
ABSTRACT
Introduction
Cerebral edema is a key contributor to death and disability in several forms of brain injury. Current treatment options are limited, reactive, and associated with significant morbidity. Targeted therapies are emerging based on a growing understanding of the molecular underpinnings of cerebral edema.
Areas Covered
We review the pathophysiology and relationships between different cerebral edema subtypes to provide a foundation for emerging therapies. Mechanisms for promising molecular targets are discussed, with an emphasis on those advancing in clinical trials, including ion and water channels (AQP4, SUR1-TRPM4) and other proteins/lipids involved in edema signaling pathways (AVP, COX2, VEGF, and S1P). Research on novel treatment modalities for cerebral edema [including recombinant proteins and gene therapies] is presented and finally, insights on reducing secondary injury and improving clinical outcome are offered.
Expert Opinion
Targeted molecular strategies to minimize or prevent cerebral edema are promising. Inhibition of SUR1-TRPM4 (glyburide/glibenclamide) and VEGF (bevacizumab) are currently closest to translation based on advances in clinical trials. However, the latter, tested in glioblastoma multiforme, has not demonstrated survival benefit. Research on recombinant proteins and gene therapies for cerebral edema is in its infancy, but early results are encouraging. These newer modalities may facilitate our understanding of the pathobiology underlying cerebral edema.
KEYWORDS:
- Cerebral edema
- SUR1-TRPM4 (sulfonylurea receptor 1- transient receptor potential cation channel subfamily M member 4)
- Glyburide / Glibenclamide
- VEGF (Vascular Endothelial Growth Factor)
- AQP4 (Aquaporin4)
- bevacizumab
- S1P (sphingosine 1 phosphate)
- Fingolimod
- Conivaptan
- AVP (Arginine Vasopressin)
- COX2 (Cyclooxygenase-2)
- Celecoxib
1. Introduction
Cerebral edema is a pathological accumulation of fluid in the brain that increases the net brain-tissue water mass [Citation1–3]. It affects almost all types of acute brain injuries (ABI) commonly seen in neurocritical care units including ischemic stroke, intracerebral hemorrhage (ICH), subarachnoid hemorrhage (SAH), cardiac arrest (CA), traumatic brain injury (TBI), meningitis/encephalitis, central nervous system (CNS) abscesses, acute liver failure, status-epilepticus, primary brain tumors, and metastases, as well as systemic diseases such as diabetic ketoacidosis and sepsis. In many of these primary pathologies, cerebral edema is an independent risk factor for unfavorable prognosis with increased morbidity and mortality, in some cases by more than 80% [Citation1,Citation2,Citation4–8]. There is some degree of molecular overlap in these diseases, with several of the same components, pathways, and networks identified as contributors to cerebral edema. However, there are also important distinctions depending on predominance of specific pathways, spatial location, timing, individual host response, and genetic predispositions. Substantial advances in identifying the underlying molecular machinery of cerebral edema have yielded exciting therapeutic targets to prevent or reduce edema generation. In isolation, these targeted anti-edema agents may not reach their full potential. However, edema therapy based on precision medicine could be optimized by early recognition and serial monitoring of key pathways that are activated/suppressed in individual patients using complementary tools like biomarkers, imaging, multimodal monitoring, and genetic predispositions.
Current clinical management of cerebral edema remains limited with non-specific temporizing strategies utilized largely to prevent the consequences of raised intracranial pressure (ICP) and impending herniation rather than targeting specific molecular pathways. The mechanism by which raised ICP causes herniation and death is explained by the Monro-Kellie doctrine (Section II-A) whereby expanding edema (or other causes of intracranial volume) is limited by the rigid skull, and after compensatory mechanisms are compensated, increased pressure on the brain tissue causes herniation. The current therapeutic armamentarium to address this devastating clinical problem commonly consists of hyperosmolar therapies, cerebrospinal fluid (CSF) drainage, steroids (primarily for peritumoral edema), therapeutic hypothermia, deep sedation with anesthetic agents, paralysis, and decompressive craniectomy. Many of these agents have been used in some form for centuries. For example, hyperosmolar therapy was described in the 1920s, trephination to relieve intracranial pressure was used in Peru (8000 BCE), ancient Egypt (1700 BCE) and China (Xinjiang province, 1000 BCE), and was formally described by Hippocrates in 400 BCE [Citation9]. While some of the more extreme therapies, e.g. mannitol, 23.4% NaCl, and craniectomy, can acutely salvage a herniating brain, these agents carry high risks of morbidity. In recent trials, craniectomy for certain conditions causing cerebral edema (like ischemic stroke/large hemispheric infarction), has improved both morbidity and mortality, whereas in other disease-types (like TBI) the benefit has predominantly been life-saving without clear benefit on functional outcome or disability [Citation10–16].
The next generation of anti-edema therapies will leverage molecular pathophysiology to prevent or minimize edema formation [Citation1–3,Citation8,Citation17]. Advances in bioinformatics, unbiased transcriptomics and proteomics, biomarker development, NextGen sequencing, gene and stem-cell therapies have already valuably informed research into cerebral edema therapies, and likely will continue to exponentially grow (). As outlined (Box 1), this article discusses physical drivers and molecular contributors to cerebral edema (Section II), and focuses on the mechanisms and therapeutic potential of emerging clinically promising targets (Section III). Newer therapeutic modalities such as recombinant proteins (Section IV) and gene therapies for cerebral edema are also briefly presented (Section V).
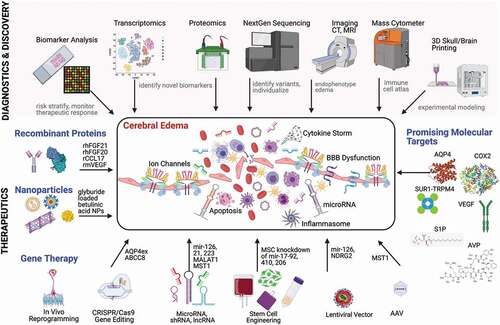
Figure 1. Emerging Therapeutic Strategies for Cerebral Edema. Schematic showing advances in diagnostics and discovery technologies (shaded pink) that have the potential to inform and identify key molecular contributors to cerebral edema. These include biomarker analysis (for risk stratification, theragnostics, and monitoring the therapeutic response), transcriptomics and proteomics (for unbiased identification of novel RNA and protein-based biomarkers and therapeutic targets), NextGen sequencing (to identify important variants that may alter host response to injury and inform individualized targets), advanced imaging (for edema endophenotyping), mass cytometry (immune cell atlas to determine cellular properties), and 3D printing (to facilitate experimental modeling). The bottom panel highlights some novel anti-cerebral edema therapies under development including recombinant proteins (rhFGF21, rhFGF20, rCCL17, rmVEGF), nanoparticles (loaded with glyburide/glibenclamide), gene therapies (including CRISPR/Cas9 gene editing, microRNA, shRNA and lncRNA targets, AAV and lentiviral vectors), and current promising molecular targets with therapeutic agents in clinical trials (AQP4, AVP, COX2, S1P, SUR1-TRPM4 and VEGF). The inset rectangle depicts various contributors to cerebral edema (ion channels, inflammasome, BBB dysfunction, and cytokine storms) as well as potential detrimental after- effects (apoptosis). Figure created with the assistance of BioRender.
2. Pathogenesis
Understanding the physical forces, physiological networks, and molecular drivers of cerebral edema is key to recognizing the rationale underlying currently available therapies. This also enables an appreciation for their limitations, and informs the development of promising targets. In this section, we summarize physical principles that drive cerebral edema, different networks/systems involved in fluid influx and efflux from the CNS, and we provide an overview of key mechanisms and molecular contributors to different cerebral edema subtypes (-B). Specific promising targets are subsequently discussed in detail in Section III on emerging therapies.
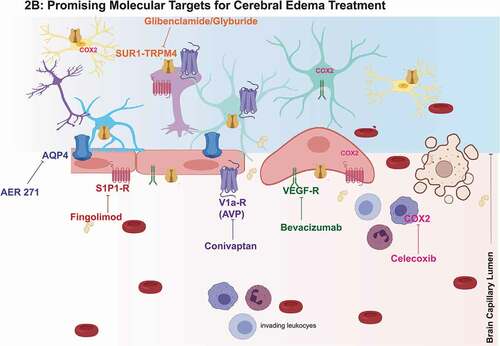
Figure 2. Promising Molecular Targets and Pathophysiologic Evolution of Cerebral Edema.
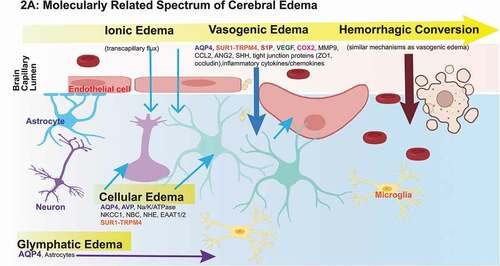
Figure 2b. Continue
2.1. Physical Drivers of Cerebral Edema
Basic Forces
The Monro-Kellie doctrine, described over two centuries ago, outlines the principle that a rigid skull necessitates a fixed sum of intracranial components (blood, CSF/interstitial fluid, brain tissue) whereby an increase in one causes a reciprocal decrease in one (or more) of the others. Once these compensatory reserves are exhausted, ICP rises and can cause herniation and death. The threshold at which this occurs varies between individuals and is a function of intracranial elastance and compliance [Citation8]. Conceptually, this has provided the basis for several current approaches to edema including CSF diversion (external ventricular drain, EVD), reduction in metabolic demand and CBF (sedation, hypothermia), and hyperosmotic agents (mannitol, hypertonic saline). At a local level, the modified Starling equation [quantifies the net flow (Jv) of water across membranes by leveraging differences in capillary (c) vs. interstitial (i) hydrostatic (P) and oncotic (p) pressures [Citation8,Citation18]. This principle explains the action/benefit of hyperosmotic therapies whereby water moves from the brain interstitium and intracellular compartments into the vasculature. The phenomenon of rebound edema can be observed with repeated administration of mannitol or hypertonic saline; it is related to breakdown of the blood–brain barrier (BBB) and accumulation of the hyperosmolar agent within the interstitium/outside an intact vasculature creating a reverse-osmolar gradient.
2.2. Cerebral Edema Pathophysiology and Systems Facilitating Fluid Movement in the Brain
The taxonomy of cerebral edema has historically resulted in separating cytotoxic/cellular edema from vasogenic edema (BBB disruption). While these distinctions are clinically informative to distinguish intracellular energy failure and cell swelling from a leaky BBB, they are also somewhat artificial in light of increasing recognition that these processes are molecularly interconnected and likely represent a spectrum of edema evolution. Although the primary sources of cerebral edema are putatively the cerebrovascular system and CSF circulation as discussed below (particularly re: ionic, cytotoxic/cellular and vasogenic edema), we also present emerging evidence of the contributions of the glymphatic system and meningeal lymphatic vessels to this process. Cerebral edema is also highly linked with neuroinflammation (reviewed elsewhere)[Citation19–22]. This subsection provides an overview of the underlying pathophysiology and key reported molecular contributors to cerebral edema subtypes.
Importantly, cerebral edema manifests differently in different disease states. For example, edema after cerebral ischemia involves early cellular/cytotoxic swelling followed by delayed opening of the BBB. This is different than manifestation in TBI where mechanical disruption of the BBB occurs almost immediately and is subsequently accompanied by cellular, vasogenic, and inflammatory contributors to edema. Furthermore, edema related to acute brain injury varies significantly from that occurring in more chronic disorders including neurodegenerative diseases like Alzheimer’s disease, or even multiple sclerosis. An exhaustive disease-specific delineation of edema mechanisms is beyond the scope of this review, however we present mechanisms that have been found to be relevant and common across the disease spectrum and in Section III (NextGen Emerging Targets), and outline promising molecular targets as well as the preclinical and clinical research on which they are based.
Cellular/Cytotoxic edema refers to intracellular fluid accumulation resulting in cell swelling after several forms of ABI [Citation1,Citation8,Citation17]. Although historically referred to as ‘edema,’ cellular edema is a redistribution of water from the interstitium to the parenchyma. In isolation, it therefore does not increase total brain water content but in the setting of an external fluid source (e.g. perfusion or the glymphatic system), it can serve as a precursor to ionic and vasogenic edema that do cause brain swelling/increase in water content.
Cellular edema has predominantly been described in perineuronal and perivascular astrocytes, immediately after the insult (hypoxia, injury) but can occur in multiple cell-types [Citation8,Citation17]. It has multiple potential triggers including energy failure and endogenous/exogenous toxins (ammonia, glutamate, arachidonic acid, cyanide, H+, K+) resulting in maladaptive ion influx. Osmolyte/ionic uptake into the cell can occur via constitutively expressed drivers (NKCC1, EAAT1/2, NHE, NBC, and mGluR4), or de novo upregulated channels (SUR1-TRPM4) [Citation23–40]. Ionic influx creates an osmotic gradient for water to follow via diffusion, water channels (AQP4, GLUT1, and SGLT1), or ion channels that co-transport water (NKCC1, EAAT1/2). When oncotic cell death from cellular swelling affects perivascular astrocyte podocytes, or endothelial cells, it contributes to vasogenic edema. Clinically, magnetic resonance imaging (MRI)-based apparent diffusion coefficient (ADC) sequences are used to identify cellular swelling [Citation41].
Ionic edema is mechanistically similar to cellular edema in that it utilizes many of the same ion transporters, but it occurs primarily at the capillary level and results in transcapillary flux of ions and water based on a the spatial distribution of ion channels [Citation8,Citation17]. Water and ions are transported inward through the luminal membrane of the capillary endothelial cell, and, in equivalent amounts, outward through the abluminal membrane. With cellular swelling, the interstitial ionic gradient (e.g. Na+) is depleted. The osmotic driving force outlined by the Starling principle () results in an influx of Na+ into the interstitium from the actively perfused capillary lumen, followed by water and negatively charged ions (Cl–) for osmotic and electrical neutrality [Citation17].
Vasogenic edema is a complex interplay of several mechanisms that results in the loss of BBB integrity and extravasation of water and plasma proteins into the CNS interstitium. It overlaps with neuroinflammation. Release of damage associated molecular patterns (DAMPs) after primary and secondary injury elicits an inflammatory immune response including the recruitment, adherence/migration and activation of inflammatory cells facilitated by various endothelial cell surface receptors (VCAM-1, ICAM-1, LFA, and VLA-4) as well as second messenger cascades involving proinflammatory cytokines (TNF-a, IL-1b, IL-6, IL-8, and TGF-b) and chemokines (CXCL-1/2)[Citation8,Citation17,Citation19,Citation42–48]. The mechanisms through which these pathways influence BBB integrity are still being elucidated. From what is known, they include increased MMP expression, downregulation of tight junction proteins, expression of permeability-increasing molecules (substance-P, bradykinin) and increasing intercellular gaps by promoting cytoskeletal reorganization [Citation1]. Cellular retraction of endothelial cells via cytoskeletal contraction by actin and myosin light chain kinase, further increases BBB permeability [Citation17].
Other pathways involved in vasogenic edema include degradation and downregulation of tight junction proteins (ZO-1, occludin, claudin 5) and extracellular matrix (laminin) by factors such as MMP9, vascular endothelial growth factor-A (VEGF), angiopoietin-2, CCL2, and NO [Citation8,Citation17,Citation46–55]. The Hedgehog pathway may also play a key role in BBB maintenance. Astrocytic sonic hedgehog (SHH) downregulation has been reported to induce BBB disruption and vasogenic edema in in vivo and in vitro models of ICH, TBI, ischemic stroke and CNS tuberculosis[Citation56–62]. Reported mechanisms in these studies include increased MMP-9 activity as well as degradation of endothelial tight junctions (ZO-1, occludin) via pathways involving Patched1 (a SHH receptor on endothelial cells), Smoothened (transmembrane receptor inhibited by Patched), and Gli-1 (transcription factor).
Arginine vasopressin (AVP, V1a receptor) and NLR family pyrin domain containing-3 (NLRP3) have also been identified as contributors to vasogenic edema via mechanisms that include AQP4, peroxisome proliferator-activated receptor (PPAR-g), and tight junction degradation [Citation63–65]. Channels such as SUR1-TRPM4 are expressed on endothelial cells and perivascular astrocytes, and cellular edema and oncotic cell death in these cell-types involved in BBB maintenance may further contribute to vasogenic edema [Citation1,Citation17].
Glymphatic System
First discovered less than a decade ago, the glymphatic system provides a route for CSF influx into the brain (via periarterial space), and interstitial fluid clearance out of the brain (perivenous spaces) with a key role of aquaporin-4 (AQP4) channels on astrocyte vascular endfeet [Citation66,Citation67]. Although this system has been extensively studied in the context of neurodegenerative disease (amyloid-b and tau clearance) and sleep pathways, recent efforts have also identified its involvement in driving cerebral edema development after ischemic stroke [Citation68,Citation69]. Using calcium imaging in transgenic mice expressing GCaMP7 in cortical neurons and astrocytes, spreading depolarizations (triggered by cessation of blood flow) were found to initiate rapid CSF influx along perivascular spaces - coinciding with edema onset and increased brain water content. In the same study, magnetic resonance and multimodal imaging with radiolabeled tracers identified CSF as a hyperacute (within minutes) contributor of both fluid and ions – inflow and spreading edema were dependent upon astrocytic AQP4 channel expression [Citation68].
Subsequent studies in SAH and TBI have also invoked glymphatic system impairment as a contributor to cerebral edema, secondary injury, and BBB dysfunction [Citation70–74]. Exogenous Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP38) reduced edema, BBB disruption, and glymphatic dysregulation after SAH via reduced expression of several recognized (and interconnected) molecular contributors to edema discussed in subsequent sections: SUR1, AQP4, and matrix metalloproteinase-9 (MMP9) [Citation74]. In this study, SUR1 expression was reduced via the negative phosphorylation effect of p-PKA, which reportedly facilitates SUR1 degradation. Taken together, the evidence suggests that the glymphatic system is a promising emerging target for cerebral edema treatment across a range of ABIs.
Meningeal Lymphatic Vessels (mLV)
The concept of CNS immune privilege has continued to evolve. Previously thought to be devoid of conventional lymphatic vasculature, the role of mLVs in cerebral edema clearance was initially described in 2015 [Citation75,Citation76]. Work in mouse models identified a lymphatic vessel network in the dura matter that absorbs CSF from subarachnoid and interstitial spaces via the glymphatic system [Citation75]. This network ultimately transports this fluid into deep cervical lymph nodes, potentially assisting in the clearance of cerebral edema [Citation75]. Recent zebrafish models suggest that cerebrovascular injury induces rapid ingrowth of mLVs into the injured parenchyma where they become lumenized and drain interstitial fluid (alleviating cerebral edema), and additionally serve as growing tracks for nascent blood vessels [Citation77]. These findings were not corroborated in rodent/primate models: transgenic mice with mLV absence had attenuated clearance of macromolecules and a compromised peripheral immune response, but no difference in brain water content; however, these mice did not have cerebral edema pathology [Citation75,Citation78]. Murine models of glioblastoma multiforme (GBM) and SAH demonstrated impaired lymphatic outflow, increased edema and unfavorable markers of neuroinflammation and apoptosis, suggesting that mLVs. may indeed play a role in both cerebral edema and secondary injury after ABI[Citation72,Citation79,Citation80]. There is emerging evidence that meningeal lymphatic drainage is functionally connected with glymphatic flow.
3. NextGen Emerging Targets
This section discusses emerging targets for cerebral edema treatment based on supporting molecular data. Although the literature is rife with reports of strategies targeting several of the molecular components described above (Section II), few have successfully translated to clinical practice. Our focus is on candidates (listed alphabetically) with promising preclinical and clinical data that are currently advancing toward/actively in clinical trials ().
Table 1. Summary of Completed and Ongoing Clinical Trials Targeting Specific Molecular Contributors to Cerebral Edema
3.1. AQP4 – AER 271
Mechanism: The role of AQP4 in cerebral edema (both cellular and vasogenic) is complex since it is related to both water influx as well as efflux and edema clearance [Citation8,Citation81]. Water transport via this channel is passive, bidirectional, and is determined by the transmembrane osmotic gradient [Citation17]. AQP4 expression has been demonstrated in several cell-types involved in brain-fluid interfaces including perivascular astrocyte endfeet, glia limitans, ependymal cells (basolateral membranes), subependymal processes [Citation1,Citation82]. It has also been shown to be upregulated in activated/reactive microglia [Citation83]. The glymphatic system discussed above, is dependent on AQP4, likely since this is a major mechanism for rapid neutralization of osmotic gradients during ion transport [Citation17]. The AQP4 M23 isoform associates in membranes and forms supramolecular assemblies by aggregating into orthogonal arrays that facilitate rapid water flux [Citation17].
AQP4 interfaces with several other known contributors to cerebral edema. Arginine vasopressin, via V1a receptor modulation of AQP4, contributes to BBB disruption [Citation63,Citation84]. AQP4 forms a hetero-multimeric complex with SUR1-TRPM4 and drives astrocyte swelling after cold injury via amplifying ion/water coupling [Citation85]. AQP4 is linked with the NLRP3 inflammasome that has been identified as a facilitator of vasogenic edema via a variety of mechanisms including regulation of AQP4 expression and distribution in ischemic stroke, tight junction degradation in TBI, and PPAR-g in TBI [Citation64,Citation65,Citation86]. In preclinical models of cerebral ischemia, water intoxication, TBI, and cardiac arrest, effects of AQP4 inhibition have been mixed, with reports of decreased cellular edema vs. no-effect/potential worsening [Citation1,Citation87–97]. This is possibly due to the divergent role of these channels in both edema generation and edema clearance, suggesting that modulation and timing may be more valuable than consistent/early inhibition.
Human Studies: AQP4 has demonstrated limited but promising potential as a clinical biomarker in patients with brain injury. Levels in CSF have been reported as being elevated after injury, and genetic polymorphisms have been associated with outcome after TBI [Citation8,Citation98–100]. Clinical agents targeting/inhibiting AQP4 include AER-271 (a selective AQP4 antagonist), and Aquaporumab (a monoclonal AQP4-specific antibody). A double-blind, placebo-controlled, sequential group, phase 1 study of AER-271 has recently been completed (NCT03804476), with results pending. There are no currently registered studies evaluating aquaporumab on clinicaltrials.gov. Nonetheless, given the strategic expression of CNS AQP4, involvement of this channel with several mechanisms of edema, and multiple relationships with other key contributors to cerebral edema, further clinical studies are warranted. Although harnessing the potential of AQP4 channels in cerebral edema management will be complex, and could be complicated by renal toxicity, it may prove to be important.
3.2. AVP – Conivaptan
Mechanism: AVP, also known as antidiuretic hormone (ADH), is a nonapeptide produced as preproAVP by the hypothalamus, in the paraventricular and supraoptic nuclei [Citation101]. After synthesis, it is packaged into neurosecretory granules, processed (cleaved into AVP), and transported to the posterior pituitary where it is released into the systemic circulation. Additional routes of distribution (especially to the brain parenchyma) have been described, including hypothalamo-extrahypophyseal pathways with direct release into ventricular CSF, central secretion by neurons and the choroid plexus epithelium, and transport across the BBB via a carrier mediated system [Citation2,Citation102–105]. Indeed after injury, several sources of AVP synthesis and release have been identified, including microglia and the cerebrovascular endothelium [Citation104].
The V1a receptor (for AVP) is widely distributed in the mammalian CNS in neurons, astrocytes, and microvascular endothelium, and is thought to be the primary mediator of cerebral edema via G protein receptor signaling [Citation1,Citation2]. Several pathways have been implicated, including BBB permeability and vasogenic edema (AQP4 regulation, PPAR-g, and tight junction degradation), modulating astrocyte volume, influencing cerebral blood flow, and affecting CSF production/absorption [Citation1,Citation2,Citation8,Citation63–65,Citation106–111]. The V2 receptor (renal duct principal cells) are involved in controlling intravascular body fluid volume. AVP signaling is involved in the syndrome of inappropriate antidiuretic hormone, and may also exacerbate cerebral edema via euvolemic hyponatremia.
In several preclinical models (ischemic stroke, cryoinjury, TBI), V1 receptors are upregulated, AVP exacerbates cerebral edema, and inhibition (genetic or pharmacological) is beneficial [Citation104,Citation112–117]. Pharmacological inhibitors include vaptans (conivaptan, tolvaptan), a small molecule selective V1a antagonist (SR 49059), and a peptide V1 receptor antagonist (V1880). Both SR 49059 and V1880 have been shown to reduce cytotoxic edema and ICP in preclinical models, but unlike vaptans, have not been tested in humans [Citation116,Citation118,Citation119].
Human Studies: Vaptans, a class of small-molecule AVP receptor inhibitors currently approved to treat hyponatremia, are now being clinically investigated as potential anti-cerebral edema agents. Specificity for different receptors subtypes varies based on the drug. Conivaptan inhibits both V1A and V2 receptors, whereas tolvaptan is V2 specific. There are case reports/series of conivaptan improving hyponatremia in the neurocritical care unit (19 patients), reducing intracranial pressure in TBI (1 patient), and decreasing cerebral edema in ischemic stroke (1 patient)[Citation120–122]. A recent single-center open-label phase 1 study of intravenous conivaptan (20 mg, every 12 hours for 2 days) was reported in 7 patients with primary spontaneous intracranial hemorrhage (NCT03000283)[Citation123]. Safe administration with the expected increase in serum sodium levels was observed (p = 0.01). None of the patients required rescue therapy with mannitol, hypertonic saline, or an external ventricular drain. There was no significant change in mean arterial pressure or heart rate to suggest significant intravascular volume depletion. The effect of conivaptan on the V2 receptor and the potential for resultant diuresis did not impact clinical hemodynamics. All patients survived until follow up.
3.3. Cyclooxygenase 2 (COX2) – Celecoxib
Mechanism: COX enzymes catalyze the formation of prostanoids (thromboxane A1, prostacyclin) from arachidonic acid, and thereby contribute to post-injury neuroinflammation (via leukocyte invasion and BBB breakdown) in ICH, ischemic stroke, TBI, SAH [Citation124–134]. Upregulated COX2 expression in these models has been reported primarily in the CNS endothelium, and in other cell types including neurons, astrocytes, microglia, and invading leukocytes [Citation126,Citation129,Citation133–135]. Inhibition in preclinical models, via COX2 antagonists (e.g. celecoxib, diclofenac, CAY10404), have demonstrated decreased lesion volume, decreased apoptosis, decreased cerebral edema, and improved neurologic outcome in ICH, ischemic stroke, SAH, and TBI [Citation124,Citation136–141]. Of note, progesterone has been reported to reduce COX2 expression, particularly in models of TBI [Citation142–145].
Human Studies: Although there may be clinical reluctance to use cyclooxygenase inhibitors, particularly in hemorrhagic CNS disease, due to the potential for decreased platelet aggregation, COX2 inhibitors (particularly celecoxib) have minimal effects on platelet function even at supratherapeutic doses (1200 mg per day) [Citation146]. Human studies on COX2 inhibitors on cerebral edema/in brain injury have primarily been in ICH. In a retrospective study (34 patients), celecoxib treatment resulted in lower edema volumes vs. controls (30.2 ± 17.7 vs. 55.5 ± 40.6 mL, p = 0.027) [Citation147]. A subsequent multicenter randomized controlled trial of ICH in Korea (44 patients) demonstrated that acute administration of celecoxib (400 mg twice a day × 14 days) reduced expansion of both perihematomal edema (p = 0.005) as well as hemorrhage progression (p = 0.046; NCT00526214) [Citation148]. No additional studies evaluating COX2 inhibition CNS disorders are currently listed on clinicaltrials.gov.
3.4. Sphingosine-1 Phosphate (S1P) – Fingolimod
Mechanism: S1P is a metabolic product of sphingolipids that can act as an intracellular second messenger, as well as an extracellular signaling ligand at G protein-coupled S1P receptors, S1P1 – S1P5[Citation149]. S1P receptors are widely expressed on neurons (S1P1, S1P3) and endothelial cells (S1P1). S1P5 receptors are expressed predominantly on oligodendrocytes (activation protects against apoptosis)[Citation149]. Currently, S1P4 receptors are not known to be expressed in the brain [Citation149]. After injury, proinflammatory cytokines affect astrocytes, which strongly increase S1P1 and S1P3 expression, which modulate astrocyte proliferation, migration of leukocytes (particularly lymphocytes) and proapoptotic proteins. S1P1 signaling facilitates egress of lymphocytes from secondary lymphoid tissues, including infiltration into cerebral blood vessels after injuries such as ischemic stroke, which can be blocked by inhibition/receptor modulation [Citation150–152]. In cerebral ischemia, S1P1, S1P2, and S1P3 affect microglial polarization toward M1 activation (proinflammatory) [Citation153–155].
S1P signaling through these various receptors is complex, as it involves immunomodulation, cell migration, vascular tone, BBB integrity, vascular cell proliferation, and direct neuroprotection. Effects may be beneficial or detrimental depending on the receptor subtype and specific cell type. For example, via S1P1, S1P reduces leukocyte adhesion (alters endothelial adhesion molecule expression), prevents endothelial apoptosis (Bcl2 activation), and stimulates tight-junction protein ZO1 thereby promoting BBB integrity. S1P1 signaling has also been reported to have direct cytoprotective effects via pathways including PI3K/Akt/FOXO3a, or suppression of neuronal autophagy via mammalian target of rapamycin (mTOR), among others [Citation156,Citation157]. S1P1 activation can also be harmful/pro-inflammatory in microglia [Citation153]. S1P2, like S1P1, is also expressed in endothelial cells. However, in contrast to the protective effects of S1P1, S1P2 contributes to pro-inflammatory phenotypes and increases permeability of vascular beds by disrupting adherens junctions [Citation158]. This has predominantly been demonstrated in lung and spleen vascular beds, but is increasingly recognized as also increasing cerebrovascular inflammation, BBB injury and permeability [Citation158].
CNS S1P receptors and inhibition have long been studied for their role in multiple sclerosis (MS). Fingolimod (previously FTY720), the only approved oral therapy for MS, modulates S1P receptors. The mechanism is complex and incompletely understood, particularly in the setting of multifaceted roles of S1P signaling in the CNS and immune system, different receptor subtypes, and different cell types. In the CNS, fingolimod is phosphorylated to FTY720-p/Fingolimod-p) by sphingosine-kinase-2 (Sphk2). Sphk2 is highly expressed in the brain parenchyma, including the microvasculature [Citation159]. Fingolimod-p is a high-affinity, non-specific S1P-receptor modulator at S1P1, 2, 4, and 5 (strongly at S1P1) with high oral bioavailability (93%) [Citation160]. Although fingolimid-p initially acts as an agonist, it is more a modulator since stimulation induced internalization of receptors also results in functional antagonism of S1P signaling [Citation149,Citation151]. Overall, preclinical data suggest a benefit of fingolimod (for edema, infarct volume, and neuroprotection) largely in models of ischemic stroke (reviewed elsewhere [Citation149]), but also in ICH, TBI, and SAH [Citation161–169].
Human Studies: Aside from MS, S1P signaling and inhibition with fingolimod has been studied in patients with ICH, ischemic stroke, and SAH. Although it is generally well tolerated, side effects include bradycardia, and decreased pulmonary function. Importantly, fingolimod has been associated with increased odds of progressive multifocal leukoencephalopathy (although less than natalizumab) [Citation170].
In a proof-of-concept study (NCT02002390; 23 subjects) of primary deep supratentorial ICH, patients treated with fingolimod within 72 hours (0.5 mg daily × 3 days) vs. clinical and imaging-matched patients had reduced perihematomal edema, neurological deficits, and improved recovery [Citation171]. In acute ischemic stroke, the same investigators demonstrated that patients treated within 72 hours (0.5 mg daily × 3 days) had lower circulating lymphocyte counts, milder neurological deficits, and better functional recovery including reduction of the NIHSS (p = 0.0001)[Citation172]. Day-7 rT1% (a marker of vascular permeability) was almost 50% lower in treated patients (p = 0.005)[Citation172]. No safety concerns were reported in either study. When co-administered with alteplase (in the delayed time window, 4.5–6 hours), a double blind randomized trial in 46 patients, fingolimod improved 24-hour NIHSS score (by 4 vs. 0 points; p = 0.004), and shifted the 90-day mRS scores towards more favorable outcome (p = 0.037) [Citation173]. Secondary endpoints were also improved with regards to decreasing the perfusion lesion (0.001), and inhibiting infarct expansion (p < 0.001), with a persistent benefit through day-7. Although fingolimod has not been studied in SAH, a prospective study identified higher S1P CSF levels in SAH patients vs. controls (36 patients; p = 0.0005), and significant correlations with hemorrhage volume, as well as 12-month mRS (sliding dichotomy; p = 0.007)[Citation174].
Several newer agents modulating S1P1 (ozanimod, ponesimod) and S1P1+S1P5 (siponimod, ceralifimod, amiselimod) are being tested, predominantly in MS. Recruitment for NCT03338998, a trial evaluating Siponimod (BAF312) in ICH, is currently paused due to the COVID19 pandemic.
3.5. SUR1-TRPM4 – Glibenclamide
Mechanism: First discovered by patch-clamp experiments in adult rat astrocytes, the SUR1-TRPM4 channel is increasingly recognized as a central contributor to cerebral edema in several neurological conditions including ischemic stroke, TBI, SAH, ICH, spinal cord injury, cardiac arrest, encephalitis, and others [Citation1,Citation17,Citation175–193]. Unique to this pathway, SUR1-TRPM4 is an octameric monovalent cation channel that is not normally expressed in the CNS, but upregulated de novo after injury [Citation1,Citation17,Citation191]. It is therefore exceptionally positioned as a potentially important biomarker, as well as target for therapeutic intervention. SUR1 is an ATP-binding cassette family regulatory protein that co-assembles with pore forming subunits, in this case (after CNS injury) with TRPM4[Citation188]. TRPM4 constitutes the pore-forming subunits of the channel, and is a constitutively expressed monovalent calcium-sensitive cation channel [Citation194,Citation195]. Post-injury co-association of SUR1-TRPM4 doubles TRPM4 calcium sensitivity, and sensitizes this inner pore to intracellular ATP depletion [Citation2,Citation188,Citation194]. Channel opening after injury results in cell depolarization from influx of Na+ (which raises the osmotic pressure gradient), oncotic swelling/blebbing and cell death [Citation1,Citation17,Citation85,Citation175,Citation176,Citation191,Citation196]. Water influx may be via associated channels like AQP4[Citation85]. The SUR1 pathway is also linked with other contributors to cerebral edema and BBB integrity including, MMP9, NOS2, and tissue plasminogen activator [Citation197–199]. SUR1-TRPM4 expression has been demonstrated in several cell types including neurons, astrocytes, microglia, and endothelial cells in multiple preclinical models of CNS injury as well as in human tissue [Citation177–179,Citation188,Citation191,Citation196,Citation200]. In addition to cellular edema, SUR1-TRPM4 mediated oncotic cell death of structures involved in BBB maintenance (endothelial cells, astrocytes) results in capillary fragmentation, worsening vasogenic edema, and ultimately can result in secondary hemorrhage progression [Citation1,Citation2,Citation18].
A large and growing body of preclinical literature suggests SUR1-TRPM4 inhibition, both via gene silencing as well as pharmacologic (glibenclamide, a.k.a. glyburide) is beneficial. It has been shown to reduce cerebral edema, hemorrhage progression/BBB disruption and to improve neurological outcome in several preclinical studies of ischemic stroke, TBI, ICH, SAH, spinal cord injury, cardiac arrest, and CNS malignancies [Citation179,Citation180,Citation183,Citation184,Citation187,Citation193,Citation201–215]. Nanoparticle-mediated delivery of glibenclamide for targeted delivery is an area of active research [Citation216].
Human Studies: SUR1 expression has been reported in human contusions, infarcted brain tissue, and brain tumors [Citation178,Citation194,Citation200,Citation217–219]. SUR1 levels in human CSF (both adult and pediatric) and possibly serum may also be informative biomarkers for secondary injury based on exploratory studies in TBI and SAH [Citation220–222]. SUR1 (encoded by ABCC8) and TRPM4 genetic variation may also impact the extent of cerebral edema, ICP, hemorrhage progression, and outcome [Citation8,Citation223–225]. The biological mechanism by which this occurs is currently unknown but may be related to transcriptional regulation and mRNA levels (Jha et al., in press) and/or channel function/efficiency.
A retrospective study of oral glibenclamide (61 patients) demonstrated greater improvement in NIHSS (p = 0.007) and mRS < 2 (p = 0.035) at discharge in patients taking oral glibenclamide [Citation226]. Benefit occurred only in patients with non-lacunar strokes. Another retrospective evaluation of diabetics with acute ischemic stroke revealed that continued use of their sulfonylurea medications was associated with reduced hemorrhagic transformation [Citation227]. Small randomized trials (40–66 subjects) of oral glibenclamide have demonstrated slower contusion expansion rate, and improved outcome after contusional TBI and diffuse axonal injury [Citation228,Citation229]. In a study of patients with ischemic stroke (NIHSS ≥ 8; 213 patients), those treated with oral glibenclamide had lower cerebral edema and a trend toward less severe disability and death vs. controls, without increased risk of hypoglycemia [Citation230]. The utility of oral glibenclamide is being evaluated in clinical trials of ischemic stroke (NCT03284463, SE-GRACE) and spinal cord injury (NCT02524379, SCING) [Citation231,Citation232]. In a retrospective review of oral glyburide (2.5 mg twice daily, 71 patients) hypoglycemia occurred in 23.9% of patients, with acute kidney injury and low body mass index being significant risk factors [Citation233].
Intravenous glibenclamide (BIIB093) has emerged as a promising anti-edema/anti-secondary injury agent, particularly in large hemispheric infarction (LHI) but also in TBI [Citation176,Citation197,Citation234–238]. In the first pilot trial (GAMES-pilot), feasibility of IV glibenclamide was reported in 10 patients with anterior circulation LHI [Citation239]. Secondary analyses of these patients demonstrated successful reduction of vasogenic edema on T2 FLAIR MRI with intravenous glyburide [Citation238]. After this initial safety/feasibility study, a phase 2 randomized controlled trial in anterior circulation LHI (n = 86, age 18–80) was completed in 2016[Citation197]. Although there was no difference in the primary endpoint (90-day mRS 0–4 without decompressive craniectomy), there was a trend toward mortality benefit (p = 0.06), and significant reduction in MMP-9 levels (p = 0.006; marker of BBB integrity) and midline shift (from 8.5 to 4.6 mm; p = 0.0006) in the treatment group [Citation197]. The inter-center variability in surgical decompression was thought to contribute to the failure to meet primary end point. Subsequent analyses of patients in this phase 2 study have yielded favorable long-term outcomes in patients <70 years old, as well as a reduction in neurological and edema-related deaths [Citation235,Citation240]. Two studies of BIIB093 (phase 3 study in LHI [CHARM, NCT02864953]; phase 2 study in contusional TBI [ASTRAL, NCT03954041]) are actively recruiting.
3.6. VEGF – Bevacizumab
Mechanism: VEGF, a secreted glycoprotein, has long been known for its role in angiogenesis/vasculogenesis and stimulating endothelial cell proliferation/increasing vascular permeability[Citation241,Citation242]. The role of VEGF in the CNS is expansive and, as reviewed elsewhere, it has been implicated in neurodegenerative disease, neuroinflammatory/demolinating disease, peripheral nerve diseases, neuromuscular disease (amyotrophic lateral sclerosis), epilepsy, stroke, TBI, ICH, SAH and both primary and metastatic CNS malignancies [Citation241,Citation242]. There are several subclasses of VEGF (A–E), and VEGF receptors (1–3). VEGF-B is less angiogenic than VEGF-A [Citation241]. VEGF-A is upregulated in astrocytes, endothelium, and macrophages after both in vitro and in vivo injury [Citation8,Citation243,Citation244]. Many effects of VEGF are related to VEGF binding to tyrosine kinases cell surface receptors and downstream signaling pathways, including mitogen activated protein kinase (MAPK; proliferation including neural stem cells and neurogenesis, migration), PI3K-Akt (cell survival), and eNOS (vascular permeability). Several molecular mechanisms of VEGF-related increased microvascular permeability have been reported, including downregulation/degradation/ubiquitination of tight junction proteins (occludin, claudin), angiogenesis via endothelial progenitor cell recruitment, activation by MMP-9, and capillary endothelial transcytosis [Citation8,Citation50,Citation242–247].
As is a common theme, effects of VEGF can be both beneficial (collateral formation, reparative angiogenesis, neuroprotection) as well as deleterious (BBB breakdown, vasogenic edema) depending on the specific pathways activated, timing, and cell-types affected. At low levels, VEGF-A is necessary for BBB integrity and endothelial cell survival; however, at high levels, it increases vascular permeability and cerebral edema [Citation241,Citation248,Citation249]. Therefore, the safety/efficacy of VEGF modulation (inhibition or activation) will depend on the dose, timing, indication, and specificity.
In mouse models of glioblastoma multiforme (GBM) VEGF-A is overexpressed and is associated with unfavorable prognosis. Inhibition with bevacizumab (a VEGF antibody) normalized the vasculature and reduced vascular permeability, reducing edema [Citation250]. Preclinical studies in ischemic stroke have demonstrated that VEGF administration worsened vascular edema and infarct size, and that blockade is beneficial [Citation248,Citation249]. However, via vasodilation, perfusion augmentation/penumbra rescue, and collateral induction, delayed VEGF treatment may also stimulate angiogenesis, neuroprotection and improve neurological function without worsening edema in both ischemic stroke and TBI models [Citation241,Citation251–253]. Delayed VEGF inhibition (bevacizumab) has been shown to exacerbate necrosis and neurological deficits in TBI, with no reduction in either vasogenic edema or BBB permeability [Citation254].
Human Studies: Bevacizumab received accelerated approval in 2009 for recurrent GBM after several trials in patients demonstrated radiographic benefit on the BBB and on vasogenic edema [Citation250,Citation255,Citation256]. However, several subsequent trials failed to show overall survival benefit in either recurrent or newly diagnosed GBM [Citation250,Citation257–262]. Other receptor tyrosine kinase inhibitors have shown similar results without improvement in overall survival (cediranib, enzastaurin) [Citation263,Citation264]. This is possibly related to the divergent effects of VEGF on neuroprotection vs. vasogenic edema.
In ischemic stroke, TBI, and SAH patients, VEGF levels have been elevated in serum as well as CSF [Citation265–267]. No clinical trials of inhibition in these populations have been reported or are currently listed on clinicaltrials.gov.
4. Recombinant Proteins
Over the last decade, hundreds of protein-based therapeutics have been approved in the US – including antibody-based drugs (‘biologics’), fusion proteins, growth factors, hormones, enzymes, and a variety of other molecular types [Citation268]. The majority are recombinant proteins and are utilized in cancers and immune disorders. Based on known signaling pathways of cerebral edema as discussed above, several recombinant proteins have been tested in preclinical models with promising results. Preclinical research on recombinant proteins that impact tight/adherens junctions (fibroblast growth factors; FGF21, FGF20), chemokines (CCL17-CCR4), and VEGF are discussed below.
FGF21 has been reported to have neuroprotective effects including enhancing angiogenesis; it is downregulated in the brain post-TBI and in cerebral ischemia [Citation269]. Recombinant human FGF21 (rhFGF21) treatment post-TBI reduced cerebral edema, BBB breakdown and improved functional recovery by forming an FGF21/FGFR1/β-klotho complex, likely via PPARγ and upregulation of tight junction proteins ZO-1, occludin, VE-cadherin, and claudin-5 [Citation270]. Recombinant human FGF20 (rhFGF20) also helped preserve BBB integrity by increasing tight and adherens junction proteins, and reducing IL1-β, IL-6 and iNOS levels via AKT/GSKβ and JNK/NFκB pathways in vitro, and after in vivo TBI [Citation271]. In this study, rhFGF20 also improved behavioral outcomes.
As discussed earlier, chemokines influence vasogenic edema, neuroinflammation, as well as cell migration for survival/recovery post-injury. CC chemokine ligand 17 (CCL17) a specific ligand for the high affinity CC chemokine receptor 4 (CCR4), is expressed in hippocampal neurons. CCR4 is expressed on microglia, neurons, astrocytes, monocytes and natural killer cells [Citation272]. CCR4 activation by CCL17 activates the downstream PI3K-Akt pathway associated with cell survival and proliferation; neuroinflammation and neuronal apoptosis were inhibited via suppression of Foxo1 (which increases pro-inflammatory cytokine production) [Citation272]. In an ICH model, intranasal recombinant CCL17 (rCCL17) dependent activation of CCR4 reduced cerebral edema, neuronal apoptosis, and neuroinflammation and improved functional outcome [Citation272]. The same group demonstrated that intranasal rCCL17 facilitated hematoma resolution and improved long-term neurobehavioral outcomes post-ICH associated with increased CD163 expression through CCR4/ERK/Nrf2 signaling pathway [Citation273].
A recent study explored the therapeutic potential of recombinant mouse VEGF-D (rmVEGF-D) in a murine model of ischemic stroke (middle cerebral artery occlusion) [Citation274]. Intranasal rmVEGF-D administration reduced infarct volume, dendrite loss, and improved functional recovery. In this study, changes on cerebral edema or BBB were not reported. Although rmVEGF-D therapy is in its infancy, it warrants further investigation. Modulation and timing may be important, particularly given the dual role of VEGF in both advancing neurogenesis/neuroprotection and worsening cerebral edema.
5. Gene Therapies
Gene therapy delivery has classically been divided into viral (adeno-, adeno associated-, lenti- retro- viral vectors) vs. non-viral approaches (CRISPR-based approaches, mesenchymal stem cell delivery, physical mediators like microbubbles/microparticles/nanoparticles)[Citation275–278]. DNA- as well as RNA-based targets have been described (including RNA interference, microRNA); the strategies have predominantly included gene augmentation, modification, blockage (of defective gene) or replacement (of a deficient gene)[Citation279]. The first clinical trial using gene therapy was reported by Rosenberg et al., in 1990 in patients with advanced melanoma [Citation280]. Since then, the role of gene therapy has exponentially grown, and although historically the majority of studies have addressed cancer and monogenic disorders, indications have now widely expanded to include cardiovascular disease, infectious disease, and neurological disease [Citation281]. This section summarizes recent advances in gene therapy pertaining to cerebral edema including microRNA targets as well as delivery mechanisms, currently these are all at the preclinical stage and not currently ready for clinical translation.
5.1. Micro RNA
MicroRNAs are endogenous small (~22nt, 19–25nt) non-coding RNAs discovered in the 1990s that bind to complementary target mRNAs via sequence complementarity, and either inhibit mRNA translation inhibition or facilitate their degradation [Citation282–284]. The latest version of miRbase (v22) contains microRNA sequences from 271 organisms, >38,589 hairpin precursors and >48,860 mature microRNAs [Citation285]. Multiple clinical trials using microRNA-based therapeutics are ongoing worldwide [Citation286–288].
Current studies suggest that circulating microRNAs can be successfully used as noninvasive surrogate biomarkers for various CNS diseases including TBI, stroke, SAH and multiple neurodevelopmental and neurodegenerative disorders [Citation289–292]. Several microRNAs (including miR-1228-5p, miR-1268a, miR-1268b, miR-4433b-3p, miR-6090, miR-6752-5p, and miR-6803-5p) have been identified as potential biomarkers predicting risk of cerebrovascular disorder before the clinical onset of ischemic stroke in human patients [Citation293]. Others have been reported as influencing (increasing/decreasing) secondary injury after TBI and ischemic stroke. Increased expression of some microRNAs (miR-93, miR-126, miR-210, and miR-130a) is associated with cerebral edema and BBB disruption in preclinical models [Citation294–297]. Downregulation of others (miR-374, miR-29b) increases infarct volume and neuronal apoptosis [Citation298–300]. Moreover, loss of miR-29b post-acute ischemic stroke contributes to neural cell death and infarct size [Citation300]. In vitro and in vivo models demonstrate that miR-126-3p and 5p are anti-inflammatory; miR-126-3p is thought to promote angiogenesis and attenuate BBB disruption, and −5p promotes endothelial proliferation and survival. A clinical study in 197 patients with ischemic stroke demonstrated markedly reduced miR-126 (and miR-30a) levels vs. 50 healthy volunteers [Citation301].
MicroRNAs have also been evaluated for their therapeutic potential in CNS disease. For example, miR-21 and miR-21-5p have been demonstrated to confer neuroprotection, improved cognitive function and alleviate BBB disruption via MAPK and PTEN/Akt signaling pathways [Citation302,Citation303]. Similarly, miR-223 has been shown to be neuroprotective by targeting glutamate receptors [Citation304]. Exosomes derived from mesenchymal stromal cells express miR-17-92 and miR-410 or mir-206 knockdown and confer neuroprotection [Citation305–307].
In addition to microRNAs, the long noncoding RNA (lncRNA) MALAT1 has been demonstrated to ameliorate TBI-induced brain edema by inhibiting AQP4 and NF-κB/IL-6 [Citation308,Citation309]. Intravenous delivery of the exosomes derived from adipose-derived stem cells containing MALAT1 lncRNA revealed that the exosomes migrated into the spleen ~1 hour after injury in a contusional model of TBI, and entered the brain several hours later. Treatment with MALAT1 containing exosomes improved motor impairment and reduced lesion volume [Citation309]. Systemic delivery of bone marrow stem cell-derived exosomes nucleofected with miR-193b-3p has been shown to attenuate neuroinflammation, suppress the activity of HDAC3, upregulate acetylation of NF-κB p65, mitigate neurological behavioral impairment, reduce brain edema and BBB injury as well as neurodegeneration after SAH [Citation310]. While there are no data in humans, such studies highlight the therapeutic potential for microRNAs in the treatment of secondary injury processes such as cerebral edema after several forms of ABI.
5.2. Viral Vectors
Studies of adenoviral and lentiviral vector delivery of different therapeutic molecules have successfully demonstrated the ability to reduce secondary injury (cerebral edema, BBB disruption, proinflammatory cytokine secretion) and improve functional outcome in preclinical models of stroke, ICH, SAH and TBI [Citation292,Citation311–317]. Not surprisingly, several of the downstream targets in these studies are familiar players, including AQP4, MMP9, NOS, inflammatory cytokines (IL1b, TNF-a), tight-junction proteins (E-selectin), and adhesion molecules (VCAM-1).
Examples of preclinical studies evaluating viral-vector-based treatment for cerebral edema are presented here. As discussed in Section V-A, miR-126-3p/5p is important for maintaining BBB integrity, angiogenesis and reducing cerebral edema, with levels being reduced after ischemic stroke. Lentiviral-mediated overexpression of miR-126-3p/5p in a mouse middle cerebral artery occlusion (MCAO) model reduced brain infarct and cerebral edema volumes, and improved behavioral outcome [Citation292]. Further analyses demonstrated that injection of lentiviral-miR-126-3p or −5p downregulated the expression of proinflammatory cytokines (IL-1b, TNFa), VCAM1, and attenuated the ischemia-induced decrease in ZO-1. In subarachnoid hemorrhage, mammalian-sterile 20 like kinase 1 (MST1) was knocked down via intraventricular injection of AAV packaged with MST1 short hairpin. This successfully reduced BBB disruption, edema and white matter injury via downstream NF-κB and MMP-9 signaling [Citation315]. N-myc downstream regulated gene 2 (NDRG2), expressed in mammalian astrocytes, can worsen cerebral edema when deficient via changing the distribution of AQP4 and the Na+/K+/ATPase pump [Citation316]. In a mouse MCAO model of transient cerebral ischemia and reperfusion, 5 days after intraventricular injection with an NDRG2 overexpressing lentivirus, brain water content was significantly decreased, as was BBB permeability [Citation316].
One disadvantage of viral vectors (particularly adeno-associated viral vectors, AAV) is the potential for eliciting a potent immune response which may limit benefit in acute CNS injury [Citation318]. It is also important to note that AAV clearance may be affected in aged and diseased brains via the glymphatic system and function of AQP4[Citation319]. Advantages include the availability of multiple serotypes, vector modification to limit tropism to a specific cell-type, lack of integration into the host genome in certain types of viral vectors, and machine learning-based creation of numerous diverse AAV capsid variants [Citation279,Citation320–322]. Although there are several ongoing clinical trials using AAV gene transfer in the CNS (Alzheimer’s, Parkinson’s), viral vector-based gene therapy is not yet being evaluated clinically in acute brain injuries or for cerebral edema [Citation323].
5.3. CRISPR
Attempts to harness the 2020 Doudna-Charpentier Nobel prize winning discovery (in chemistry) of CRISPR to treat human disease are ongoing, with phase 1 reports emerging in cancer [Citation324–327]. However, unlike cancer or monogenic disorders, cerebral edema in many cases is a reactive and acute condition. Thus, permanent genetic modification may not be desirable. Off target effects and limited efficiency may further limit current clinical utility, although ongoing research has demonstrated improved editing efficiency in several cell types in vitro and in vivo by using different Cas9 proteins. Moreover, off-target effects in both mouse and human cell lines have been improved by several techniques including optimizing single guide RNA sequence/design rules [Citation328–331].
Despite the unclear role of CRISPR/Cas9 technology in clinical treatment of cerebral edema, given the limitations above, it is being used to further understand edema pathobiology in preclinical models. Recently, AQP4ex (the extended isoform of AQP4a containing a 29 amino-acid C terminal extension), has been identified in vitro as the potential modulator of supramolecular organization and regulation of water channel activity [Citation332]. CRISPR/Cas9 technology was used to generate an AQP4ex–/ – mouse. In these mice, although total AQP4 protein expression was unchanged, removal of AQP4ex completely suppressed the location of AQP4 at the astrocyte endfeet [Citation333]. Similarly, although the number of AQP4 orthogonal arrays of particles (OAP) remained unchanged, the OAP size was markedly reduced. This has important implications for cerebral edema generation and may valuably inform future targeted therapies. Both homozygous and heterozygous ABCC8 (encoding SUR1) mutant cell lines have been generated using CRISPR/Cas9 technology[Citation334,Citation335]. These were generated to evaluate effects of ABCC8 genetic mutations on congenital hyperinsulinism and revealed defective KATP channels unresponsive to diazoxide. Nonetheless, this technique may be valuable for understanding the biological consequences of important ABCC8 genetic variation contributing to cerebral edema and hemorrhage progression (described in Section III-E).
6. Conclusions
We have reviewed fundamental principles underlying the clinically relevant and molecularly complex phenomenon of cerebral edema. Unchecked, it causes morbidity and mortality across several different types of acute and chronic brain injury. Basic physical principles such as the Monro-Kellie doctrine and Starling-forces have long been recognized as the drivers of edema across intracranial compartments – the former having particular bearing on the development of intracranial hypertension and herniation. While the cerebrovascular and CSF circulation systems are putatively major contributors to edema development, there is emerging evidence of key involvement from the glymphatic system and meningeal lymphatic vessels. The spectrum of cerebral edema can be classified as cellular/cytotoxic edema, ionic edema, and vasogenic edema – with overlapping molecular mechanisms. The clinical manifestation of cerebral edema however, varies by disease type and acuity – e.g., the timeline, extent, and predominant components of edema in stroke may vary from TBI despite having some shared molecular channels and mechanisms. Preclinical and clinical research has identified several promising candidates for targeted inhibition including AQP4, AVP, COX2, S1P, SUR1-TRPM4, and VEGF- these targets have demonstrated importance in edema pathogenesis across several different types of brain injury. Other emerging therapies include recombinant proteins (e.g. FGF21 and CCL17) as well as gene therapies (micro-RNA, viral vectors, and CRISPR). Ultimately, a nuanced and precision-medicine approach that endophenotypes the constellation of molecular drivers of cerebral edema in different patients may guide targeted modulation of the complex underlying pathophysiology and thereby improve outcomes for patients across a wide range of affected diseases.
7. Expert Opinion
Cerebral edema is a complex pathobiological phenomenon, and a key determinant of secondary injury that occurs in almost all types of acute neurological insults. Untreated, cerebral edema can be fatal. The landscape of cerebral edema diagnosis and management is evolving. Current practices are reactive; they address existing edema and attempt to minimize the downstream deleterious consequences such as intracranial hypertension and herniation. Recent key developments in this area include emerging therapies that focus primarily on molecularly derived approaches to prevent or minimize edema formation by targeting key pathways/contributors discussed in this review including AQP4, AVP, COX2, S1P, SUR1-TRPM4, and VEGF. These targets were identified based on data from in vitro, in vivo preclinical and clinical studies. Inhibition of SUR1-TRPM4 and VEGF are currently the most advanced in terms of clinical trials and translation to humans. However, VEGF inhibition with bevacizumab, despite encouraging anti-edema effects radiographically, has not demonstrated improved survival in GBM. Alternative modalities such as recombinant proteins and various gene-based therapies are being developed (many with similar/known molecular targets involved in edema pathophysiology); these newer modalities are currently in their infancy with regards to clinical translation, but early preclinical reports are encouraging. However, despite this exponential growth in research, several gaps remain including a comprehensive understanding of how these mechanisms interact with and influence each other, disease heterogeneity, translation across species, molecularly endophenotyping patients using biomarkers (fluid, imaging, and genetics) to identify key pathways activated in individuals. It is therefore crucial, for researchers in this space, to continue rigorous, reproducible and blinded preclinical studies of therapeutics in different models of brain injury, and across species. Similar to the field of oncology, it is likely that combination therapies may be required depending on individual profiles/signatures of cerebral edema. Therefore, in addition to drug development, a parallel focus on biomarkers and deep endophenotyping of cerebral edema is likely crucial for successful translation. Over the next few years, the advent of technologies such as single cell gene expression and spatial transcriptomics may further inform identification of key/novel contributors to cerebral edema and important differences based on variables such as age, sex, race, and types of primary brain injury. Given the potential teleological role of edema, it may also be important to harness components that are necessary for repair/regeneration while minimizing the harmful processes and impact on intracranial pressure. Given the complex interplay between harm and benefit/neuroprotection of many of the discussed molecular targets involved in edema generation, a nuanced modulation of dose and timing based on both the individual and the specific disease/pathways will likely be critical to reducing this form of secondary injury and improving clinical outcome.
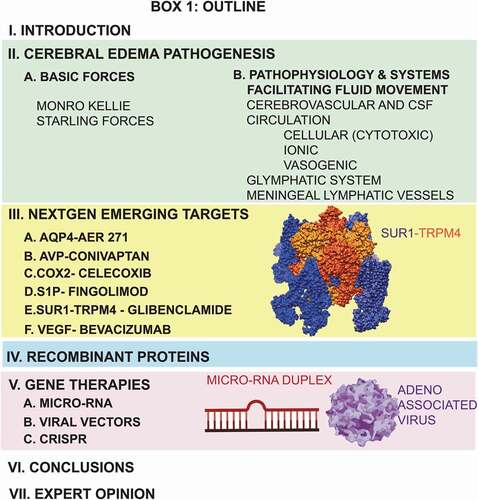
Box 1. Review Article Outline.
Article Highlights
Cerebral edema is a complex and heterogeneous process that contributes to morbidity and mortality in several different types of acute and chronic brain injury.
The relationship of cerebral edema with intracranial pressure is nuanced and related via the Monro-Kellie doctrine as well as principles of intracranial compliance/elastance and autoregulation.
Current treatment modalities while potentially life-saving (like decompressive craniectomy), are reactive, non-specific, invasive, and carry significant risks to patients.
The cerebrovascular system and CSF are major sources of cerebral edema contributing to the spectrum ionic, cellular/cytotoxic and vasogenic edema – with overlapping contributory molecular mechanisms. Emerging evidence suggests key involvement of the glymphatic system and meningeal lymphatic vessels to the development of cerebral edema.
Recent research has identified promising molecular targets to treat and potentially prevent cerebral edema – those that are advancing in clinical trials are discussed here, including AQP4, AVP, COX2, S1P, SUR1-TRPM4, VEGF. These targets include both ion and water channels as well as proteins/lipids involved in edema signaling pathways. Of these, SUR1-TRPM4 inhibition is actively in phase-2 and-3 clinical trials for TBI and large hemispheric infarction respectively.
Several treatment modalities being developed include recombinant proteins (e.g. FGF21, CCL17) and different types of gene therapies (micro-RNA, viral vectors, CRISPR). These are currently in their infancy with regards to clinical translation but are gaining momentum in preclinical studies.
Ongoing challenges for successful translation include identifying different molecular ‘signatures’ of cerebral edema to inform combination therapies, rigor/reproducibility across species, parallel development of biomarkers (fluid, imaging, physiologic neuromonitoring, genetics), and identifying differences in pathophysiology based on important variables like age, sex, race, and disease type.
A granular precision-medicine and combination-therapy approach may be important for mitigating the deleterious effects of cerebral edema and improving clinical outcomes. This strategy includes molecularly endophenotyping drivers of cerebral edema to inform individualized targeted modulation of this complex process – reducing the harmful effects while maintaining components important for neuro-regeneration and repair.
This box summarizes key points contained in the article.
Declaration of interest
R.M.J. provides consulting services for Biogen and is on the advisory board for ASTRAL. J.M.S. holds a US patent (7,285,574), ‘A novel non-selective cation channel in neural cells and methods for treating brain swelling.’ J.M.S. is a member of the Board of Directors and holds shares in Remedy Pharmaceuticals and is a paid consultant for Biogen. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Jha RM, Kochanek PM, Simard JM. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology. 2019;145:230–246.
- Stokum JA, Gerzanich V, Sheth KN, et al. Emerging Pharmacological Treatments for Cerebral Edema: Evidence from Clinical Studies. Annu Rev Pharmacol Toxicol. 2020 Jan;60(1):291–309.
- Robert SM, Reeves BC, Alper SL, et al. New drugs on the horizon for cerebral edema: what’s in the clinical development pipeline? Expert Opin Investig Drugs. 2020 Oct;29(10):1099–1105.
- Battey TWK, Karki M, Singhal AB, et al. Brain edema predicts outcome after nonlacunar ischemic stroke. Stroke. 2014 Dec;45:3643–3648.
- Morimoto Y, Kemmotsu O, Kitami K, et al. Acute brain swelling after out-of-hospital cardiac arrest: pathogenesis and outcome. Crit Care Med. 1993 Jan;21(1):104–110.
- Arima H, Wang JG, Huang Y, et al. INTERACT Investigators. Significance of perihematomal edema in acute intracerebral hemorrhage: the INTERACT trial. Neurology. 2009 Dec;73:1963–1968.
- Wu C-X, Lin G-S, Lin Z-X, et al. Peritumoral edema shown by MRI predicts poor clinical outcome in glioblastoma. World J Surg Oncol. 2015 Mar;13:97.
- Rm J, Pm K. A Precision Medicine Approach to Cerebral Edema and Intracranial Hypertension after Severe Traumatic Brain Injury: Quo Vadis? Curr Neurol Neurosci Rep. 2018 Nov;18:105.
- Zusman BE, Kochanek PM, Jha RM. Cerebral edema in traumatic brain injury: a historical framework for current therapy. Curr Treat Options Neurol. 2020 Mar;22:9.
- Cooper DJ, Rosenfeld JV, Murray L, et al. DECRA Trial Investigators, Australian and New Zealand Intensive Care Society Clinical Trials Group. Decompressive craniectomy in diffuse traumatic brain injury. N Engl J Med. 2011 Apr;364(16):1493–1502.
- Shutter LA, Timmons SD. Intracranial Pressure Rescued by Decompressive Surgery after Traumatic Brain Injury. N Engl J Med. 2016 Sep;375(12):1183–1184.
- Hutchinson PJ, Kolias AG, Timofeev IS, et al. RESCUEicp Trial Collaborators. Trial of decompressive craniectomy for traumatic intracranial hypertension. N Engl J Med. 2016 Sep;375(12):1119–1130.
- Juttler E, Schwab S, Schmiedek P, et al. the DESTINY Study Group for. Decompressive Surgery for the Treatment of Malignant Infarction of the Middle Cerebral Artery (DESTINY): A Randomized, Controlled Trial. Stroke. 2007 Aug;38(9):2518–2525.
- Vahedi K, Hofmeijer J, Juettler E, et al. DECIMAL, DESTINY, and HAMLET investigators. Early decompressive surgery in malignant infarction of the middle cerebral artery: a pooled analysis of three randomised controlled trials. Lancet Neurol. 2007 Mar;6(3):215–222.
- Jüttler E, Unterberg A, Woitzik J, et al. DESTINY II Investigators. Hemicraniectomy in older patients with extensive middle-cerebral-artery stroke. N Engl J Med. 2014 Mar;370(12):1091–1100.
- Hofmeijer J, Kappelle LJ, Algra A, et al. HAMLET investigators. Surgical decompression for space-occupying cerebral infarction (the Hemicraniectomy After Middle Cerebral Artery infarction with Life-threatening Edema Trial [HAMLET]): a multicentre, open, randomised trial. Lancet Neurol. 2009 Apr;8(4):326–333.
- Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab. 2016 Mar;36(3):513–538.
- Simard JM, Kent TA, Chen M, et al. Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 2007 Mar;6(3):258–268.
- Simon DW, McGeachy MJ, Bayır H, et al. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017 Feb;13(3):171–191.
- Yang -Q-Q, Zhou J-W. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia. 2019;67(6):1017–1035.
- Ransohoff RM, Schafer D, Vincent A, et al. Neuroinflammation: ways in which the immune system affects the brain. Neurotherapeutics. 2015 Oct;12(4):896–909.
- Sulhan S, Lyon KA, Shapiro LA, et al. Neuroinflammation and blood-brain barrier disruption following traumatic brain injury: Pathophysiology and potential therapeutic targets. J Neurosci Res. 2020;98(1):19–28.
- Hamann S, Herrera-Perez JJ, Zeuthen T, et al. Cotransport of water by the Na+-K+-2Cl(-) cotransporter NKCC1 in mammalian epithelial cells. J. Physiol. (Lond.). 2010 Nov;588(21):4089–4101. https://doi.org/10.1113/jphysiol.2010.194738
- Su G, Kintner DB, Sun D. Contribution of Na(+)-K(+)-Cl(−)cotransporter to high-[K(+)](o)- induced swelling and EAA release in astrocytes. Am J Physiol Cell Physiol. 2002 May;282(5):C1136–46.
- Su G, Kintner DB, Flagella M, et al. Astrocytes from Na(+)-K(+)-Cl(-) cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am J Physiol Cell Physiol. 2002 May;282(5):C1147–60.
- Hansson E, Muyderman H, Leonova J, et al. Astroglia and glutamate in physiology and pathology: aspects on glutamate transport, glutamate-induced cell swelling and gap-junction communication. Neurochem Int. 2000 Sep;37(2–3):317–329.
- Haugeto O, Ullensvang K, Levy LM, et al. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 1996 Nov;271(44):27715–27722.
- Tanaka K, Watase K, Manabe T, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997 Jun;276(5319):1699–1702.
- Hinson SR, Roemer SF, Lucchinetti CF, et al. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J Exp Med. 2008 Oct;205(11):2473–2481.
- Bender AS, Schousboe A, Reichelt W, et al. Ionic mechanisms in glutamate-induced astrocyte swelling: role of K+ influx. J Neurosci Res. 1998 May;52(3):307–321.
- Illarionova NB, Gunnarson E, Li Y, et al. Functional and molecular interactions between aquaporins and Na,K-ATPase. Neuroscience. 2010 Jul;168(4):915–925.
- Douglas RM, Schmitt BM, Xia Y, et al. Sodium-hydrogen exchangers and sodium-bicarbonate co-transporters: ontogeny of protein expression in the rat brain. Neuroscience. 2001;102(1):217–228.
- Jakubovicz DE, Klip A. Lactic acid-induced swelling in C6 glial cells via Na+/H+ exchange. Brain Res. 1989 Apr;485(2):215–224.
- Kintner DB, Su G, Lenart B, et al. Increased tolerance to oxygen and glucose deprivation in astrocytes from Na(+)/H(+) exchanger isoform 1 null mice. Am J Physiol Cell Physiol. 2004 Jul;287(1):C12–21.
- Kitayama J, Kitazono T, Yao H, et al. Inhibition of Na+/H+ exchanger reduces infarct volume of focal cerebral ischemia in rats. Brain Res. 2001 Dec;922(2):223–228.
- Bevensee MO, Weed RA, Boron WF. Intracellular pH regulation in cultured astrocytes from rat hippocampus. I. Role Of HCO3-. J. Gen. Physiol. 1997 Oct;110(4):453–465. https://doi.org/10.1085/jgp.110.4.453
- Jayakumar AR, Valdes V, Tong XY, et al. Sulfonylurea receptor 1 contributes to the astrocyte swelling and brain edema in acute liver failure. Transl Stroke Res. 2014 Feb;5(1):28–37.
- Jayakumar AR, Tong XY, Ruiz-Cordero R, et al. Activation of NF-κB mediates astrocyte swelling and brain edema in traumatic brain injury. J Neurotrauma. 2014 Jul;31(14):1249–1257.
- Jayakumar AR, Panickar KS, Curtis KM, et al. Na-K-Cl cotransporter-1 in the mechanism of cell swelling in cultured astrocytes after fluid percussion injury. J Neurochem. 2011 May;117(3):437–448.
- Jayakumar AR, Liu M, Moriyama M, et al. Na-K-Cl Cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J Biol Chem. 2008 Dec;283(49):33874–33882.
- Sevick RJ, Kanda F, Mintorovitch J, et al. Cytotoxic brain edema: assessment with diffusion-weighted MR imaging. Radiology. 1992 Dec;185(3):687–690.
- Corrigan F, Mander KA, Leonard AV, et al. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J Neuroinflammation. 2016 Oct;13(1):264.
- Song L, Pachter JS. Monocyte chemoattractant protein-1 alters expression of tight junction-associated proteins in brain microvascular endothelial cells. Microvasc Res. 2004 Jan;67(1):78–89.
- Yamagata K, Tagami M, Takenaga F, et al. Hypoxia-induced changes in tight junction permeability of brain capillary endothelial cells are associated with IL-1beta and nitric oxide. Neurobiol Dis. 2004 Dec;17(3):491–499.
- Nag S, Kapadia A, Stewart DJ. Review: molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathol Appl Neurobiol. 2011 Feb;37(1):3–23.
- Winkler EA, Minter D, Yue JK, et al. Cerebral edema in traumatic brain injury: pathophysiology and prospective therapeutic targets. Neurosurg Clin N Am. 2016 Oct;27(4):473–488.
- Yang C, Hawkins KE, Doré S, et al. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol. 2019 Feb;316(2):C135–C153.
- Keep RF, Andjelkovic AV, Xiang J, et al. Brain endothelial cell junctions after cerebral hemorrhage: Changes, mechanisms and therapeutic targets. J Cereb Blood Flow Metab. 2018 May;38(8):1255–1275.
- Rosenberg GA, Yang Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus. 2007 May;22(5):E4.
- Wang W, Dentler WL, Borchardt RT. VEGF increases BMEC monolayer permeability by affecting occludin expression and tight junction assembly. Am J Physiol Heart Circ Physiol. 2001 Jan;280(1):H434–40.
- Mankertz J, Tavalali S, Schmitz H, et al. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J Cell Sci. 2000 Jun;113(Pt 11):2085–2090.
- Fischer S, Wobben M, Marti HH, et al. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res. 2002 Jan;63(1):70–80.
- Schoch HJ, Fischer S, Marti HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002 Nov;125(11):2549–2557.
- Yang Y, Estrada EY, Thompson JF, et al. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007 Apr;27(4):697–709.
- Yang Y, Rosenberg GA. MMP-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol Biol. 2011;762:333–345.
- Xing G, Zhao T, Zhang X, et al. Astrocytic Sonic Hedgehog Alleviates Intracerebral Hemorrhagic Brain Injury via Modulation of Blood-Brain Barrier Integrity. Front. Cell Neurosci. 2020 Dec;14:575690.
- Brilha S, Ong CWM, Weksler B, et al. Matrix metalloproteinase-9 activity and a downregulated Hedgehog pathway impair blood-brain barrier function in an in vitro model of CNS tuberculosis. Sci Rep. 2017 Nov;7:16031.
- Liu S, Chang L, Wei C. The sonic hedgehog pathway mediates Tongxinluo capsule-induced protection against blood-brain barrier disruption after ischaemic stroke in mice. Basic Clin Pharmacol Toxicol. 2019 Jun;124:660–669.
- Michinaga S, Inoue A, Sonoda K, et al. Down-regulation of Astrocytic Sonic Hedgehog by Activation of Endothelin ETB Receptors: Involvement in Traumatic Brain Injury-induced Disruption of Blood Brain Barrier in a Mouse Model. Neurochem Int. 2021;146 ;105042.
- Chechneva OV, Mayrhofer F, Daugherty DJ, et al. A Smoothened receptor agonist is neuroprotective and promotes regeneration after ischemic brain injury. Cell Death Dis. 2014 Oct;5:e1481.
- Xia Y, He Q, Li Y, et al. Recombinant human sonic hedgehog protein regulates the expression of ZO-1 and occludin by activating angiopoietin-1 in stroke damage. PLoS One. 2013 Jul;8:e68891.
- Alvarez JI, Dodelet-Devillers A, Kebir H, et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011 Dec;334:1727–1731.
- Manaenko A, Fathali N, Khatibi NH, et al. Arginine-vasopressin V1a receptor inhibition improves neurologic outcomes following an intracerebral hemorrhagic brain injury. Neurochem Int. 2011 Mar;58:542–548.
- Yi HJ, Lee JE, Lee DH, et al. The role of NLRP3 in traumatic brain injury and its regulation by pioglitazone. J Neurosurg. 2019 Sep;1–9.
- Wang H, Chen H, Jin J, et al. Inhibition of the NLRP3 inflammasome reduces brain edema and regulates the distribution of aquaporin-4 after cerebral ischaemia-reperfusion. Life Sci. 2020 Jun;251:117638.
- Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012 Aug;4:147ra111.
- Mestre H, Mori Y, Nedergaard M. The brain’s glymphatic system: current controversies. Trends Neurosci. 2020 May;43:458–466.
- Mestre H, Du T, Sweeney AM, et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science. 2020Mar;367.
- Ji C, Yu X, Xu W, et al. The role of glymphatic system in the cerebral edema formation after ischemic stroke. Exp Neurol. 2021 Mar;113685.
- Liu E, Sun L, Zhang Y, et al. Aquaporin4 Knockout Aggravates Early Brain Injury Following Subarachnoid Hemorrhage Through Impairment of the Glymphatic System in Rat Brain. Acta Neurochir Suppl. 2020;127:59–64.
- Christensen J, Wright DK, Yamakawa GR, et al. Repetitive mild traumatic brain injury alters glymphatic clearance rates in limbic structures of adolescent female rats. Sci Rep. 2020 Apr;10:6254.
- Pu T, Zou W, Feng W, et al. Persistent malfunction of glymphatic and meningeal lymphatic drainage in a mouse model of subarachnoid hemorrhage. Exp Neurobiol. 2019 Feb;28:104–118.
- Liu E, Peng X, Ma H, et al. The Involvement of Aquaporin-4 in the Interstitial Fluid Drainage Impairment Following Subarachnoid Hemorrhage. Front Aging Neurosci. 2020;12:611494.
- Fang Y, Shi H, Ren R, et al. Pituitary Adenylate Cyclase-Activating Polypeptide Attenuates Brain Edema by Protecting Blood-Brain Barrier and Glymphatic System After Subarachnoid Hemorrhage in Rats. Neurotherapeutics. 2020 Sep;17:1954–1972.
- Aspelund A, Antila S, Proulx ST, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015 Jun;212:991–999.
- Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015 Jul;523:337–341.
- Chen J, He J, Ni R, et al. Cerebrovascular Injuries Induce Lymphatic Invasion into Brain Parenchyma to Guide Vascular Regeneration in Zebrafish. Dev Cell. 2019 Jun;49:697–710.e5.
- Wojciechowski S, Virenque A, Vihma M, et al. Developmental Dysfunction of the Central Nervous System Lymphatics Modulates the Adaptive Neuro-Immune Response in the Perilesional Cortex in a Mouse Model of Traumatic Brain Injury. Front Immunol. 2020;11:559810.
- Chen J, Wang L, Xu H, et al. Meningeal lymphatics clear erythrocytes that arise from subarachnoid hemorrhage. Nat Commun. 2020 Jun;11:3159.
- Ma Q, Schlegel F, Bachmann SB, et al. Lymphatic outflow of cerebrospinal fluid is reduced in glioma. Sci Rep. 2019 Oct;9:14815.
- Clément T, Rodriguez-Grande B, Badaut J. Aquaporins in brain edema. J Neurosci Res. 2020;98:9–18.
- Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013 Apr;14:265–277.
- Tomás-Camardiel M, Venero JL, de Pablos RM, et al. In vivo expression of aquaporin-4 by reactive microglia. J Neurochem. 2004 Nov;91:891–899.
- Nakayama S, Migliati E, Amiry-Moghaddam M, et al. Osmotherapy With Hypertonic Saline Attenuates Global Cerebral Edema Following Experimental Cardiac Arrest via Perivascular Pool of Aquaporin-4. Crit Care Med. 2016 Aug;44:e702–10.
- Stokum JA, Kwon MS, Woo SK, et al. SUR1-TRPM4 and AQP4 form a heteromultimeric complex that amplifies ion/water osmotic coupling and drives astrocyte swelling. Glia. 2018;66:108–125.
- Xu X, Yin D, Ren H, et al. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol Dis. 2018 May;117:15–27.
- Manley GT, Fujimura M, Ma T, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000 Feb;6:159–163.
- Taya K, Marmarou CR, Okuno K, et al. Effect of secondary insults upon aquaporin-4 water channels following experimental cortical contusion in rats. J Neurotrauma. 2010 Jan;27:229–239.
- Papadopoulos MC, Manley GT, Krishna S, et al. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004 Aug;18:1291–1293.
- Wallisch JS, Janesko-Feldman K, Alexander H, et al. The aquaporin-4 inhibitor AER-271 blocks acute cerebral edema and improves early outcome in a pediatric model of asphyxial cardiac arrest. Pediatr Res. 2019 Mar;85:511–517.
- Wallisch J, Jha R, Vagni V, et al. Effect of the novel aquaporin-4 antagonist AER-271 in combined TBI plus hemorrhagic shock in mice. Crit Care Med. 2015 Dec;43:6–7.
- Chen J-Q, Zhang -C-C, Jiang S-N, et al. Effects of aquaporin 4 knockdown on brain edema of the uninjured side after traumatic brain injury in rats. Med Sci Monit. 2016 Dec;22:4809–4819.
- Fukuda AM, Adami A, Pop V, et al. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J Cereb Blood Flow Metab. 2013 Oct;33:1621–1632.
- Higashida T, Kreipke CW, Rafols JA, et al. The role of hypoxia-inducible factor-1α, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 2011 Jan;114:92–101.
- Kiening KL, van Landeghem FKH, Schreiber S, et al. Decreased hemispheric Aquaporin-4 is linked to evolving brain edema following controlled cortical impact injury in rats. Neurosci Lett. 2002 May;324:105–108.
- Ren Z, Iliff JJ, Yang L, et al. “Hit & Run” model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J Cereb Blood Flow Metab. 2013 Jun;33:834–845.
- Yao X, Uchida K, Papadopoulos MC, et al. Mildly Reduced Brain Swelling and Improved Neurological Outcome in Aquaporin-4 Knockout Mice following Controlled Cortical Impact Brain Injury. J Neurotrauma. 2015 Oct;32:1458–1464.
- Hu H, Yao H, Zhang W, et al. Increased expression of aquaporin-4 in human traumatic brain injury and brain tumors. J Zhejiang Univ Sci B. 2005 Jan;6:33–37.
- Lo Pizzo M, Schiera G, Di Liegro I, et al. Aquaporin-4 distribution in control and stressed astrocytes in culture and in the cerebrospinal fluid of patients with traumatic brain injuries. Neurol Sci. 2013 Aug;34:1309–1314.
- Dardiotis E, Paterakis K, Tsivgoulis G, et al. AQP4 tag single nucleotide polymorphisms in patients with traumatic brain injury. J Neurotrauma. 2014 Dec;31(23):1920–1926.
- Rotondo F, Butz H, Syro LV, et al. Arginine vasopressin (AVP): a review of its historical perspectives, current research and multifunctional role in the hypothalamo-hypophysial system. Pituitary. 2016 Aug;19(4):345–355.
- Zlokovic BV, Hyman S, McComb JG, et al. Kinetics of arginine-vasopressin uptake at the blood-brain barrier. Biochim Biophys Acta. 1990 Jun;1025(2):191–198.
- Chodobski A, Loh YP, Corsetti S, et al. The presence of arginine vasopressin and its mRNA in rat choroid plexus epithelium. Brain Res. Mol. Brain Res. 1997 Aug;48(1):67–72. https://doi.org/10.1016/S0169-328X(97)00079-X
- Szmydynger-Chodobska J, Zink BJ, Chodobski A. Multiple sites of vasopressin synthesis in the injured brain. J Cereb Blood Flow Metab. 2011 Jan;31(1):47–51.
- Buijs RM, Swaab DF, Dogterom J, et al. Intra- and extrahypothalamic vasopressin and oxytocin pathways in the rat. Cell Tissue Res. 1978 Jan;186(3):423–433.
- Raichle ME, Grubb RL. Regulation of brain water permeability by centrally-released vasopressin. Brain Res. 1978 Mar;143(1):191–194.
- Sarfaraz D, Fraser CL. Effects of arginine vasopressin on cell volume regulation in brain astrocyte in culture. Am J Physiol. 1999;276(3):E596–601.
- Latzkovits L, Cserr HF, Park JT, et al. Effects of arginine vasopressin and atriopeptin on glial cell volume measured as 3-MG space. Am J Physiol. 1993 Mar;264(3):C603–8.
- Faraci FM, Mayhan WG, Heistad DD. Effect of vasopressin on production of cerebrospinal fluid: possible role of vasopressin (V1)-receptors. Am J Physiol. 1990 Jan;258(1 Pt 2):R94–8.
- Seckl JR, Lightman SL. Intracerebroventricular vasopressin reduces CSF absorption rate in the conscious goat. Exp Brain Res. 1991;84(1):173–176.
- Filippidis AS, Liang X, Wang W, et al. Real-time monitoring of changes in brain extracellular sodium and potassium concentrations and intracranial pressure after selective vasopressin-1a receptor inhibition following focal traumatic brain injury in rats. J Neurotrauma. 2014 Jul;31(14):1258–1267.
- Liu X, Nakayama S, Amiry-Moghaddam M, et al. Arginine-vasopressin V1 but not V2 receptor antagonism modulates infarct volume, brain water content, and aquaporin-4 expression following experimental stroke. Neurocrit Care. 2010 Feb;12(1):124–131.
- Krieg SM, Trabold R, Plesnila N. Time-Dependent Effects of Arginine-Vasopressin V1 Receptor Inhibition on Secondary Brain Damage after Traumatic Brain Injury. J Neurotrauma. 2017 Apr;34(7):1329–1336.
- Kleindienst A, Dunbar JG, Glisson R, et al. The role of vasopressin V1A receptors in cytotoxic brain edema formation following brain injury. Acta Neurochir (Wien). 2013 Jan;155(1):151–164.
- Pascale CL, Szmydynger-Chodobska J, Sarri JE, et al. Traumatic brain injury results in a concomitant increase in neocortical expression of vasopressin and its V1a receptor. J Physiol Pharmacol. 2006 Nov;57(Suppl 11):161–167.
- Rauen K, Trabold R, Brem C, et al. Arginine Vasopressin V1a Receptor-Deficient Mice Have Reduced Brain Edema and Secondary Brain Damage following Traumatic Brain Injury. J Neurotrauma. 2013 Aug;30(16):1442–1448.
- Vakili A, Kataoka H, Plesnila N. Role of Arginine Vasopressin V1 and V2 Receptors for Brain Damage After Transient Focal Cerebral Ischemia. J Cereb Blood Flow Metab. 2005 Aug;25(8):1012–1019.
- Krieg SM, Sonanini S, Plesnila N, et al. Effect of Small Molecule Vasopressin V1a and V2 Receptor Antagonists on Brain Edema Formation and Secondary Brain Damage following Traumatic Brain Injury in Mice. J Neurotrauma. 2015 Feb;32(4):221–227
- Marmarou CR, Liang X, Abidi NH, et al. Selective vasopressin-1a receptor antagonist prevents brain edema, reduces astrocytic cell swelling and GFAP, V1aR and AQP4 expression after focal traumatic brain injury. Brain Res. 2014 Sep;1581:89–102.
- Dhar R, Murphy-Human T. A bolus of conivaptan lowers intracranial pressure in a patient with hyponatremia after traumatic brain injury. Neurocrit Care. 2011 Feb;14(1):97–102.
- Hedna VS, Bidari S, Gubernick D, et al. Treatment of stroke related refractory brain edema using mixed vasopressin antagonism: a case report and review of the literature. BMC Neurol. 2014 Nov;14(1):213.
- Murphy T, Dhar R, Diringer M. Conivaptan bolus dosing for the correction of hyponatremia in the neurointensive care unit. Neurocrit Care. 2009 Jan;11(1):14–19.
- Corry JJ, Asaithambi G, Shaik AM, et al. Conivaptan for the reduction of cerebral edema in intracerebral hemorrhage: A safety and tolerability study. Clin Drug Investig. 2020 May;40(5):503–509.
- Yang C, Yang Y, DeMars KM, et al Genetic Deletion or Pharmacological Inhibition of Cyclooxygenase-2 Reduces Blood-Brain Barrier Damage in Experimental Ischemic Stroke. Front Neurol. 2020 Aug;11:887.
- Wang J, Doré S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007 May;27(5):894–908.
- Gong C, Ennis SR, Hoff JT, et al. Inducible cyclooxygenase-2 expression after experimental intracerebral hemorrhage. Brain Res. 2001 May;901(1–2):38–46.
- Strauss KI. Antiinflammatory and neuroprotective actions of COX2 inhibitors in the injured brain. Brain Behav Immun. 2008 Mar;22(3):285–298.
- Liu H, Rose ME, Miller TM, et al. COX2-derived primary and cyclopentenone prostaglandins are increased after asphyxial cardiac arrest. Brain Res. 2013 Jun;1519:71–77.
- Wu T, Wu H, Wang J, et al. Expression and cellular localization of cyclooxygenases and prostaglandin E synthases in the hemorrhagic brain. J Neuroinflammation. 2011 Mar;8(1):22.
- Zhang H-B, Tu X-K, Chen Q, et al. Propofol Reduces Inflammatory Brain Injury after Subarachnoid Hemorrhage: Involvement of PI3K/Akt Pathway. J Stroke Cerebrovasc Dis. 2019 Dec;28(12):104375.
- Cernak I, O’Connor C, Vink R. Activation of cyclo-oxygenase-2 contributes to motor and cognitive dysfunction following diffuse traumatic brain injury in rats. Clin Exp Pharmacol Physiol. 2001 Nov;28(11):922–925.
- Strauss KI, Barbe MF, Marshall RM, et al. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J Neurotrauma. 2000 Aug;17(8):695–711.
- Munakata A, Naraoka M, Katagai T, et al. Role of Cyclooxygenase-2 in Relation to Nitric Oxide and Endothelin-1 on Pathogenesis of Cerebral Vasospasm After Subarachnoid Hemorrhage in Rabbit. Transl Stroke Res. 2016 Apr;7(3):220–227.
- Tran Dinh YR, Jomaa A, Callebert J, et al. Overexpression of cyclooxygenase-2 in rabbit basilar artery endothelial cells after subarachnoid hemorrhage. Neurosurgery. 2001 Mar;48(3):626–633. discussion 633.
- Dash PK, Mach SA, Moore AN. Regional expression and role of cyclooxygenase-2 following experimental traumatic brain injury. J Neurotrauma. 2000 Jan;17(1):69–81.
- Dehlaghi Jadid K, Davidsson J, Lidin E, et al. COX-2 Inhibition by Diclofenac Is Associated With Decreased Apoptosis and Lesion Area After Experimental Focal Penetrating Traumatic Brain Injury in Rats. Front Neurol. 2019 Jul;10:811.
- Cernak I, O’Connor C, Vink R. Inhibition of cyclooxygenase 2 by nimesulide improves cognitive outcome more than motor outcome following diffuse traumatic brain injury in rats. Exp Brain Res. 2002 Nov;147(2):193–199.
- Ayer R, Jadhav V, Sugawara T, et al. The neuroprotective effects of cyclooxygenase-2 inhibition in a mouse model of aneurysmal subarachnoid hemorrhage. Acta Neurochir Suppl. 2011;111:145–149.
- Sinn D-I, Lee S-T, Chu K, et al. Combined neuroprotective effects of celecoxib and memantine in experimental intracerebral hemorrhage. Neurosci Lett. 2007 Jan;411(3):238–242.
- Chu K, Jeong S-W, Jung K-H, et al. Celecoxib induces functional recovery after intracerebral hemorrhage with reduction of brain edema and perihematomal cell death. J Cereb Blood Flow Metab. 2004 Aug;24(8):926–933.
- Santos-Galdiano M, Pérez-Rodríguez D, Anuncibay-Soto B, et al. Celecoxib Treatment Improves Neurologic Deficit and Reduces Selective Neuronal Loss and Glial Response in Rats after Transient Middle Cerebral Artery Occlusion. J Pharmacol Exp Ther. 2018 Oct;367(3):528–542.
- Si D, Li J, Liu J, et al. Progesterone protects blood-brain barrier function and improves neurological outcome following traumatic brain injury in rats. Exp Ther Med. 2014 Sep;8(3):1010–1014.
- Wang X, Zhang J, Si D, et al. Progesterone inhibits the expression of cycloxygenase-2 and interleukin-1β in neonatal rats with hypoxic ischemic brain damage. Int J Neurosci. 2014 Jan;124(1):42–48.
- Cutler SM, Cekic M, Miller DM, et al. Progesterone improves acute recovery after traumatic brain injury in the aged rat. J Neurotrauma. 2007 Sep;24(9):1475–1486.
- Si D, Wang H, Wang Q, et al. Progesterone treatment improves cognitive outcome following experimental traumatic brain injury in rats. Neurosci Lett. 2013 Oct;553:18–23.
- Leese PT, Hubbard RC, Karim A, et al. Effects of celecoxib, a novel cyclooxygenase-2 inhibitor, on platelet function in healthy adults: a randomized, controlled trial. J Clin Pharmacol. 2000 Feb;40(2):124–132.
- Park H-K, Lee S-H, Chu K, et al. Effects of celecoxib on volumes of hematoma and edema in patients with primary intracerebral hemorrhage. J Neurol Sci. 2009 Apr;279(1–2):43–46.
- Lee SH, Park HK, Ryu WS, et al. Effects of celecoxib on hematoma and edema volumes in primary intracerebral hemorrhage: a multicenter randomized controlled trial. Eur J Neurol. 2013 Aug;20(8):1161–1169.
- Naseh M, Vatanparast J, Rafati A, et al. The emerging role of FTY720 as a sphingosine 1-phosphate analog for the treatment of ischemic stroke: The cellular and molecular mechanisms. Brain Behav. 2021 May;11(6):e02179.
- Kraft P, Göb E, Schuhmann MK, et al. FTY720 ameliorates acute ischemic stroke in mice by reducing thrombo-inflammation but not by direct neuroprotection. Stroke. 2013 Nov;44(11):3202–3210.
- Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004 Jan;427(6972):355–360.
- Pham THM, Okada T, Matloubian M, et al. S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity. 2008 Jan;28(1):122–133.
- Gaire BP, Lee C-H, Sapkota A, et al. Identification of Sphingosine 1-Phosphate Receptor Subtype 1 (S1P1) as a Pathogenic Factor in Transient Focal Cerebral Ischemia. Mol Neurobiol. 2018;55(3):2320–2332.
- Sapkota A, Gaire BP, Kang M-G, et al. S1P2 contributes to microglial activation and M1 polarization following cerebral ischemia through ERK1/2 and JNK. Sci Rep. 2019 Aug;9(1):12106.
- Gaire BP, Song M-R, Choi JW. Sphingosine 1-phosphate receptor subtype 3 (S1P3) contributes to brain injury after transient focal cerebral ischemia via modulating microglial activation and their M1 polarization. J Neuroinflammation. 2018 Oct;15(1):284.
- Li X, Wang M-H, Qin C, et al. Fingolimod suppresses neuronal autophagy through the mTOR/p70S6K pathway and alleviates ischemic brain damage in mice. PLoS One. 2017 Nov;12(11):e0188748.
- Safarian F, Khallaghi B, Ahmadiani A, et al. Activation of S1P₁ receptor regulates PI3K/Akt/FoxO3a pathway in response to oxidative stress in PC12 cells. J Mol Neurosci. 2015 May;56(1):177–187.
- Xiang P, Chew WS, Seow WL, et al. The S1P2 receptor regulates blood-brain barrier integrity and leukocyte extravasation with implications for neurodegenerative disease. Neurochem Int. 2021 Jun;146:105018.
- Wacker BK, Park TS, Gidday JM. Hypoxic preconditioning-induced cerebral ischemic tolerance: role of microvascular sphingosine kinase 2. Stroke. 2009 Oct;40(10):3342–3348.
- Roy R, Alotaibi AA, Freedman MS. Sphingosine 1-Phosphate Receptor Modulators for Multiple Sclerosis. CNS Drugs. 2021 Apr;35(4):385–402.
- Xu H-L, Pelligrino DA, Paisansathan C, et al. Protective role of fingolimod (FTY720) in rats subjected to subarachnoid hemorrhage. J Neuroinflammation. 2015 Jan;12(1):16.
- Tschoe C, Bushnell CD, Duncan PW, et al. Neuroinflammation after Intracerebral Hemorrhage and Potential Therapeutic Targets. J. Stroke. 2020 Jan;22(1):29–46. DOI:https://doi.org/10.5853/jos.2019.02236
- Napier J, Rose L, Adeoye O, et al. Modulating acute neuroinflammation in intracerebral hemorrhage: the potential promise of currently approved medications for multiple sclerosis. Immunopharmacol Immunotoxicol. 2019 Feb;41(1):7–15.
- Gao C, Qian Y, Huang J, et al. A Three-Day Consecutive Fingolimod Administration Improves Neurological Functions and Modulates Multiple Immune Responses of CCI Mice. Mol Neurobiol. 2017 Dec;54(10):8348–8360.
- Sun N, Shen Y, Han W, et al. Selective Sphingosine-1-Phosphate Receptor 1 Modulation Attenuates Experimental Intracerebral Hemorrhage. Stroke. 2016 May;47(7):1899–1906.
- Rolland WB, Lekic T, Krafft PR, et al. Fingolimod reduces cerebral lymphocyte infiltration in experimental models of rodent intracerebral hemorrhage. Exp Neurol. 2013 Mar;241:45–55.
- Rolland WB, Manaenko A, Lekic T, et al. FTY720 is neuroprotective and improves functional outcomes after intracerebral hemorrhage in mice. Acta Neurochir Suppl. 2011;111:213–217.
- Wang Y, Zhou S, Han Z, et al Fingolimod administration improves neurological functions of mice with subarachnoid hemorrhage. Neurosci Lett. 2020 Sep;736:135250.
- Zhang L, Ding K, Wang H, et al. Traumatic Brain Injury-Induced Neuronal Apoptosis is Reduced Through Modulation of PI3K and Autophagy Pathways in Mouse by FTY720. Cell Mol. Neurobiol. 2016 Jan;36(1):131–142. https://doi.org/10.1007/s10571-015-0227-1
- Oshima Y, Tanimoto T, Yuji K, et al. Drug-associated progressive multifocal leukoencephalopathy in multiple sclerosis patients. Mult Scler. 2019;25(8):1141–1149.
- Fu Y, Hao J, Zhang N, et al. Fingolimod for the treatment of intracerebral hemorrhage: a 2-arm proof-of-concept study. JAMA Neurol. 2014 Sep;71(9):1092–1101.
- Fu Y, Zhang N, Ren L, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci USA. 2014 Dec;111(51):18315–18320.
- Tian D-C, Shi K, Zhu Z, et al. Fingolimod enhances the efficacy of delayed alteplase administration in acute ischemic stroke by promoting anterograde reperfusion and retrograde collateral flow. Ann Neurol. 2018 Nov;84(5):717–728.
- Männer A, Thomas D, Wagner M, et al. Sphingosine 1-phosphate levels in cerebrospinal fluid after subarachnoid hemorrhage. Neurol. Res. Pract. 2020 Nov;2(1):49. https://doi.org/10.1186/s42466-020-00093-x
- Chen M, Simard JM. Cell Swelling and a Nonselective Cation Channel Regulated by Internal Ca2+ and ATP in Native Reactive Astrocytes from Adult Rat Brain. J Neurosci. 2001 Sep;21(17):6512–6521.
- Jha RM, Bell J, Citerio G, et al. Role of sulfonylurea receptor 1 and glibenclamide in traumatic brain injury: A review of the evidence. Int J Mol Sci. 2020 Jan;21.
- Woo SK, Tsymbalyuk N, Tsymbalyuk O, et al. SUR1-TRPM4 channels, not KATP, mediate brain swelling following cerebral ischemia. Neurosci Lett. 2020 Jan;718:134729.
- Gerzanich V, Stokum JA, Ivanova S, et al. Sulfonylurea receptor 1, transient receptor potential cation channel subfamily M member 4, and kir6.2: rolein hemorrhagic progression of contusion. J Neurotrauma. 2019 Apr;36(7):1060–1079.
- Simard JM, Chen M, Tarasov KV, et al. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med. 2006 Apr;12(4):433–440. DOI:https://doi.org/10.1038/nm1390.
- Boggs DH, Simard JM, Steven A, et al. Potential of glyburide to reduce intracerebral edema in brain metastases. Expert Rev Neurother. 2014 Apr;14(4):379–388.
- Kurland D, Hong C, Aarabi B, et al. Hemorrhagic progression of a contusion after traumatic brain injury: a review. J Neurotrauma. 2012 Jan;29(1):19–31.
- Nakayama S, Taguchi N, Isaka Y, et al. Glibenclamide and therapeutic hypothermia have comparable effect on attenuating global cerebral edema following experimental cardiac arrest. Neurocrit Care. 2018;29(1):119–127.
- Jha RM, Mondello S, Bramlett HM, et al. Glibenclamide treatment in traumatic brain injury: operation brain trauma therapy. J Neurotrauma. 2021 Mar;38(5):628–645.
- Jha RM, Molyneaux BJ, Jackson TC, et al. Glibenclamide Produces Region-Dependent Effects on Cerebral Edema in a Combined Injury Model of Traumatic Brain Injury and Hemorrhagic Shock in Mice. J Neurotrauma. 2018 Sep;35(17):2125–2135.
- Simard JM, Tsymbalyuk O, Ivanov A, et al. Endothelial sulfonylurea receptor 1-regulated NC Ca-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J Clin Invest. 2007 Aug;117(8):2105–2113.
- Tsymbalyuk O, Gerzanich V, Mumtaz A, et al. SUR1, newly expressed in astrocytes, mediates neuropathic pain in a mouse model of peripheral nerve injury. Mol Pain. 2021 Dec;17:17448069211006604.
- Simard JM, Popovich PG, Tsymbalyuk O, et al. Spinal cord injury with unilateral versus bilateral primary hemorrhage–effects of glibenclamide. Exp Neurol. 2012 Feb;233(2):829–835.
- Woo SK, Kwon MS, Ivanov A, et al. The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. J Biol Chem. 2013 Feb;288(5):3655–3667.
- Simard JM, Woo SK, Norenberg MD, et al. Brief Suppression of Abcc8 Prevents Autodestruction of Spinal Cord After Trauma. Sci Transl Med. 2010 Apr;2(28):28ra29.
- Makar TK, Gerzanich V, Nimmagadda VKC, et al. Silencing of Abcc8 or inhibition of newly upregulated Sur1-Trpm4 reduce inflammation and disease progression in experimental autoimmune encephalomyelitis. J Neuroinflammation. 2015 Nov;12(1):210.
- Simard JM, Woo SK, Schwartzbauer GT, et al. Sulfonylurea receptor 1 in central nervous system injury: a focused review. J Cereb Blood Flow Metab. 2012 Sep;32(9):1699–1717.
- Gerzanich V, Makar TK, Guda PR, et al. Salutary effects of glibenclamide during the chronic phase of murine experimental autoimmune encephalomyelitis. J Neuroinflammation. 2017 Sep;14(1):177.
- Tata S, Zusman BE, Kochanek PM, et al. Abcc8 (Sulfonylurea Receptor-1) Impact on Brain Atrophy after Traumatic Brain Injury Varies by Sex. J Neurotrauma. 2021 May;38(17):2473–2485.
- Mehta RI, Tosun C, Ivanova S, et al. Sur1-Trpm4 Cation Channel Expression in Human Cerebral Infarcts. J Neuropathol Exp Neurol. 2015 Aug;74(8):835–849.
- Nilius B, Prenen J, Tang J, et al. Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J Biol Chem. 2005 Feb;280(8):6423–6433.
- Chen M, Dong Y, Simard JM. Functional coupling between sulfonylurea receptor type 1 and a nonselective cation channel in reactive astrocytes from adult rat brain. J Neurosci. 2003 Sep;23(24):8568–8577.
- Sheth KN, Elm JJ, Molyneaux BJ, et al. Safety and efficacy of intravenous glyburide on brain swelling after large hemispheric infarction (GAMES-RP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2016 Oct;15(11):1160–1169. DOI:https://doi.org/10.1016/S1474-4422(16)30196-X.
- Kurland DB, Gerzanich V, Karimy JK, et al. The Sur1-Trpm4 channel regulates NOS2 transcription in TLR4-activated microglia. J Neuroinflammation. 2016 Jun;13(1):130.
- Gerzanich V, Kwon MS, Woo SK, et al. SUR1-TRPM4 channel activation and phasic secretion of MMP-9 induced by tPA in brain endothelial cells. PLoS One. 2018 Apr;13(4):e0195526.
- Martínez-Valverde T, Vidal-Jorge M, Martínez-Saez E, et al. Sulfonylurea Receptor 1 in Humans with Post-Traumatic Brain Contusions. J Neurotrauma. 2015 Oct;32(19):1478–1487.
- Huang K, Gu Y, Hu Y, et al. Glibenclamide improves survival and neurologic outcome after cardiac arrest in rats. Crit Care Med. 2015 Sep;43(9):e341–9.
- Xu F, Shen G, Su Z, et al. Glibenclamide ameliorates the disrupted blood-brain barrier in experimental intracerebral hemorrhage by inhibiting the activation of NLRP3 inflammasome. Brain Behav. 2019 Mar;9(4):e01254.
- Stokum JA, Keledjian K, Hayman E, et al. Glibenclamide pretreatment protects against chronic memory dysfunction and glial activation in rat cranial blast traumatic brain injury. Behav Brain Res. 2017 Aug;333:43–53.
- Zweckberger K, Hackenberg K, Jung CS, et al. Glibenclamide reduces secondary brain damage after experimental traumatic brain injury. Neuroscience. 2014 Jul;272:199–206.
- Igarashi T, Sastre C, Wolcott Z, et al. Continuous Glibenclamide Prevents Hemorrhagic Transformation in a Rodent Model of Severe Ischemia-Reperfusion. J Stroke Cerebrovasc Dis. 2021 Mar;30(3):105595.
- Ortega FJ, Jolkkonen J, Mahy N, et al. Glibenclamide enhances neurogenesis and improves long-term functional recovery after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2013 Mar;33(3):356–364.
- Jiang B, Li L, Chen Q, et al. Role of glibenclamide in brain injury after intracerebral hemorrhage. Transl Stroke Res. 2017;8(2):183–193.
- Wang X, Chang Y, He Y, et al. Glimepiride and glibenclamide have comparable efficacy in treating acute ischemic stroke in mice. Neuropharmacology. 2020 Jan;162:107845.
- Zhang G, Lin X, Zhang S, et al. Role of Glibenclamide in Inflammation-Associated Injury. Mediators Inflamm. 2017 Jun;2017:3578702.
- Z-m X, Yuan F, Y-l L, et al. Glibenclamide Attenuates Blood-Brain Barrier Disruption in Adult Mice after Traumatic Brain Injury. J Neurotrauma. 2017 Feb;34(4):925–933.
- Simard JM, Tsymbalyuk N, Tsymbalyuk O, et al. Glibenclamide is superior to decompressive craniectomy in a rat model of malignant stroke. Stroke. 2010 Mar;41(3):531–537.
- Thompson EM, Pishko GL, Muldoon LL, et al. Inhibition of SUR1 decreases the vascular permeability of cerebral metastases. Neoplasia. 2013 May;15(5):535–543.
- Tosun C, Koltz MT, Kurland DB, et al. The protective effect of glibenclamide in a model of hemorrhagic encephalopathy of prematurity. Brain Sci. 2013 Mar;3(3):215–238.
- Simard JM, Geng Z, Woo SK, et al. Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2009 Feb;29(2):317–330.
- Tosun C, Kurland DB, Mehta R, et al. Inhibition of the Sur1-Trpm4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage. Stroke. 2013 Dec;44(12):3522–3528.
- Deng G, Ma C, Zhao H, et al. Anti-edema and antioxidant combination therapy for ischemic stroke via glyburide-loaded betulinic acid nanoparticles. Theranostics. 2019 Sep;9(23):6991–7002.
- Castro L, Noelia M, Vidal-Jorge M, et al. Kir6.2, the Pore-Forming Subunit of ATP-Sensitive K+ Channels, Is Overexpressed in Human Posttraumatic Brain Contusions. J Neurotrauma. 2019 Jan;36(1):165–175.
- Mehta RI, Ivanova S, Tosun C, et al. Sulfonylurea receptor 1 expression in human cerebral infarcts. J Neuropathol Exp Neurol. 2013 Sep;72(9):871–883.
- Thompson EM, Halvorson K, McLendon R. Sulfonylurea receptor 1 expression is variable in adult and pediatric brain tumors. Clin Neuropathol. 2018;37(5):221–227.
- Zusman BE, Kochanek PM, Bell MJ, et al. Cerebrospinal Fluid Sulfonylurea Receptor-1 Is Associated with Intracranial Pressure and Outcome after Pediatric TBI: An Exploratory Analysis of the Cool Kids Trial. J Neurotrauma. 2021 Feb;38(12):1615–1619.
- Jha RM, Puccio AM, Chou SH-Y, et al. Sulfonylurea Receptor-1: A Novel Biomarker for Cerebral Edema in Severe Traumatic Brain Injury. Crit Care Med. 2017;45(3):e255–e264.
- Dundar TT, Abdallah A, Yurtsever I, et al. Serum SUR1 and TRPM4 in patients with subarachnoid hemorrhage. Neurosurg Rev. 2020 Dec;43(6):1595–1603.
- Jha RM, Desai SM, Zusman BE, et al. Downstream TRPM4 Polymorphisms Are Associated with Intracranial Hypertension and Statistically Interact with ABCC8 Polymorphisms in a Prospective Cohort of Severe Traumatic Brain Injury. J Neurotrauma. 2019 Feb;36(11):1804–1817.
- Jha RM, Puccio AM, Okonkwo DO, et al. ABCC8 Single Nucleotide Polymorphisms are Associated with Cerebral Edema in Severe TBI. Neurocrit Care. 2017;26(2):213–224.
- Jha RM, Koleck TA, Puccio AM, et al. Regionally clustered ABCC8 polymorphisms in a prospective cohort predict cerebral oedema and outcome in severe traumatic brain injury. J Neurol Neurosurg Psychiatry. 2018 Nov;89(11):1152–1162.
- Kunte H, Schmidt S, Eliasziw M, et al. Sulfonylureas improve outcome in patients with type 2 diabetes and acute ischemic stroke. Stroke. 2007 Sep;38(9):2526–2530.
- Kunte H, Busch MA, Trostdorf K, et al. Hemorrhagic transformation of ischemic stroke in diabetics on sulfonylureas. Ann Neurol. 2012 Nov;72(5):799–806.
- Khalili H, Derakhshan N, Niakan A, et al. Effects of Oral Glibenclamide on Brain Contusion Volume and Functional Outcome of Patients with Moderate and Severe Traumatic Brain Injuries: A Randomized Double-Blind Placebo-Controlled Clinical Trial. World Neurosurg. 2017 May;101:130–136.
- Zafardoost P, Ghasemi AA, Salehpour F, et al. Evaluation of the effect of glibenclamide in patients with diffuse axonal injury due to moderate to severe head trauma. Trauma Mon. 2016 Nov;21(5):e25113. https://doi.org/10.5812/traumamon.25113
- Huang K, Hu Y, Wu Y, et al. Exploratory analysis of oral glibenclamide in acute ischemic stroke. Acta Neurol Scand. 2019 Sep;140(3):212–218.
- Huang K, Ji Z, Wu Y, et al. Safety and efficacy of glibenclamide combined with rtPA in acute cerebral ischemia with occlusion/stenosis of anterior circulation (SE-GRACE): study protocol for a randomized controlled trial. BMC Neurol. 2020 Jun;20(1):239.
- Minnema AJ, Mehta A, Boling WW, et al. SCING-Spinal Cord Injury Neuroprotection with Glyburide: a pilot, open-label, multicentre, prospective evaluation of oral glyburide in patients with acute traumatic spinal cord injury in the USA. BMJ Open. 2019 Oct;9(10):e031329.
- Armahizer MJ, Howard AK, Seung H, et al. Risk Factors for Hypoglycemia with the Use of Enteral Glyburide in Neurocritical Care Patients. World Neurosurg. 2021 Mar;147:e63–e68.
- Vorasayan P, Bevers MB, Beslow LA, et al. Intravenous glibenclamide reduces lesional water uptake in large hemispheric infarction. Stroke. 2019 Sep;50(11):3021–3027.
- Sheth KN, Petersen NH, Cheung K, et al. Long-Term Outcomes in Patients Aged ≤70 Years With Intravenous Glyburide From the Phase II GAMES-RP Study of Large Hemispheric Infarction: An Exploratory Analysis. Stroke. 2018 May;49(6):1457–1463.
- Za K, Kn S, Wt K, et al. Profile of intravenous glyburide for the prevention of cerebral edema following large hemispheric infarction: evidence to date. Drug Des Devel Ther. 2018 Aug;12:2539–2552.
- Sheth KN, Kimberly WT, Elm JJ, et al. Exploratory analysis of glyburide as a novel therapy for preventing brain swelling. Neurocrit Care. 2014 Aug;21(1):43–51.
- Kimberly WT, Battey TWK, Pham L, et al. Glyburide is associated with attenuated vasogenic edema in stroke patients. Neurocrit Care. 2014 Apr;20(2):193–201.
- Sheth KN, Kimberly WT, Elm JJ, et al. Pilot study of intravenous glyburide in patients with a large ischemic stroke. Stroke. 2014 Jan;45(1):281–283.
- Kimberly WT, Bevers MB, Von Kummer R, et al. Effect of IV glyburide on adjudicated edema endpoints in the GAMES-RP Trial. Neurology. 2018 Dec;91(23):e2163–e2169.
- Lange C, Storkebaum E, de Almodóvar CR, et al. Vascular endothelial growth factor: a neurovascular target in neurological diseases. Nat Rev Neurol. 2016 Jul;12(8):439–454.
- Shim JW, Madsen JR. VEGF signaling in neurological disorders. Int J Mol Sci. 2018 Jan;19.
- Chodobski A, Chung I, Koźniewska E, et al. Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience. 2003;122(4):853–867.
- Suzuki R, Fukai N, Nagashijma G, et al. Very early expression of vascular endothelial growth factor in brain oedema tissue associated with brain contusion. Acta Neurochir Suppl. 2003;86:277–279.
- Argaw AT, Gurfein BT, Zhang Y, et al. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci USA. 2009 Feb;106(6):1977–1982.
- Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem. 2009 Jul;284(31):21036–21046.
- Salehi A, Zhang JH, Obenaus A. Response of the cerebral vasculature following traumatic brain injury. J Cereb Blood Flow Metab. 2017 Jul;37(7):2320–2339.
- Zhang ZG, Zhang L, Jiang Q, et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000 Oct;106(7):829–838.
- van Bruggen N, Thibodeaux H, Palmer JT, et al. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest. 1999 Dec;104(11):1613–1620.
- Kim MM, Umemura Y, Leung D. Bevacizumab and glioblastoma: past, present, and future directions. Cancer J. 2018;24(4):180–186.
- Svensson B, Peters M, König H-G, et al. Vascular endothelial growth factor protects cultured rat hippocampal neurons against hypoxic injury via an antiexcitotoxic, caspase-independent mechanism. J Cereb Blood Flow Metab. 2002 Oct;22(10):1170–1175.
- Greenberg DA, Jin K. Vascular endothelial growth factors (VEGFs) and stroke. Cell Mol Life Sci. 2013 May;70(10):1753–1761.
- Thau-Zuchman O, Shohami E, Alexandrovich AG, et al. Vascular endothelial growth factor increases neurogenesis after traumatic brain injury. J Cereb Blood Flow Metab. 2010 May;30(5):1008–1016.
- Tado M, Mori T, Fukushima M, et al. Increased expression of vascular endothelial growth factor attenuates contusion necrosis without influencing contusion edema after traumatic brain injury in rats. J Neurotrauma. 2014 Apr;31(7):691–698.
- Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009 Oct;27(28):4733–4740.
- Tn K, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009 Feb;27(5):740–745.
- Field KM, Simes J, Nowak AK, et al. CABARET/COGNO investigators, Rosenthal MA. Randomized phase 2 study of carboplatin and bevacizumab in recurrent glioblastoma. Neuro Oncol. 2015 Nov;17(11):1504–1513.
- Taal W, Oosterkamp HM, Walenkamp AME, et al. van den Bent MJ. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol. 2014 Aug;15(9):943–953.
- Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014 Feb;370(8):699–708.
- Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014 Feb;370(8):709–722.
- Herrlinger U, Schäfer N, Steinbach JP, et al. Bevacizumab Plus Irinotecan Versus Temozolomide in Newly Diagnosed O6-Methylguanine-DNA Methyltransferase Nonmethylated Glioblastoma: The Randomized GLARIUS Trial. J Clin Oncol. 2016 May;34(14):1611–1619.
- Lombardi G, Pambuku A, Bellu L, et al. Effectiveness of antiangiogenic drugs in glioblastoma patients: A systematic review and meta-analysis of randomized clinical trials. Crit Rev Oncol Hematol. 2017 Mar;111:94–102.
- Batchelor TT, Mulholland P, Neyns B, et al. van den Bent M. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013 Sep;31(26):3212–3218.
- Wick W, Vk P, Chamberlain MC, et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol. 2010 Mar;28(7):1168–1174.
- Slevin M, Krupinski J, Slowik A, et al. Serial measurement of vascular endothelial growth factor and transforming growth factor-beta1 in serum of patients with acute ischemic stroke. Stroke. 2000 Aug;31(8):1863–1870.
- Mellergård P, Sjögren F, Hillman J. Release of VEGF and FGF in the extracellular space following severe subarachnoidal haemorrhage or traumatic head injury in humans. Br J Neurosurg. 2010 Jun;24(3):261–267.
- Pikula A, Beiser AS, Chen TC, et al. Serum brain-derived neurotrophic factor and vascular endothelial growth factor levels are associated with risk of stroke and vascular brain injury: Framingham Study. Stroke. 2013 Oct;44(10):2768–2775.
- Dimitrov DS. Therapeutic proteins. Methods Mol Biol. 2012;899:1–26.
- Shahror RA, Linares GR, Wang Y, et al. Transplantation of mesenchymal stem cells overexpressing fibroblast growth factor 21 facilitates cognitive recovery and enhances neurogenesis in a mouse model of traumatic brain injury. J Neurotrauma. 2020 Jan;37(1):14–26.
- Chen J, Hu J, Liu H, et al. FGF21 Protects the Blood-Brain Barrier by Upregulating PPARγ via FGFR1/β-klotho after Traumatic Brain Injury. J Neurotrauma. 2018 Sep;35(17):2091–2103.
- Chen J, Wang X, Hu J, et al. FGF20 protected against BBB disruption after traumatic brain injury by upregulating junction protein expression and inhibiting the inflammatory response. Front Pharmacol. 2021;11:590669.
- Deng S, Jin P, Sherchan P, et al. Recombinant CCL17-dependent CCR4 activation alleviates neuroinflammation and neuronal apoptosis through the PI3K/AKT/Foxo1 signaling pathway after ICH in mice. J Neuroinflammation. 2021 Mar;18(1):62.
- Deng S, Sherchan P, Jin P, et al. Recombinant CCL17 enhances hematoma resolution and activation of ccr4/erk/nrf2/cd163 signaling pathway after intracerebral hemorrhage in mice. Neurotherapeutics. 2020;17(4):1940–1953.
- Mauceri D, Buchthal B, Hemstedt TJ, et al. Nasally delivered VEGFD mimetics mitigate stroke-induced dendrite loss and brain damage. Proc Natl Acad Sci USA. 2020 Apr;117(15):8616–8623.
- Yahya EB, Alqadhi AM. Recent trends in cancer therapy: A review on the current state of gene delivery. Life Sci. 2021 Mar;269:119087.
- Ferrari G, Thrasher AJ, Aiuti A. Gene therapy using haematopoietic stem and progenitor cells. Nat Rev Genet. 2021;22(4):216–234.
- Matharu N, Ahituv N. Modulating gene regulation to treat genetic disorders. Nat Rev Drug Discov. 2020 Oct;19(11):757–775.
- Ghaemi A, Bagheri E, Abnous K, et al. CRISPR-cas9 genome editing delivery systems for targeted cancer therapy. Life Sci. 2021 Feb;267:118969.
- Bulcha JT, Wang Y, Ma H, et al. Viral vector platforms within the gene therapy landscape. Signal Transduct Target Ther. 2021 Feb;6(1):53.
- Rosenberg SA, Aebersold P, Cornetta K, et al. Gene transfer into humans–immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990 Aug;323(9):570–578.
- Ginn SL, Amaya AK, Alexander IE, et al. Gene therapy clinical trials worldwide to 2017: An update. J Gene Med. 2018 Apr;20(5):e3015.
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004 Jan;116(2):281–297.
- Sun K, Lai EC. Adult-specific functions of animal microRNAs. Nat Rev Genet. 2013 Aug;14(8):535–548.
- Reinhart BJ, Slack FJ, Basson M, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000 Feb;403(6772):901–906.
- Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019 Jan;47(D1):D155–D162.
- Mueller C, Berry JD, McKenna-Yasek DM, et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N Engl J Med. 2020 Jul;383(2):151–158. DOI:https://doi.org/10.1056/NEJMoa2005056.
- Täubel J, Hauke W, Rump S, et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J. 2021 Jan;42(2):178–188.
- Esrick EB, Lehmann LE, Biffi A, et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N Engl J Med. 2021 Jan;384(3):205–215. DOI:https://doi.org/10.1056/NEJMoa2029392.
- Eyileten C, Sharif L, Wicik Z, et al. The Relation of the Brain-Derived Neurotrophic Factor with MicroRNAs in Neurodegenerative Diseases and Ischemic Stroke. Mol Neurobiol. 2021 Jan;58(1):329–347.
- Raikwar SP, Thangavel R, Ahmed ME, et al. Real-Time Noninvasive Bioluminescence, Ultrasound and Photoacoustic Imaging in NFκB-RE-Luc Transgenic Mice Reveal Glia Maturation Factor-Mediated Immediate and Sustained Spatio-Temporal Activation of NFκB Signaling Post-Traumatic Brain Injury in a Gender-Specific Manner. Cell Mol. Neurobiol. 2020 Aug;41(8):1687–1706. https://doi.org/10.1007/s10571-020-00937-9
- Guedes VA, Devoto C, Leete J, et al. Extracellular vesicle proteins and micrornas as biomarkers for traumatic brain injury. Front Neurol. 2020 Jul;11:663.
- Pan J, Qu M, Li Y, et al. MicroRNA-126-3p/-5p Overexpression Attenuates Blood-Brain Barrier Disruption in a Mouse Model of Middle Cerebral Artery Occlusion. Stroke. 2020;51(2):619–627.
- Sonoda T, Matsuzaki J, Yamamoto Y, et al. Serum MicroRNA-Based Risk Prediction for Stroke. Stroke. 2019 Apr;50(6):1510–1518.
- Shang Y, Dai S, Chen X, et al. MicroRNA-93 regulates the neurological function, cerebral edema and neuronal apoptosis of rats with intracerebral hemorrhage through TLR4/NF-κB signaling pathway. Cell Cycle. 2019 Nov;18(22):3160–3176.
- Ma Q, Dasgupta C, Li Y, et al. MicroRNA-210 Suppresses Junction Proteins and Disrupts Blood-Brain Barrier Integrity in Neonatal Rat Hypoxic-Ischemic Brain Injury. Int J Mol Sci. 2017 Jun;18.
- Fish JE, Santoro MM, Morton SU, et al. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008 Aug;15(2):272–284.
- Harris TA, Yamakuchi M, Ferlito M, et al. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci USA. 2008 Feb;105(5):1516–1521.
- Xing F, Liu Y, Dong R, et al. miR-374 improves cerebral ischemia reperfusion injury by targeting Wnt5a. Exp Anim. 2021 Feb;70(1):126–136.
- Wang Y, Wang M-D, Xia Y-P, et al. MicroRNA-130a regulates cerebral ischemia-induced blood-brain barrier permeability by targeting Homeobox A5. FASEB J. 2018 Jan;32(2):935–944.
- Khanna S, Rink C, Ghoorkhanian R, et al. Loss of miR-29b following acute ischemic stroke contributes to neural cell death and infarct size. J Cereb Blood Flow Metab. 2013 Aug;33(8):1197–1206.
- Long G, Wang F, Li H, et al. Circulating miR-30a, miR-126 and let-7b as biomarker for ischemic stroke in humans. BMC Neurol. 2013 Nov;13(1):178.
- Gao X, Xiong Y, Li Q, et al. Extracellular vesicle-mediated transfer of miR-21-5p from mesenchymal stromal cells to neurons alleviates early brain injury to improve cognitive function via the PTEN/Akt pathway after subarachnoid hemorrhage. Cell Death Dis. 2020 May;11(5):363.
- Yao X, Wang Y, Zhang D. microRNA-21 Confers Neuroprotection Against Cerebral Ischemia-Reperfusion Injury and Alleviates Blood-Brain Barrier Disruption in Rats via the MAPK Signaling Pathway. J Mol Neurosci. 2018 May;65(1):43–53.
- Harraz MM, Eacker SM, Wang X, et al. MicroRNA-223 is neuroprotective by targeting glutamate receptors. Proc Natl Acad Sci USA. 2012 Nov;109(46):18962–18967.
- Xin H, Liu Z, Buller B, et al. MiR-17-92 enriched exosomes derived from multipotent mesenchymal stromal cells enhance axon-myelin remodeling and motor electrophysiological recovery after stroke. J Cereb Blood Flow Metab. 2020 Aug;271678X20950489.
- Han J, Yang S, Hao X, et al. Extracellular Vesicle-Derived microRNA-410 From Mesenchymal Stem Cells Protects Against Neonatal Hypoxia-Ischemia Brain Damage Through an HDAC1-Dependent EGR2/Bcl2 Axis. Front Cell Dev Biol. 2020;8:579236.
- Zhao H, Li Y, Chen L, et al. HucMSCs-Derived miR-206-Knockdown Exosomes Contribute to Neuroprotection in Subarachnoid Hemorrhage Induced Early Brain Injury by Targeting BDNF. Neuroscience. 2019 Oct;417:11–23.
- Zhang Y, Wang J, Zhang Y, et al. Overexpression of long noncoding RNA Malat1 ameliorates traumatic brain injury induced brain edema by inhibiting AQP4 and the NF-κB/IL-6 pathway. J Cell Biochem. 2019 Jun;120(10):17584–17592.
- Patel NA, Moss LD, Lee J-Y, et al. Long noncoding RNA MALAT1 in exosomes drives regenerative function and modulates inflammation-linked networks following traumatic brain injury. J Neuroinflammation. 2018 Jul;15(1):204.
- Lai N, Wu D, Liang T, et al. Systemic exosomal miR-193b-3p delivery attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage in mice. J Neuroinflammation. 2020 Feb;17(1):74.
- Zhang S, Hu Z-W, Luo H-Y, et al. AAV/BBB-Mediated Gene Transfer of CHIP Attenuates Brain Injury Following Experimental Intracerebral Hemorrhage. Transl Stroke Res. 2020;11(2):296–309.
- Ni H-Y, Song Y-X, Lin Y-H, et al. Dissociating nNOS (Neuronal NO Synthase)-CAPON (Carboxy-Terminal Postsynaptic Density-95/Discs Large/Zona Occludens-1 Ligand of nNOS) Interaction Promotes Functional Recovery After Stroke via Enhanced Structural Neuroplasticity. Stroke. 2019;50(3):728–737.
- Li F, Yang B, Li T, et al. HSPB8 over-expression prevents disruption of blood-brain barrier by promoting autophagic flux after cerebral ischemia/reperfusion injury. J Neurochem. 2019;148(1):97–113.
- Cabral-Miranda F, Nicoloso-Simões E, Adão-Novaes J, et al. rAAV8-733-Mediated Gene Transfer of CHIP/Stub-1 Prevents Hippocampal Neuronal Death in Experimental Brain Ischemia. Mol Ther. 2017 Feb;25(2):392–400.
- Qu J, Zhao H, Li Q, et al. MST1 Suppression Reduces Early Brain Injury by Inhibiting the NF-κB/MMP-9 Pathway after Subarachnoid Hemorrhage in Mice. Behav Neurol. 2018 Jun;2018:6470957.
- Guo H, Yin A, Ma Y, et al. Astroglial N-myc downstream-regulated gene 2 protects the brain from cerebral edema induced by stroke. Glia. 2021 Feb;69:281–295.
- Degeorge ML, Marlowe D, Werner E, et al. Combining glial cell line-derived neurotrophic factor gene delivery (AdGDNF) with L-arginine decreases contusion size but not behavioral deficits after traumatic brain injury. Brain Res. 2011 Jul;1403:45–56.
- Shirley JL, de Jong YP, Terhorst C, et al. Immune responses to viral gene therapy vectors. Mol Ther. 2020 Mar;28(3):709–722.
- Murlidharan G, Crowther A, Reardon RA, et al. Glymphatic fluid transport controls paravascular clearance of AAV vectors from the brain. JCI Insight. 2016 Sep;1(14):e88034.
- Bryant DH, Bashir A, Sinai S, et al. Deep diversification of an AAV capsid protein by machine learning. Nat Biotechnol. 2021 Feb;39(6):691–696.
- Fakhiri J, Grimm D. Best of most possible worlds: Hybrid gene therapy vectors based on parvoviruses and heterologous viruses. Mol Ther. 2021 Apr. DOI:https://doi.org/10.1016/j.ymthe.2021.04.005
- Chatterjee D, Marmion DJ, McBride JL, et al. Enhanced CNS transduction from AAV.PHP.eB infusion into the cisterna magna of older adult rats compared to AAV9. Gene Ther. 2021 Mar. DOI:https://doi.org/10.1038/s41434-021-00244-y
- Hudry E, Vandenberghe LH. Therapeutic AAV gene transfer to the nervous system: A clinical reality. Neuron. 2019 Mar;101(5):839–862.
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014 Nov;346(6213):1258096.
- Cohen J. CRISPR, the revolutionary genetic ‘scissors,’ honored by Chemistry Nobel. Science. 2020 Oct.
- Stadtmauer EA, Fraietta JA, Davis MM, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020 Feb;368(6489):367.
- Lu Y, Xue J, Deng T, et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med. 2020 Apr;26(5):732–740.
- Doench JG, Fusi N, Sullender M, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016 Feb;34(2):184–191.
- Ran FA, Hsu PD, Lin C-Y, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013 Sep;154(6):1380–1389.
- Kleinstiver BP, Pattanayak V, Prew MS, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016 Jan;529(7587):490–495.
- Hou Z, Zhang Y, Propson NE, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA. 2013 Sep;110(39):15644–15649.
- De Bellis M, Pisani F, Mola MG, et al. Translational readthrough generates new astrocyte AQP4 isoforms that modulate supramolecular clustering, glial endfeet localization, and water transport. Glia. 2017 Feb;65:790–803.
- Palazzo C, Buccoliero C, Mola MG, et al. AQP4ex is crucial for the anchoring of AQP4 at the astrocyte end-feet and for neuromyelitis optica antibody binding. Acta Neuropathol Commun. 2019 Apr;7(1):51.
- Guo D, Liu H, Gao G, et al. Generation of an Abcc8 heterozygous mutation human embryonic stem cell line using CRISPR/Cas9. Stem Cell Res. 2016 Nov;17(3):670–672.
- Guo D, Liu H, Gao G, et al. Generation of an Abcc8 homozygous mutation human embryonic stem cell line using CRISPR/Cas9. Stem Cell Res. 2016 Nov;17(3):640–642.