable target for metabolic modulation therapy to mend the failing heart and explore options for manipulating CD36 presence and/or function by natural compounds and chemical drugs.
2. CD36 functions in myocardial fatty acid uptake and in lipid signaling
Cluster of Differentiation 36 (CD36), officially designated scavenger receptor B2 (SR-B2) but also known as fatty acid translocase (FAT), was first described in 1977 and implicated in platelet aggregation and the adherence of erythrocytes infected with the malaria parasite [Citation8]. Subsequently, it was established that CD36 functions as a scavenger receptor for oxidized LDL on macrophages [Citation9]. In 1993, CD36 was shown to facilitate cellular uptake of long-chain fatty acids, the predominant fuel for many tissues throughout the body with a high rate of fatty acid metabolism, such as cardiac muscle [Citation10,Citation11]. CD36 facilitates both transendothelial fatty acid transport [Citation12] and fatty acid uptake into cardiomyocytes [Citation6,Citation7] and is responsible for >70% of the rate of fatty acid uptake and oxidation in contracting myocardium [Citation6]. CD36 is a transmembrane protein that undergoes extensive co-translational and post-translational modifications, including glycosylation, palmitoylation, ubiquitination, and phosphorylation [Citation13]. Despite many years of research, the protein structure and membrane topology were only elucidated in 2016. The fatty acid facilitator function of CD36 involves its interaction with other membranes and cytoplasmic proteins () [Citation14].
Figure 1. Cartoon illustrating the sequential steps involved in the uptake of long-chain fatty acids by cells. 1. Release of fatty acids from (interstitial) albumin; 2. Binding in the hydrophobic cavity of CD36 which can accommodate up to two fatty acids at a time; 3. Guidance of the fatty acid through the CD36 ectodomain interior to pass the unstirred water layer and be exposed to the plasma membrane surface; 4. Exit of the fatty acid from CD36 to the outer leaflet of the phospholipid bilayer; 5. Transmembrane translocation (‘flip-flop’) of single fatty acids; 6. Desorption of fatty acids from the inner leaflet of the phospholipid bilayer and binding to the interior of FABP that is anchored by binding to the intracellular part of CD36; 7. Diffusion into the soluble cytoplasm of the fatty acid–FABP complex toward sites of intracellular fatty acid metabolism. Note that proteins and membranes, and their putative mutual interactions are not drawn to scale. Reproduced with permission from Glatz JFC, Luiken JJFP. Time for a détente in the war on the mechanism of cellular fatty acid uptake. J Lipid Res. 2020;61:1300–1303 [Citation14]
![Figure 1. Cartoon illustrating the sequential steps involved in the uptake of long-chain fatty acids by cells. 1. Release of fatty acids from (interstitial) albumin; 2. Binding in the hydrophobic cavity of CD36 which can accommodate up to two fatty acids at a time; 3. Guidance of the fatty acid through the CD36 ectodomain interior to pass the unstirred water layer and be exposed to the plasma membrane surface; 4. Exit of the fatty acid from CD36 to the outer leaflet of the phospholipid bilayer; 5. Transmembrane translocation (‘flip-flop’) of single fatty acids; 6. Desorption of fatty acids from the inner leaflet of the phospholipid bilayer and binding to the interior of FABP that is anchored by binding to the intracellular part of CD36; 7. Diffusion into the soluble cytoplasm of the fatty acid–FABP complex toward sites of intracellular fatty acid metabolism. Note that proteins and membranes, and their putative mutual interactions are not drawn to scale. Reproduced with permission from Glatz JFC, Luiken JJFP. Time for a détente in the war on the mechanism of cellular fatty acid uptake. J Lipid Res. 2020;61:1300–1303 [Citation14]](/cms/asset/2f082b23-a44e-4a73-9660-5add74d17608/iett_a_1941865_f0001_oc.jpg)
Insight into the molecular mechanism underlying the functioning of CD36 was dramatically enhanced when it was discovered that CD36 recycles between intracellular endosomes and the plasma membrane [Citation15]. Translocation from endosomes to the plasma membrane is triggered by hormones, such as insulin, and by an increase in muscle contraction, which takes place within minutes. Conversely, when these triggers disappear, CD36 quickly is internalized. In addition to the sarcolemma, CD36 also translocates to mitochondria to support fatty acid oxidation [Citation16]. In this way, CD36 not only facilitates but also governs on a short-term timescale the rate of cellular fatty acid uptake and metabolismin particular, mitochondrial oxidation. This subcellular recycling mechanism has been delineated in much detail during the past few years, with major roles for endosomal H+-ATPase and for CD36 ubiquitination [reviewed in six]. Importantly, this molecular mechanism has been confirmed independently by several investigators. It is of interest that the mechanism shows a high degree of similarity, besides specific differences, with the dynamic regulation of cellular glucose uptake by glucose transporters (GLUTs) [Citation17].
By virtue of its gatekeeper role for cellular fatty acid entry, CD36 also is involved in lipid signaling. Fatty acids are known to be a ligand for nuclear receptorsin particular, peroxisome proliferator-activated receptor (PPAR), thereby influencing the expression of proteins involved in cellular lipid metabolism [Citation18]. In this way, CD36 facilitates the regulation of its own expression. Changes in its expression level contribute to the long-term (mal)adaptation of cells to chronic alterations in lipid metabolism. Exciting more recent studies have revealed that CD36 signaling also regulates the activity of AMP-activated kinase to coordinate fatty acid uptake with its oxidation, and influences insulin action [Citation19]. For example, through a functional interaction with the insulin receptor, CD36 promotes tyrosine phosphorylation of the insulin receptor by Fyn kinase and enhances downstream insulin signaling. This latter process is suppressed by fatty acids [Citation19]. These findings suggest a role for CD36 in optimal insulin responsiveness, and thereby also in the regulation of cardiac glucose metabolism.
The dynamic regulation of sarcolemmal CD36, both on a short-term basis (CD36 subcellular recycling) and a long-term basis (CD36 gene transcription), aimed at properly governing the rate of myocardial fatty acid uptake, is schematically depicted in [Citation20].
Figure 2. Schematic presentation of both the facilitatory and regulatory roles of CD36 in (long-chain) fatty acid uptake into cardiomyocytes. Middle part of figure: At sarcolemmal lipid rafts, CD36, presumably in interaction with the peripheral membrane protein FABPpm and, at the intracellular site, with cytoplasmic FABP (FABPc), facilitates the entry of fatty acids into the cell. Left part of figure: Short-term regulation (i.e. minutes) of the rate of cellular fatty acid uptake occurs by reversible intracellular recycling (by vesicular transport) of CD36 from an endosomal storage compartment to the sarcolemma, which is triggered by changes in the frequency of muscle contraction or by plasma insulin. These latter triggers are mediated by the AMPK-activated and insulin signaling cascades, respectively, which converge at AS160. Right part of figure: Long-term regulation of cellular fatty acid uptake occurs via changes in CD36 gene transcription, mediated amongst others by fatty acid-induced PPAR activation, HIF-1, and C/EBP. Reproduced with permission from Glatz JFC, Luiken JJFP. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie. 2017;136:21–26 [Citation20]
![Figure 2. Schematic presentation of both the facilitatory and regulatory roles of CD36 in (long-chain) fatty acid uptake into cardiomyocytes. Middle part of figure: At sarcolemmal lipid rafts, CD36, presumably in interaction with the peripheral membrane protein FABPpm and, at the intracellular site, with cytoplasmic FABP (FABPc), facilitates the entry of fatty acids into the cell. Left part of figure: Short-term regulation (i.e. minutes) of the rate of cellular fatty acid uptake occurs by reversible intracellular recycling (by vesicular transport) of CD36 from an endosomal storage compartment to the sarcolemma, which is triggered by changes in the frequency of muscle contraction or by plasma insulin. These latter triggers are mediated by the AMPK-activated and insulin signaling cascades, respectively, which converge at AS160. Right part of figure: Long-term regulation of cellular fatty acid uptake occurs via changes in CD36 gene transcription, mediated amongst others by fatty acid-induced PPAR activation, HIF-1, and C/EBP. Reproduced with permission from Glatz JFC, Luiken JJFP. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie. 2017;136:21–26 [Citation20]](/cms/asset/442c4e52-34a4-4216-9756-45028b7b955f/iett_a_1941865_f0002_oc.jpg)
3. CD36 in the pathogenesis of cardiometabolic diseases
Several chronic diseases are characterized by a perturbed fatty acid or lipid metabolism. These include (i) obesity-induced cardiac dysfunction and diabetic cardiomyopathy, (ii) cardiac hypertrophy and failure, and (iii) ischemia/reperfusion.
Obesity is associated with disturbances in lipid metabolism resulting in deleterious triacylglycerol accumulation in ectopic tissues including the heart, and subsequently in insulin resistance and cardiac contractile dysfunction (together referred to as diabetic cardiomyopathy) [Citation2]. The molecular mechanism underlying this latter pathological development has been disclosed in much detail and has shown a key role for CD36. Retention of CD36 in the endosomes was found to depend on proper acidification of the organelle, which is maintained by the proton-pumping activity of the endosomal enzyme vacuolar H+-ATPase (v-ATPase). The v-ATPase multimeric protein complex consists of two subcomplexes, i.e. a membrane-associated subcomplex, and a cytoplasmic subcomplex. Prolonged exposure of cardiomyocytes to excess lipid supply induces disassembly of v-ATPase which impairs proton pumping into the endosomes. The resulting alkalinization of endosomes is followed by enhanced vesicular trafficking of CD36 to the plasma membrane and finally a further increased rate of lipid uptake [Citation21,Citation22]. Chronic lipid oversupply of the cardiomyocytes eventually inhibits insulin signaling and causes cardiac contractile dysfunction. Importantly, in isolated rat cardiomyocytes it was found that immunochemical inhibition or deletion of CD36 prevents this entire sequence of events and thus protects against high-fat diet-induced insulin resistance and contractile dysfunction [Citation23].
In the hypertrophic heart, myocardial energy provision has shifted toward an increased utilization of glucose at the expense of fatty acids [Citation5]. When the hypertrophy progresses to heart failure, this is accompanied by a further reliance on glucose and a further decline of fatty acid utilization. Such chronic fuel shift toward a single type of substrate is closely linked with cardiac contractile dysfunction [Citation24]. Therefore, rebalancing substrate supply (glucose versus fatty acids) is an effective strategy to treat the failing heart, while a further fuel shift toward a single type of substrate will increasingly impair cardiac function. This concept is illustrated by the observation that cardiomyocyte-specific ablation of CD36 in mice [Citation25] or CD36 deficiency in humans [Citation26,Citation27] decreases fatty acid uptake and accelerates the progression from compensated hypertrophy to heart failure. These findings suggest that restoring or stimulating CD36 function may be useful to prevent or treat, respectively, hypertrophic heart failure.
Finally, CD36 has been implicated in the metabolic changes occurring in cardiac ischemia and reperfusion, both acute and chronic. Following the onset of ischemia, there is a reorganization of sarcolemmal substrate transporters with CD36 moving away and GLUT4 moving toward the sarcolemma, resulting in a shift away from fatty acid metabolism toward (more oxygen-efficient) glucose metabolism [Citation28]. During reperfusion, the transporters initially remain relocated to allow glucose to be used for replenishment of glycogen stores while preventing intracellular lipid overload so as to protect the heart should another ischemic insult occur. Chronic ischemia of the heart causes a downregulation of CD36 expression [Citation7,Citation29], which is generally reported as being harmful for functional recovery from ischemic episodes [Citation29–31]. Although not the total cellular pool of CD36, but rather the portion present at the sarcolemma determines the rate of fatty acid uptake, a small CD36 pool may limit the dynamic interplay of substrate transporters (in particular CD36 and GLUT4) and that compromise the rapid changes in myocellular substrate preference required to sustain the phases of an ischemic insult (i.e. ischemia, reperfusion, and normal perfusion).
Taken together, CD36 has emerged as a pivotal protein both for the homeostatic control of myocellular fatty acid uptake and utilization (and indirectly of that of other fuels such as glucose), and for the development of pathological changes leading to myocardial dysfunction, suggesting that manipulation of sarcolemmal CD36 presence or functioning is an effective therapeutic approach to fight metabolic heart disease.
4. Modulation of sarcolemmal CD36 and cardiac disease
The dynamic subcellular recycling of CD36 permits a temporal and accurate adjustment of the presence of CD36 in the plasma membrane and, thereby, of the rate of cellular fatty acid uptake and utilization. Evidence accumulating in the last few years strongly indicates the applicability of the subcellular recycling of CD36 to manipulate myocardial substrate preference. In general, normalizing cardiac energy provision aimed at rebalancing the use of fatty acids versus glucose through manipulation of CD36 restores contractile function of the failing heart [reviewed in 24].
Specific examples of modulation of sarcolemmal CD36 to mend the failing heart are:
Decreasing CD36: Ablation of CD36 in an experimental model of lipotoxic cardiomyopathy (cardiac-specific PPARα overexpression) normalized cardiac fatty acid and glucose utilization and rescued contractile dysfunction [Citation32].
Increasing CD36: Mice with a cardiac-specific overexpression of protein kinase-D1, which leads to a higher utilization of glucose combined with a lower utilization of fatty acids and to cardiac hypertrophy, could be rescued by feeding a high-fat diet upon which both the CD36-mediated fatty acid utilization rate and contractile function normalized [Citation33].
Inhibition of CD36: Mice treated with daily injections of the selective CD36 inhibitor EP-80,317 showed reduced myocardial fatty acid uptake and were protected against cardiac injury and dysfunction elicited by experimental ischemia/reperfusion [Citation34].
These examples from experimental studies provide proof-of-principle for CD36 to be an effective target for normalizing cardiac substrate metabolism aimed at improving or restoring cardiac contractile function [Citation1]. This approach is part of the so-called metabolic modulation therapy [Citation35].
5. How to manipulate CD36 and its therapeutic effect?
Manipulating the presence or activity of CD36 at the sarcolemma could be achieved by several approaches, including (i) altering the expression of the CD36 gene and/or influence its post-translational modification, (ii) interfering with the recycling of CD36 between endosomes and the sarcolemma either to increase or decrease its sarcolemmal presence, and (iii) functional inhibiting CD36 present at the sarcolemmal membrane (). Most efforts described in the literature comprise this last option. Examples of known compounds affecting CD36 function are listed in .
Figure 3. Illustration of the four distinct levels at which the functioning of CD36 in cardiomyocyte fatty acid uptake can be modulated. 1. Intervention of signaling pathways affecting CD36 gene expression; 2. Intervention in co-translational modification of CD36, in particular its glycosylation; 3. Modulation of reversible vesicular transport of CD36 between an endosomal storage compartment and the sarcolemma (subcellular recycling); 4. Influencing CD36 palmitoylation, its insertion into the sarcolemma, and putative activation (e.g. interaction with other membrane-associated proteins)
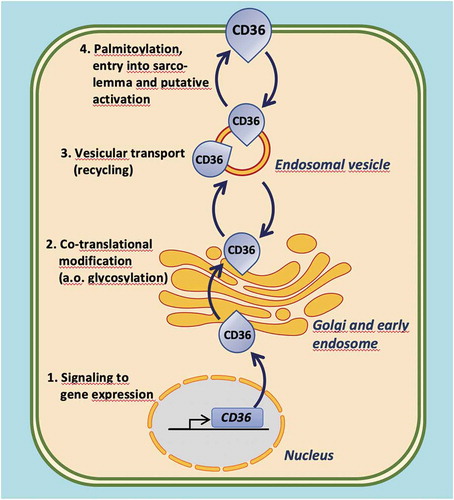
Table 1. Compounds affecting CD36 function or gene expression and their reported effects
Various compounds have been described that inhibit CD36 functioning at the plasma membrane. The best-known examples are sulfo-N-succinimidyl derivatives of long-chain fatty acids, in particular palmitate (SSP) or oleate (SSO), which compounds have been instrumental for the discovery of the fatty acid-binding properties of CD36 [Citation10]. SSO is widely applied to inhibit cellular fatty acid uptake in experimental studies in vitro [Citation36–39]. SSO binds in the fatty acid-binding pocket of CD36 by covalent interaction with amino acid residue Lys-164 thus acts as an irreversible inhibitor of CD36, thereby preventing the binding of long-chain fatty acids [Citation37]. However, the short half-life of SSP or SSO when in aqueous solution limits their use in vivo [Citation36].
As mentioned above, EP80317 is a selective synthetic peptide ligand of CD36 that was reported to protect the heart against post-ischemic myocardial injury in mice [Citation34]. Specifically, the pretreatment of mice with subcutaneous injections of EP80317 followed by experimentally elicited infarction showed reduced injury and dysfunction when compared to sham injections, while no effect of treatment was observed in CD36 null mice [Citation34]. Likewise, in rat studies, hexarelin, a synthetic growth hormone-releasing peptide that binds to the growth hormone receptor but also to CD36 was found to protect the heart from ischemia/reperfusion injury [Citation40,Citation41]. Exenatide, a long half-life analog of glucagon-like peptide 1 (GLP-1) also improves post-ischemic cardiac function in rats [Citation42]. Interestingly, exenatide does not inhibit CD36 but suppresses its translocation to the sarcolemma while simultaneously increasing the translocation of GLUT4 and GLUT1 from the intracellular stores to the sarcolemma [Citation42].
Chemical compounds with anti-CD36 action were also identified by high-throughput screening of large chemical libraries. Using a CD36-expressing HEK-293 cell-line Geloen et al. [Citation43] identified the small molecular mass compounds AP5055 and AP5258 as CD36 inhibitors that block cellular lipid uptake and, when applied in vivo in rodents, can reduce postprandial hypertriglyceridemia, atherosclerosis, and be beneficial for type 2 diabetes.
Puerarin, a natural isoflavonoid compound isolated from the root of the plant Pueraria lobata and known to have anti-diabetic activity [Citation44] was found to alleviate cardiac lipotoxicity in mice both in vitro and in vivo [Citation45]. Interestingly, in the skeletal muscles of diabetic rats puerarin decreased the trafficking of CD36 to the sarcolemma to reduce the rate of cellular fatty acid uptake [Citation46]. Other natural compounds affecting CD36 function include the plant alkoid berberine [Citation47] and the sesquiterpene lactone endoperoxide artemisinin [Citation48]. These latter two compounds lower cellular fatty acid uptake by suppressing CD36 gene expression, but most likely also influence the expression of other proteins and enzymes involved in lipid metabolism.
More recently, the pharmacological sodium-glucose cotransporter 2 (SGLT2) inhibitor Empagliflozin was shown in type 2 diabetes to significantly reduce myocardial CD36 and cardiotoxic lipids (specifically ceramides), explaining its beneficial action to restore cardiac function in type 2 diabetes patients [Citation49]. Finally, monoclonal antibodies, specifically directed against CD36 have also been used to inhibit fatty acid uptake by isolated cardiomyocytes [Citation23].
6. Expert opinion
Research performed in recent years has clearly established a pivotal role for the membrane protein CD36 in cellular lipid signaling and metabolism with particular reference to the heart. The underlying molecular mechanisms of CD36 functioning have been largely disclosed and proof-of-concept has been obtained that its manipulation alters lipid metabolism effectively and can be applied to modulate cardiac metabolism so as to improve or restore cardiac contractile functioning. Furthermore, in the past two decades several compounds were found to be ligands of CD36, and a number of these were shown to act as inhibitor for CD36-mediated cellular lipid uptake as illustrated in . Finally, small molecule inhibitors of CD36 have been generated and when administered to animal models in vivo were shown to inhibit CD36 [Citation34,Citation36,Citation37,Citation43] or affect its subcellular recycling [Citation42] thereby altering lipid metabolism. While the suppression of CD36 confers protection against obesity-induced cardiac dysfunction and diabetic cardiomyopathy, stimulation of CD36 is useful for the alleviation of cardiac hypertrophy and heart failure. These exciting developments identify CD36 as an exceptionally promising and effective treatment option to correct cardiac energy metabolism and cure heart disease. However, a number of aspects need further consideration.
Of major importance is the specificity of the approach to alter CD36 functioning. First, CD36 is a scavenger receptor with a broad cell-type expression, being found not only in cardiac and skeletal myocytes, but also in adipocytes, intestinal enterocytes, macrophages, and monocytes, while its expression in the liver is induced upon high fat consumption [Citation50]. Second, CD36 not only binds long-chain fatty acids, but also is a receptor for oxidized low-density lipoproteins, thrombospondin, and collagen, apoptotic cells, and erythrocytes infected with Plasmodium falciparum [reviewed in 50]. As a result, CD36 is a multifunctional protein involved in a variety of (mostly lipid-related) pathways. Therefore, when altering CD36 presence or functioning by direct inhibition of the protein, the possibility of introducing unwanted side effects should always be considered and evaluated. For instance, it has been found that blocking CD36 inhibits macrophage phagocytosis of dead myocytes and neutrophils resulting in impaired removal of debris and adverse left ventricular remodeling post-myocardial infarction [Citation51]. However, this limitation may be overcome by indirect approaches to alter CD36 presence in the plasma membrane, i.e. by targeting the subcellular recycling of CD36 between endosomes and the plasma membrane, and do so in a tissue-specific manner [Citation52] as explained below.
The subcellular vesicular recycling of membrane proteins and receptors is a highly specific process in which some 50–60 distinct proteins are involved, including coat proteins, adaptor proteins, bilayer destabilizing kinases, vesicle-associated membrane proteins (VAMPs), and motor proteins [Citation53,Citation54]. Particular sets of proteins participate in the intracellular trafficking of specific membrane proteins, such as CD36, thereby providing tissue and cell-type specificity. Although the CD36-dedicated vesicular trafficking machinery has not yet been fully elucidated, a recent study has identified a VAMP isoform (VAMP4) to be specifically participating in myocardial CD36 traffic [Citation55]. Silencing of this VAMP isoform in cardiomyocytes simultaneously decreased stimulus-induced CD36 translocation to the sarcolemma and the rate of cellular fatty acid uptake without affecting GLUT4-mediated glucose uptake [Citation55]. This finding indicates the feasibility of using VAMP4 as target to manipulate specifically CD36-mediated cellular fatty acid uptake without affecting glucose uptake. The latter is crucial to avoid an energy deficit of the heart.
Alternative manners of manipulating CD36 sarcolemmal presence and/or function are targeting intrinsic mechanisms regulating the expression of CD36 [Citation47,Citation48] or of the post-translational modification of CD36 () [Citation7,Citation13]. However, known agents that affect these processes are commonly not specific for CD36 because these are directed against pathways regulating sets of lipid metabolic proteins and/or enzymes. For instance, in addition to the natural and pharmacological compounds already discussed (), selective PPAR δ activation increases the expression of myocardial lipid transport and metabolism genes, including CD36, to rebalance cardiac substrate metabolism and reduce hypertrophy and failure [Citation56]. It should be emphasized that the sarcolemmal content of CD36 is not strictly dependent on the total pool size of CD36 although a minimal amount will be required to allow fluctuations in CD36 recycling. Interestingly, there is preliminary evidence that once at the sarcolemma, CD36 needs to be ‘activated’ to facilitate cellular long-chain fatty acid uptake [Citation23]. The nature of this activation is not yet known (it putatively involves interaction with other membrane-associated or cytoplasmic proteins [Citation6]), but may represent a further option to specifically manipulate the rate of cellular fatty acid uptake.
Being a heavily glycosylated transmembrane protein, the molecular structure of CD36 has long remained undissolved. However, with newer technologies and by comparison with other scavenger receptors, a detailed structure of the spatial organization of the CD36 molecule and its membrane topology are now available [Citation57]. This knowledge will dramatically accelerate the design of small-molecule compounds to specifically influence CD36 functioning. The subsequent evaluation of such compounds may use a high-throughput fatty acid uptake assay (using fluorescently labeled fatty acids). An alternative high-throughput screening assay using an immunochemical approach has been described by Wang et al. [Citation58]. Please note that a robust compound testing flow scheme is also available to identify compounds that would affect CD36 recycling [Citation55].
Finally, the impact of CD36 manipulation on myocardial metabolism and contractile function will be complicated and difficult to predict in the many patients who suffer from two, or more cardiac pathologies at the same time (e.g. diabetes and ischemia/myocardial infarction). Further research is needed to investigate such cases. Likewise, the insights obtained for myocardium should be advocated to be explored for their applicability to other tissues as well as other conditions associated with altered metabolism such as cancer.
Article highlights
The transmembrane protein CD36 is a master regulator of cellular lipid metabolism.
In cardiac muscle, the uptake of long-chain fatty acids is facilitated by CD36 and regulated by the reversible subcellular recycling of CD36 between endosomes and the sarcolemma.
Chronic cardiac diseases are commonly associated with specific changes in energy metabolismin particular, the uptake and utilization of fatty acids versus that of glucose.
In experimental models of cardiac disease, manipulation of the presence of CD36 in the sarcolemma was found to normalize myocardial energy metabolism and improve or restore cardiac contractile function.
CD36 is an excellent target for metabolic modulation therapy to fight chronic cardiac diseases; this warrants the search for small molecule inhibitors of CD36 to combat lipid overload or for agents specifically affecting the (substrate and tissue-specific) subcellular recycling process of CD36 to either increase or decrease myocardial lipid uptake.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers in this manuscript have no relevant financial or other relationships to disclose
Additional information
Funding
References
- Wende AR, Brahma MK, McGinnis GR, et al. Metabolic origins of heart failure. JACC Basic Transl Science. 2017;2:297–310.
- Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocrinol Metabol Disorders. 2010;11(1):31–39.
- Noordali H, Loudon BL, Frenneaux MP, et al. Cardiac metabolism – a promising therapeutic target for heart failure. Pharmacol Therap. 2018;182:95–114.
- Zuurbier CJ, Bertrand L, Beualoye CR, et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J Cell Mol Med. 2020;24(11):5937–5954.
- Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85(3):1093–1129.
- Glatz JFC, Luiken JJFP. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J Lipid Res. 2018;59(7):1084–1093.
- Shu H, Peng Y, Hang W, et al. The role of CD36 in cardiovascular disease. Cardiovasc Res. 2021; in press. DOI:10.1093/cvr/cvaa319.
- Clemetson KJ, Pfueller ST, Luscher EF, et al. Isolation of the membrane glycoproteins of human platelets by lectin affinity chromatography. Biochim Biophys Acta. 1977;464(3):493–508.
- Endemann G, Stanton LW, Madden KS, et al. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem. 1993;268(16):11811–11816.
- Abumrad NA, El-Maghrabi MR, Amri EZ, et al. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J Biol Chem. 1993;268(24):17665–17668.
- Van Nieuwenhoven FA, Verstijnen CP, Abumrad NA, et al. Putative membrane fatty acid translocase and cytoplasmic fatty acid-binding protein are co-expressed in rat heart and skeletal muscles. Biochem Biophys Res Commun. 1995;207(2):747–752.
- Son N-H, Basu D, Samovski D, et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J Clin Invest. 2018;128(10):4329–4342.
- Luiken JJFP, Chanda D, Nabben M, et al. Post-translational modifications of CD36 (SR-B2): implications for regulation of myocellular fatty acids uptake. Biochim Biophys Acta. 2016;1862(12):2253–2258.
- Glatz JFC, Luiken JJFP. Time for a détente in the war on the mechanism of cellular fatty acid uptake. J Lipid Res. 2020;61(9):1300–1303.
- Bonen A, Luiken JJFP, Arumugam Y, et al. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem. 2000;275(19):14501–14508.
- Smith BK, Jain SS, Rimbaud S, et al. FAT/CD36 is located on the outer mitochondrial membrane, upstream of long-chain acyl-CoA synthetase, and regulates palmitate oxidation. Biochem J. 2011;437(1):125–134.
- Klip A, McGraw TE, James DE. Thirty sweet years of GLUT4. J Biol Chem. 2019;294:11369–11381.
- Neels JG, Grimaldi PA. Physiological functions of peroxisome proliferator-activated receptor β. Physiol Rev. 2014;94(3):795–858.
- Samovski D, Dhule P, Pietka T, et al. Regulation of insulin receptor pathway and glucose metabolism by CD36 signaling. Diabetes. 2018;67(7):1272–1284.
- Glatz JFC, Luiken JJFP. From fat to FAT (CD36/SR-B2): understanding the regulation of cellular fatty acid uptake. Biochimie. 2017;136:21–26.
- Liu Y, Steinbusch LKM, Nabben M, et al. Palmitate-induced vacuolar-Type H+-ATPase inhibition feeds forward into insulin resistance and contractile dysfunction. Diabetes. 2017;66(6):1521–1534.
- Zhu B, Li MY, Lin Q, et al. Lipid oversupply induces CD36 sarcolemmal translocation via dual modulation of PKCζ and TBC1D1: an early event prior to insulin resistance. Theranostics. 2020;10(3):1332–1354.
- Angin Y, Steinbusch LKM, Simons PJ, et al. CD36 inhibition prevents lipid accumulation and contractile dysfunction in rat cardiomyocytes. Biochem J. 2012;448(1):43–53.
- Glatz JFC, Nabben M, Young ME, et al. Re-balancing cellular energy substrate metabolism to mend the failing heart. Biochim Biophys Acta. 2020;1866(5):165579.
- Sung MM, Byrne NJ, Kim TT, et al. Cardiomyocyte-specific ablation of CD36 accelerates the progression from compensated cardiac hypertrophy to heart failure. Am J Physiol. 2017;312:H552–H560.
- Tanaka T, Nakata T, Oka T, et al. Defect in human myocardial long-chain fatty acid uptake is caused by FAT/CD36 mutations. J Lipid Res. 2001;2001(42):751–759.
- Miyazaki M, Suematsu Y, Kato S, et al. Type 1 cluster of differentiation 36 deficiency-related cardiomyopathy accelerates heart failure with co-existing mitral valve prolapse: a case report. Eur Heart J Case Rep. 2019;3:1–5.
- Heather LC, Pates KM, Atherton HJ, et al. Differential translocation of the fatty acid transporter, FAT/CD36, and the glucose transporter, GLUT4, coordinates changes in cardiac substrate metabolism during ischemia and reperfusion. Circ Heart Fail. 2013;6(5):1058–1066.
- Nedvedova I, Kolar D, Neckar J, et al. Cardioprotective regimen of adaptation to chronic hypoxia diversely alters myocardial gene expression in SHR and SHR-mtBN conplastic rat strains. Front Endocrinol. 2019;9:809.
- Irie H, Krukenkamp IB, Brinkmann JFF, et al. Myocardial recovery from ischemia is impaired in CD36-null mice and restored by myocyte CD36 expression or medium-chain fatty acids. Proc Natl Acad Sci USA. 2003;100(11):6819–6824.
- Neckár J, Silhavy J, Zídek V, et al. CD36 overexpression predisposes to arrhythmias but reduces infarct size in spontaneously hypertensive rats: gene expression profile analysis. Physiol Genomics. 2012;44(2):173–182.
- Yang J, Sambandam N, Han X, et al. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100(8):1208–1217.
- Dirkx E, Van Eys GJJM, Schwenk RW, et al. Protein kinase-D1 overexpression prevents lipid-induced cardiac insulin resistance. J Mol Cell Cardiol. 2014;76:208–217.
- Bessi VL, Labbé SM, Huynh DN, et al. EP 80317, a selective CD36 ligand, shows cardioprotective effects against post-ischaemic myocardial damage in mice. Cardiovasc Res. 2012;96(1):99–108.
- Revenco D, Morgan JP. Metabolic modulation and cellular therapy of cardiac dysfunction and failure. J Cell Mol Med. 2009;13(5):811–825.
- Coort SLM, Willems J, Coumans WA, et al. Sulfo-N-succinimidyl esters of long chain fatty acids specifically inhibit fatty acid translocase (FAT/CD36)-mediated cellular fatty acid uptake. Mol Cell Biochem. 2002;239(1/2):213–219.
- Kuda O, Pietka TA, Demianova Z, et al. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 Lysine 164. J Biol Chem. 2013;288(22):15547–15555.
- Mansor LS, da Luz Sousa Fialho M, Yea G, et al. Inhibition of sarcolemmal FAT/CD36 by sulfo-N-succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation. Cardiovasc Res. 2017;113(7):737–748.
- Khan S, Kowluru A. CD36 mediates lipid accumulation in pancreatic beta cells under the duress of glucolipotoxic conditions: novel roles of lysine deacetylases. Biochem Biophys Res Commun. 2018;495(3):2221–2226.
- Mao Y, Tokudome T, Kishimoto I. The cardiovascular action of hexarelin. Review J Geriatr Cardiol. 2014;11(3):253–258.
- Huang J, Li Y, Zhang J, et al. The growth hormone secretagogue hexarelin protects rat cardiomyocytes from in vivo ischemia/reperfusion injury through interleukin-1 signaling pathway. Int Heart J. 2017;58(2):257–263.
- Zheng A, Cao L, Qin S, et al. Exenatide regulates substrate preferences through the p38 γ MAPK pathway after ischaemia/reperfusion injury in a rat heart. Heart Lung Circ. 2017;26(4):404–412.
- Geloen A, Helin L, Geeraert B, et al. CD36 inhibitors reduce postprandial hypertriglyceridemia and protect against diabetic dyslipidemia and atherosclerosis. PLoS ONE. 2012;7(5):e37633.
- Bai L, Li X, He L, et al. Antidiabetic potential of flavonoids from traditional Chinese medicine: a review. Am J Chin Med. 2019;47(5):933–957.
- Qin H, Zhang Y, Wang R, et al. Puerarin suppresses Na+-K+-APTase-mediated systemic inflammation and CD36 expression, and alleviates cardiac lipotoxicity in vitro and in vivo. J Cardiovasc Pharmacol. 2016;68(6):465–472.
- Chen X-F, Wang L, Wu Y-Z, et al. Effect of puerarin in promoting fatty acid oxidation by increasing mitochondrial oxidative capacity and biogenesis in skeletal muscle in diabetic rats. Nutr Diab. 2018;8(1):1.
- Chen Y, Li Y, Wang Y, et al. Berberine improves free-fatty-acid-induced insulin resistance in L6 myotubes through inhibiting peroxisome proliferator-activated receptor γ and fatty acid transferase expression. Metab Clin Exper. 2009;58(12):1694–1702.
- Wang J, Wang S, Zhang X, et al. Artemisinin reduces lipid accumulation in hepatocytes by inhibition of CD36 expression. Indian J Pharmaceut Educ Res. 2017;51(3):393–400.
- Aragón-Herrera A, Feijóo-Bandín S, Otero Santiago M, et al. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats. Biochem Pharmacol. 2019;170:113677.
- Febbraio M, Hajjar CP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108(6):785–791.
- Lindsey ML, Jung M, Yabluchanskiy A, et al. Exogenous CXCL4 infusion inhibits macrophage phagocytosis by limiting CD36 signalling to enhance post-myocardial infarction cardiac dilation and mortality. Cardiovasc Res. 2019;115(2):395–408.
- Glatz JFC, Luiken JJFP, Nabben M. CD36 (SR-B2) as a target to treat lipid overload-induced cardiac dysfunction. J Lipid Atheroscl. 2020;9(1):66–78.
- Chi RJ, Harrison MS, Burd CG. Biogenesis of endosome-derived transport carriers. Cell Mol Life Sci. 2015;72(18):3441–3455.
- Larance M, Ramm G, Stöckli J, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280(45):37803–37813.
- Schwenk RW, Dirkx E, Coumans WA, et al. Requirement for distinct vesicle-associated membrane proteins in insulin- and AMP-activated protein kinase (AMPK)-induced translocation of GLUT4 and CD36 in cultured cardiomyocytes. Diabetologia. 2010;53(10):2209–2219.
- Jucker BM, Doe CP, Schnackenberg CG, et al. PPAR δ activation normalizes cardiac substrate metabolism and reduces right ventricular hypertrophy in congestive heart failure. J Cardiovasc Pharmacol. 2007;50(1):25–34.
- Hsieh FL, Turner L, Bolla JR, et al. The structural basis for CD36 binding by the malaria parasite. Nat Commun. 2016;7(1):12837.
- Wang L, Bao Y, Yang Y, et al. Discovery of antagonists for human scavenger receptor CD36 via an ELISA-like high-throughput screening assay. J Biomol Screening. 2010;15(3):239–250.