
ABSTRACT
Introduction
Cardiac hypertrophy is associated with adverse outcomes across cardiovascular disease states. Despite strides over the last three decades in identifying molecular and cellular mechanisms driving hypertrophy, the link between pathophysiological stress stimuli and specific myocyte/heart growth profiles remains unclear. Moreover, the optimal strategy for preventing pathology in the setting of hypertrophy remains controversial.
Areas Covered
This review discusses molecular mechanisms underlying cardiac hypertrophy with a focus on factors driving the orientation of myocyte growth and the impact on heart function. We highlight recent work showing a novel role for the spectrin-based cytoskeleton, emphasizing regulation of myocyte dimensions but not hypertrophy per se. Finally, we consider opportunities for directing the orientation of myocyte growth in response to hypertrophic stimuli as an alternative therapeutic approach. Relevant publications on the topic were identified through Pubmed with open-ended search dates.
Expert Opinion
To define new therapeutic avenues, more precision is required when describing changes in myocyte and heart structure/function in response to hypertrophic stimuli. Recent developments in computational modeling of hypertrophic networks, in concert with more refined experimental approaches will catalyze translational discovery to advance the field and further our understanding of cardiac hypertrophy and its relationship with heart disease.
1. Introduction
It has been over 3 decades since the Framingham Heart Study identified left ventricular hypertrophy as a major risk factor for adverse cardiovascular outcomes in the general population [Citation1]. In the wake of this landmark observation, the field has made tremendous strides in defining molecular pathways driving hypertrophy in response to pathophysiological stimuli [Citation2]. However, a consensus about whether or not suppression of hypertrophy per se is the optimal primary target to prevent disease remains elusive [Citation3–5]. Trials aiming to rescue the organ from hypertrophic response to sustained pressure overload have shown mixed results. Conflicting data have been reported on how targeting the same signaling intermediates impact hypertrophy, cardiac function, and survival [Citation6–9]. A major challenge when attempting to parse the pathophysiological roles for hypertrophy stems in part from a general lack of precision in how, as a field, we talk about hypertrophy [Citation10]. At the most fundamental level, cardiac hypertrophy refers to an increase in cardiac myocyte size with concomitant increase in heart mass and altered structure [Citation11]. Beyond this general definition, however, the precise cell and organ-level changes involved in the hypertrophic response vary dramatically depending on context, including the nature of the stress stimulus, genetic factors, co-morbidities or environmental factors [Citation10]. While there is a general appreciation that hypertrophy can take different forms with implications for function, there is a gap in our understanding of how specific stimuli alter myocyte structure/function (especially in adult myocytes) and how these changes manifest at the organ level. Many studies are conducted in neonatal myocytes or model systems and studies in adult myocytes/tissue tend to report crude measures like myocyte cross-sectional area as a surrogate for hypertrophy. Few studies provide detailed information on how exactly the myocyte changes in size with pathophysiological stress in terms of geometry (e.g. length vs. width or aspect ratio) and how these changes relate to myocyte contractility, metabolism, or other functional measures. In preparing for this review, our literature search encompassing hundreds of publications in the field uncovered only a select handful that provide the level of data that we believe is necessary for resolving the functional consequences of hypertrophic growth. A central premise of this opinion piece is that these granular data are needed to clarify the pathophysiological role for hypertrophy and ultimately design improved strategies that tune the hypertrophic response for therapeutic benefit.
2. Cardiac hypertrophy as a physiological response to changes in metabolic demand
The heart has evolved a robust system for responding to acute and chronic changes in demand. Assuming that efficiency of blood as an O2 carrier is a constant (not true in all cases, elite athletes, for example), the heart can meet increased demand by either increasing heart rate or stroke volume [cardiac output is the product of heart rate and stroke volume]. The prevailing paradigm is that the heart is sensitive to changes in both preload (an external mechanical stress applied in the axial direction of the muscle fibers), and afterload (the mechanical impedance of the vasculature). Acute, physiological changes in load are negotiated in part through passive reflexes that increase stroke volume without requiring changes in cardiac structure – the Frank-Starling mechanism describes a length–tension relationship observed in striated muscle that impacts intrastroke contractility. Application of a preload to a muscle fiber can vary the tension generated by that fiber. For each fiber there exists an optimal length that maximizes the inotropic state [Citation12], the result of this being an organ that can produce a response compensatory to its volumetric state. The underlying mechanisms are not yet fully understood but are believed to be related to stretch-activated regulation of calcium [Citation13]. The Anrep effect is a similar compensatory mechanism that is observed following an increase in afterload. When mechanical impedance of the vasculature is increased, on the timescale of 10–15 minutes an increase in calcium transient amplitude is observed, also described as the slow force response [Citation14]. A third intrinsic inotropy-regulating mechanism exists, known as the Bowditch effect. Where the Frank-Starling and Anrep effects are regulated through mechanical strain, the Bowditch effect is instead dependent on heart rate, and describes an increase in force generation as a function of increased rate.
As with biomechanical stimuli, the heart has a myriad of signaling pathways for increasing heart rate and/or contractility in response to acute, physiological neurohumoral stimuli without changing structure. Neurohumoral stimulation includes sympathetic innervation, and many hormonal factors, such as those in the renin-angiotensin-aldosterone system (RAAS), vasopressin, and atrial natriuretic peptide (ANP). Sympathetic innervation is able to regulate heart rate and contractility [Citation15] and can activate pathways such as those initiated by β-adrenergic receptors to further regulate inotropy [Citation16]. RAAS is a homeostatic regulator that impacts many vital systems, but is especially important in the heart where angiotensin II regulates contractility, as well as arterial tension (a major contributor to afterload) [Citation17]. In the event that a stimulus is not resolved (chronic time course) and/or is of a specific pathological nature, the heart must engage additional systems to increase cardiac output. Increasing heart rate is an important component of the response to acute stress but is not sustainable or sufficient in the case of chronic/pathological stress. Without further recourse, hypertrophy may be viewed as the heart’s attempt to increase stroke volume (and thereby output), by either increasing ventricular chamber volume and/or the fraction of blood volume ejected with each contraction.
Cardiac myocytes terminally differentiate soon after birth [Citation18], resulting in limited ability for heart mass to increase through myocyte proliferation. Instead, the current understanding is that any appreciable increase in cardiac mass occurs through myocyte growth [Citation19]. At the myocyte level, hypertrophy occurs through the addition of contractile units (sarcomeres) to existing myofibrils responsible for generating contractile force. Depending on the precise nature of the hypertrophic stimulus, sarcomeres may be added in series with, or parallel to the existing myofibrils, with implications for the resulting changes in organ structure and function.
3. Classifying hypertrophy based on structure: concentric vs. eccentric remodeling
Hypertrophy is commonly classified to be either concentric or eccentric according to the structural changes at the cell and organ level (). As mentioned in the previous section, hypertrophic growth involves the addition of sarcomeres within the cardiomyocytes, which induces a morphological change at the level of the cardiac muscle depending on how exactly sarcomeres are added (i.e. parallel to or in series with the existing fibers). In concentric hypertrophy, the myocardial tissue grows in a manner preferential to increased output. While some dilation may occur, generally, the increase in ventricular wall thickness constitutes the major structural change (). At the cell level, concentric hypertrophy is thought to occur from preferential addition of sarcomeres in parallel leading to disproportionate increases in cardiomyocyte width and left ventricular (LV) wall thickness. Eccentric hypertrophy, in contrast, results in an expanded ventricular cavity having an elliptical geometry, but without the significant thickening of the ventricular wall seen in concentric hypertrophy (). This is because the sarcomeres are added along the length of existing fibers, lengthening the myocytes and extending the chamber. Given that these forms of hypertrophy differ in structural arrangement, they necessarily alter the mechanics of cardiac contractility and can result in differing consequences on organ performance, further differentiated by adaptive and maladaptive states of MH. In fact, it is suggested that changes in myocyte shape represents an additional mechanism of adaptability to optimize force generation, with improvements observed in concentric-orientated growth, further reinforcing the significance of concentric versus eccentric states [Citation20].
Figure 1. Overview of the hypertrophic response to pathophysiological stress. Although hypertrophy, in general, results in increased cardiomyocyte size and heart mass, the details depend on the nature of the stimulus (e.g. physiological vs. pathological, pressure overload vs. volume overload). Changes associated with physiological remodeling are highlighted in blue. Pathological hypertrophy changes (red) are further defined according to concentric (Orange) and eccentric (purple) types. Modified from ref [Citation21] with permission. Nakamura M and Sadoshima J, Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 2018;15:387–407.
![Figure 1. Overview of the hypertrophic response to pathophysiological stress. Although hypertrophy, in general, results in increased cardiomyocyte size and heart mass, the details depend on the nature of the stimulus (e.g. physiological vs. pathological, pressure overload vs. volume overload). Changes associated with physiological remodeling are highlighted in blue. Pathological hypertrophy changes (red) are further defined according to concentric (Orange) and eccentric (purple) types. Modified from ref [Citation21] with permission. Nakamura M and Sadoshima J, Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 2018;15:387–407.](/cms/asset/545c900f-5e2d-43c1-83b2-03d2c9b85330/iett_a_2031974_f0001_oc.jpg)
4. Functional designations for hypertrophy: adaptive vs. maladaptive responses
Hypertrophy is observed in response to a host of pathophysiological stimuli and is generally viewed to play an adaptive or maladaptive role depending on many factors, including type/duration of stress, existing pathology, and individual predisposition. Physiological hypertrophy includes postnatal growth as well as growth induced by pregnancy and exercise and is generally viewed to be supportive of cardiac health [Citation19]. Physiological hypertrophy is characterized by little to no increase in interstitial fibrosis as well as a slight (10–20%) increase in heart mass [Citation21]. As hypertrophy develops, sarcomeres are added to the existing muscle, and constitutes the increase in mass described by hypertrophy’s classical definition. The orientation of this addition plays a key role in the ability of the heart to maintain its function. In physiological hypertrophy, also referred to as adaptive hypertrophy, these sarcomeres are added in series and in parallel to the existing structure in the cardiomyocyte, resulting in a proportional enlargement of the cell [Citation21]. This results in a maintained, or increased cardiac output, which is reversible when cardiac demand is normalized () [Citation22].
Pathological hypertrophy is instead an increase in heart mass typically observed in the setting of disease [Citation19] and can also be referred to as maladaptive hypertrophy. The presentation of pathological hypertrophy can be diverse and depends upon the disease context and physiological changes that lead into its development. For example, the hyperglycemic conditions that promote remodeling in the setting of diabetic cardiomyopathy will differ from those in hypertensive cardiomyopathy [Citation23,Citation24]. In the setting of pressure overload, the initial phase of this hypertrophy is thought to be compensatory or adaptive because for a period, the result is a preservation or increase in cardiac function. During the compensatory phase, sarcomeres are added in a parallel orientation, leading to a concentric form of hypertrophy. As previously discussed, the resulting geometry is an increase in wall thickness without significant chamber enlargement, increasing contractility with preserved systolic function. The progression of pathological hypertrophy in this disease model eventually results in a dilated chamber with thinned walls, and a decreased output [Citation25]. Volume overload is another commonly studied disease setting, which instead generally results in an eccentric hypertrophic response at the outset [Citation26]. Differentiating physiological from pathological hypertrophy is primarily achieved by metrics of cardiac output, reflecting the disease state of the heart. While the differences are certainly reflected in the changing structure of the heart and thus linked to the differentiation between concentric and eccentric hypertrophy, not enough is currently known about the relationships between cardiac structure and function to clearly define how one impacts the other.
5. Describing myocyte hypertrophy at a more precise level: orientation of myocyte growth
Decades of research have uncovered a vast network of receptors, signaling molecules and effector proteins involved in the hypertrophic response to stress. Excellent reviews have been written on hypertrophic signaling to which the reader is directed for a comprehensive overview on the general topic [Citation27,Citation28]. Here, we instead focus on those signaling pathways where there is evidence of a preservation of the concentric hypertrophic state in the absence of heart failure (). While some studies publish data on how hypertrophy is regulated across scales from the myocyte (specifically, orientation of growth and not just surface area) to the structural and functional consequences at the organ level, additional data are needed to create a broader theoretical framework for understanding the link between stress, specific forms of hypertrophy and cardiac function. Our discussion is placed in the context of whether or not hypertrophy per se is a viable therapeutic target and whether we can develop strategies to tune rather than completely block hypertrophy [Citation3,Citation4,Citation29].
Figure 2. Simplified cardiomyocyte signaling network linking biomechanical/neurohumoral stress to gene expression changes and hypertrophic remodeling. Although the complete signaling web of known modulators of hypertrophy is much more expansive, here we focus on a subset of molecules with data supporting a preserved state of concentric hypertrophy without cardiac dysfunction in response to chronic stress. Abbreviations defined in ()
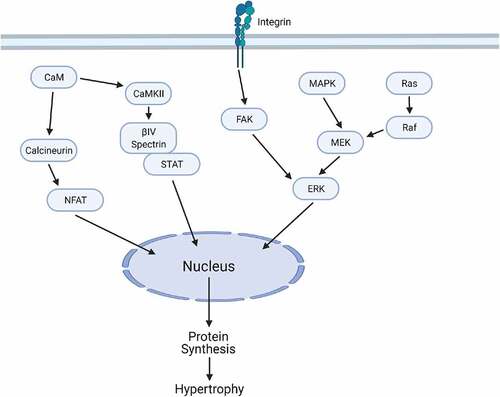
Table 1. Table of abbreviations
5.1. Calcineurin/NFAT signaling as a master regulator of cardiac hypertrophy
Heart failure is characterized by pronounced defects in intracellular Ca2+ cycling, including alterations in diastolic and systolic Ca2+ concentrations [Citation30]. Calcineurin is a Ca2+ and calmodulin-sensitive phosphatase and critical player in hypertrophic signaling pathways in cardiac myocytes (). The prevailing paradigm is that calcineurin is a master regulator of the hypertrophic response but not necessarily a regulator of the orientation of myocyte growth, although the data are incomplete in this regard. In support of this view, attenuation of calcineurin signaling (and downstream effectors) has been shown to halt hypertrophic remodeling in the heart in response to chronic stress, although the ultimate effect on outcomes like survival are less clear.
Calcineurin is known to become activated early in response to pressure overload with sustained response [Citation31]. Although the calcineurin inhibitor cyclosporin has been shown to inhibit the development of hypertrophy, it is not known to completely suppress pressure-overload induced gene expression [Citation32]. Calcineurin’s integral roles prevent complete knockout animal models from being viable experimental strategies. Martinez-Martinez et al. discuss a model of conditional Cnb1 deletion that prevents angiotensin II–induced hypertrophy in the knockout case, demonstrating a requirement for calcineurin in hypertension-driven hypertrophy [Citation33]. Later, discussion shows a protective effect in this inhibition case, potentially indicating that some gene expression may be necessary, but the upregulation from calcineurin could be deleterious.
NFAT activity describes a number of transcription factors implicated in calcineurin signaling that have been shown to participate differentially in the initiation of hypertrophy, respectively, to physiological and pathological phenotype. Wilkins et al. have shown that NFAT/calcineurin signaling is involved in pathological but not physiological forms of hypertrophy [Citation34]. Further, members of the NFAT family may upregulate IL-6 cytokines that are well known to induce hypertrophy [Citation35]. NFAT has been shown to be phosphorylated by MAPK leading to deactivation of calcineurin-NFAT signaling, and alternatively has been shown to be negatively regulated by activated cytoplasmic CaMKII [Citation36,Citation37]. The potential for cross-talk would need to be investigated in pursuing therapeutic targets, but may pose a viable target for therapy, particularly when framed around the significance of MAPK and CaMKII signaling discussed later on.
Calcineurin-NFAT signaling has received much attention due to its ability to abrogate overall hypertrophic drive in the heart (). However, as these studies are unable to consistently demonstrate pro-survival outcomes, the efficacy of this strategy remains unclear and it is necessary that further work be done to better characterize the functional implications of the remodeling that occurs under control of these signaling modalities [Citation3,Citation4,Citation23]. The appropriate selection of a therapeutic target for the treatment of hypertrophy must negotiate a balance between a preservation of the protective response and an amelioration of the detrimental impacts of a phenotype that moves toward an eccentric state. In the rest of this section, we consider pathways where there is evidence for preserved concentric hypertrophy (as opposed to complete inhibition of hypertrophy) as an alternative strategy for modulating cardiac function in response to chronic stress.
5.2. Mitogen-activated protein kinase cascades cast a wide net of yet-unresolved, but generally concentric-hypertrophic signals
Mitogen-activated protein kinase kinase (MEK), and extracellular signal-related kinase (ERK) proteins are critical nodes in the mitogen-activated protein kinase (MAPK) cascades, which have been tightly linked to concentric forms of hypertrophic remodeling. Canonical activation of this pathway involves the phosphorylation of rat sarcoma GTPase (Ras) and subsequent phosphorylation of rapidly acting fibrosarcoma kinase (Raf), MEK, and ERK1/2 (). Activated ERK1/2 is then able to phosphorylate target proteins and transcription factors. These transcription factors can alter cell growth, proliferation, differentiation, and apoptosis [Citation38].
Mounting evidence supports the MEK-ERK1/2 pathway as a critical regulator of a specific concentric orientation of the hypertrophic response. For example, transgenic mice expressing an activated form of MEK1 in the heart were characterized by concentric hypertrophy, hyperdynamic function, and anti-apoptotic effects. Furthermore, ERK1/2 activation in vivo promoted stable concentric hypertrophy and enhanced cardiac function in mice up to 12 months of age where it is hypothesized ERK1/2 may directly protect cardiac myocytes from apoptosis to induce stable concentric hypertrophy [Citation39]. Consistent with this proposed mode of action, decreased phosphorylation of ERK1/2 during the late phase of chronic pressure overload is associated with increased myocyte apoptosis [Citation40], in addition to the associated structural changes in transitioning to an eccentric growth profile. Most interesting, however, was that inhibition of ERK1/2 activity by overexpression of the targeting phosphatase DUSP6 led to cardiac dilation, decompensation, reduced fractional shortening, and actually a heightened hypertrophic response, illustrating the profound importance of ERK1/2 signaling not necessarily in driving hypertrophy, but more the orientation of hypertrophy. At the same time, however, CaMKII, an additional ‘key player’ discussed later, has been shown to play a regulatory role in the ERK cascade, and a blockade of this interaction by CaMKII inhibition successfully limit the extent of cardiac hypertrophy [Citation41,Citation42].
Whereas MEK-ERK activation is seen to induce concentric hypertrophy, overexpression of constitutively active Ras has shown to exhibit a cardiomyopathic phenotype. While Ras is an upstream activator of Raf and hence MEK-ERK, it is also involved in other signaling pathways. These observations support that while MEK-ERK activation appears to be involved in signaling a beneficial form of cardiac hypertrophy, Ras seems to signal for maladaptive hypertrophy through a MEK/ERK-independent mechanism [Citation43].
While these findings generally indicate a cardioprotective effect of the MAPK cascades by differentiating concentric from eccentric forms of hypertrophic outcomes, there is a general lack of information on the corresponding detailed structural changes at the cellular level to evaluate how sarcomeric reorganization occurs with respect to these signals. We discuss in section 5.4 a specific example of a directional impact seen with ERK1/2 targeting. Gathering these data will allow us to better understand the specific changes in remodeling that these signaling molecules promote, and more effectively target appropriate therapeutic solutions.
5.3. FAK/integrins serve a critical role producing a cardioprotective response to increased mechanotransduction
Upstream of the MAPK cascades, focal adhesion kinase (FAK) is a non-receptor tyrosine kinase responsible for regulating cell adhesion and migration with demonstrated ability to modulate hypertrophic signaling with preservation of cardiac function [Citation44]. FAK plays an important role in the accumulation of ECM and in integrin signaling, closely linking it to the mechanical response of cardiac tissue [Citation45]. FAK/integrin complexes serve as mechanotranducers in normal heart development and in pathophysiology, and emerging evidence implicates that the FAK mediates other downstream targets in hypertrophy (). One major pathway for inducing hypertrophy involves FAK and ERK1/2. ET-1 mediated ECM remodeling and mechanical stress lead to activation of FAK (S910 and Y397 residues)-ERK1/2 pathways [Citation46]. Osteoprotegerin (OPG) has also been shown to play a role in activation of FAK and ERK in inducing autophagy and hypertrophy [Citation47,Citation48]. Inhibition of autophagy reduced hypertrophy in this model. Conversely, induction of autophagy increased hypertrophy. When FAK was silenced using siRNA, OPG-induced changes in autophagy status and cellular hypertrophy were diminished [Citation48]. This correlates with other studies that have investigated FAK phosphorylation of Beclin 1, an autophagy protein. Beclin1 is required for FAK-mediated autophagy suppression, and this mechanism may be responsible for the cardioprotection in response to transaortic constriction (TAC) [Citation47]. Interestingly, there is evidence that FAK regulation can promote both eccentric and concentric hypertrophy. One study used a cardiac specific FAK transgenic mouse model and found that overexpression of FAK induces concentric hypertrophy with preserved cardiac function. Myocytes isolated from baseline FAK overexpression models showed an increased cross-sectional area but similar lengths to WT mice. When subjected to chronic pressure overload, the FAK overexpression model showed similar fractional shortening, left ventricular end diastolic diameters, and posterior wall thickness to the WT TAC model [Citation49]. However, when studies have used cardiac specific FAK KO mouse models, they found that they developed eccentric cardiac hypertrophy and decreased cardiac function following TAC or angiotensin II stimulation [Citation50]. Overexpression of FAK yields moderate concentric hypertrophy, while full KO increases the propensity for eccentric hypertrophy and loss of function, indicating that a detailed balance of FAK activity is necessary for normal cardiac function and the FAK may have some role in regulating the direction of hypertrophy. Furthermore, these data illustrate the importance of distinguishing hypertrophic profiles, and not just hypertrophy in general, in parsing the beneficial from detrimental characteristics in the hypertrophic response.
5.4. Serum response factor as a downstream effector of ERK in cardiac hypertrophy
Recent studies have identified the transcription factor serum response factor (SRF) as a potential downstream target of ERK1/2 activity and a necessary component for enacting concentric hypertrophic growth [Citation51]. As described above, the MAP-kinase dependent pathway is a significant contributor to hypertrophic remodeling in the heart. However, despite this profound phenotype, there is little understanding of the downstream ERK1/2 effectors that enable this phenotypic change. SRF is one such example of a downstream effector which may be targeted to drive the hypertrophic response in a beneficial manner. Specifically, ERK1/2 is proposed to target the kinase RSK3 (p90 ribosomal S6 kinase type 3), which is then directly responsible for SRF phosphorylation, activation, and recruitment of distinct gene expression profiles. As such, both RSK3 and SRF phosphorylation are consistently observed in various models of hypertrophic activation associated with increased myocyte width. Importantly, RSK3 knock-out or blockade of SRF phosphorylation was able to impair the hypertrophic growth of cardiac width associated with early TAC remodeling. Conversely, targeted dephosphorylation of SRF was instead found to associate with conditions of eccentric myocyte growth, including human samples of dilated cardiomyopathy. The significance of this relationship was investigated by therapeutically preserving SRF phosphorylation in an eccentrically remodeling mouse MI model, resulting in restoration of heart wall thickness and preservation of cardiac function. Excitingly, as we begin to understand with greater clarity the nuances of cardiac hypertrophy, these observations illustrate the significance and therapeutic opportunities dependent on hypertrophic orientation, as well as a greater understanding of the unique pathways myocyte utilized to direct the hypertrophic response.
5.5. G-Protein receptor kinases are powerful regulators of signal transduction
G-protein receptor kinases (GRKs) are classically understood to serve as signal attenuators for G-protein coupled receptors (GPCRs), the largest family of human membrane proteins with over 800 members [Citation52,Citation53]. Recent data support GRKs as another way to modulate the orientation of hypertrophy and preserve cardiac function without completely shutting down the hypertrophic response.
GPCRs serve as the point of cellular entry for many hormonal, neurotransmitter, and ionic signaling pathways and as such, their function is critical in many processes. Because of this, GPCRs and their modulators have been studied extensively as therapeutic targets for a range of disease states, many beyond the cardiac context [Citation54]. In their role as attentuators, GRKs phosphorylate occupied receptors, an event followed by recruitment of β-arrestins, which separate the G-protein, uncoupling the receptor-binding events from downstream activity [Citation55]. β-arrestin binding additionally targets the receptors for internalization [Citation56]. At scale, these processes result in desensitization and dowregulation respectfully, attenuating the signal transduction through the membrane with high specificity.
Because of the powerful ability GRKs have to modulate such a versatile signaling apparatus, this class of proteins has become a popular area of study for therapeutic targets with a demonstrated ability to halt or direct hypertrophic signaling [Citation57]. One of these recently published studies has shown that mice expressing a truncated GRK2 variant did not exhibit the expected transition to heart failure after chronic pressure overload [Citation58] and instead, produced a case of preserved concentric hypertrophy. Similarly, targeting of GRK5 has shown promise. While a global GRK5 KO model demonstrated complete inhibition of hypertrophy under induction by phenylephrine, a conditional, cardiac-specific knockout KO demonstrated cardiac growth, but significantly attenuated hypertrophy relative to control at 12 weeks post-TAC with no drop in function [Citation59]. Following up on these findings, several studies have shown that the amino-terminal domain of GRK5 (GRK5-NT) binds calmodulin and may account for the cardioprotective role of GRK5 activity [Citation60,Citation61] and another recent publication has shown promise in a small molecule inhibitor of GRK5, KR39038, demonstrating attenuation for the duration of the 2 week endpoint [Citation62]. Activity of NF-κB, a transcription factor associated with regulation of hypertrophic growth [Citation63] has additionally been linked to GRKs with experimental models showing decreased NF-κB activity following selective inhibition of GRK2 [Citation64]. Given these experimental successes, further study of the GRK family and interactions is warranted and may prove crucial for the development of a therapeutic attenuating the detrimental progression of dilatative hypertrophy.
5.6. CaMKII/βIV spectrin/STAT3 as a novel axis for regulation of hypertrophy
The notion of hypertrophic orientation as a critical determinant of cardiac function is illustrated further by recent work investigating a macromolecular complex consisting of the cytoskeletal protein βIV-spectrin, the Ca2+/calmodulin-dependent protein kinase II (CaMKII), and the transcription factor signal transducer and activator of transcription 3 (STAT3) [Citation65]. Independently, both CaMKII and STAT3 have an abundant history in hypertrophic remodeling. CaMKII is a serine/threonine kinase activated by heightened levels of Ca2+/calmodulin, oxidative stress, and glycosylation induced by elevated levels of glucose, leading to the phosphorylation of a significant number of target proteins [Citation66–69]. These include a number of ion handling proteins contributing to heightened inotropic drive but also increased risk for electrical dysfunction and arrhythmias. Other target proteins like the class 2 histone deacetylases 4 and 5 (HDAC4/5) are direct contributors of hypertrophic drive by relieving inhibition of the transcription factor MEF2 [Citation67]. Therefore, it is unsurprising CaMKII activation has been observed to be sufficient for establishing hypertrophic remodeling. Mouse models of transgenic overexpression drive hypertrophy and the development of dilated cardiomyopathy [Citation70–72], while human patients with cardiac hypertrophy have increased CaMKII expression and activity [Citation68,Citation72]. Alternatively, mouse models which utilize both pharmacologic or peptide inhibitors or direct CaMKII deletion show success in attenuating heart failure development following TAC [Citation73–75], neurohumoral activation [Citation76–78], or myocardial infarction [Citation79].
The role of STAT3 in the development of hypertrophic remodeling and heart failure is less straight forward. STAT3 is a transcriptional activator instrumental in driving an array of gene programs important for inflammation, hypertrophy, proliferation, differentiation, migration, extracellular matrix synthesis, fibrosis, and cell survival [Citation80–82]. Its canonical activation is largely recruited through receptor tyrosine kinases responding to cytokine and/or neurohumoral ligands, leading to STAT3 phosphorylation, dimerization, and nuclear translocation [Citation80]. Owing to these observations, investigations tested the impact cardiac specific STAT3 deletion, which unexpectedly found significant cardiac fibrosis, impaired cardiac function, ventricular remodeling, developing heart failure, and increased mortality [Citation83,Citation84]. Alternatively, transgenic expression of constitutively active STAT3 led to increased mortality in mice following MI through enhanced inflammation, increased wall rupture, scar dilation, and heart failure [Citation85], indicating that unregulated activity of this protein can also be pathologic. Studies which achieve a more balanced state of STAT3 activity through ligand activation following acute ischemic injury instead show cardioprotective effects through reduced fibrosis and increased myocyte survival [Citation86–89]. However, under more chronic conditions, such as TAC, the inhibition of STAT3 through pharmacologic inhibition or genetic deletion of the upstream cytokine activator, IL-6, also contributes to reduced fibrosis and hypertrophy [Citation65,Citation90,Citation91].
The significance of achieving balanced STAT3 activity has also emerged in investigations for the cytoskeletal protein βIV-spectrin. βIV-spectrin is an adapter protein, linking membrane associated proteins with the cytoskeleton [Citation92]. Initially characterized as a passive element for maintaining membrane shape and elasticity, our understanding of βIV-spectrin has expanded to include dynamic roles in protein localization and signal transduction, with particular focus on cardiac fibrosis and hypertrophy [Citation65,Citation93,Citation94]. In myocytes, βIV-spectrin localizes to the intercalated disc, where it contributes to localization of CaMKII and STAT3 within the same domain () [Citation65]. It was identified that cardiac stress induces the degradation of βIV-spectrin in response to activated CaMKII activity, contributing to more diffuse cytosolic and nuclear localization and heightened STAT3 transcriptional activity. Genetic ablation of the βIV-spectrin/CaMKII interaction, however, protected βIV-spectrin from TAC-induced degradation, preserving its, and consequently STAT3, signal at the intercalated disc, effectively limiting STAT3 activity and related gene expression. This contributed to significantly reduced fibrosis in response to TAC, but most interestingly led to sustained concentric hypertrophy and cardiac function, whereas WT mice developed decompensated heart failure (eccentric hypertrophy) characterized by ventricular dilation and impaired contractility.
Whereas most instances of cardioprotection in TAC models often attenuate hypertrophy, this instance instead preserved the hypertrophic response but maintained myocardial wall thickness, suggesting the role for concentric hypertrophy more as an adaptive rather than pathologic state, again illustrating that orientation of hypertrophy is an influential determinant of cardiac function.
6. Existing therapies that modulate hypertrophy
Currently, there are several strategies that aim to reduce the extent of hypertrophy by targeting cell-surface receptors with the goal of minimizing activation of the neurohumoral stress response. The renin-angiotensin-aldosterone system (RAAS) is a major regulator of this response and target of these therapies. Angiotensin converting enzyme (ACE) inhibitors, β-adrenergic receptor blockers, and angiotensin receptor blockers fall under this umbrella and have been clinically administered demonstrating favorable outcomes [Citation95,Citation96]. An additional route often explored is reduction of the pressure overload that stimulates this stress response. Often termed antihypertensives, these treatments can include the RAAS treatments above, and similarly aim to reduce activation of stress response. More recently, therapeutic strategies have focused on action within the cell. A promising class of these therapies are histone de-acetylase (HDAC) inhibitors. These compounds have shown promise in reducing the growth of cardiomyocytes and reducing the effects of hypertrophy within the context of increased load [Citation8,Citation97,Citation98]. Schiattarella et al. even posit using these agents in a targeted manner to ‘sculpt’ ventricular mass through the progression of disease [Citation4].
Some promising preliminary studies have explored the blockade of cell-surface receptors and signaling pathways as a method of inhibiting hypertrophy [Citation99–101]. Among these, calcineurin has shown to be a promising target. Its involvement in the induction of hypertrophy was shown to be critical by Sussman et al. in their work using the calcineurin inhibitors cyclosporin and FK506 [Citation99]. This study showed an ability to block development of hypertrophy in a genetically predisposed murine model. Work by Akhter et al. has shown antagonists of the Gaq receptor subunit can similarly inhibit hypertrophy in the context of pressure overload.
Additional studies, however, have shown similar success blocking hypertrophy, but not without detrimental effect. An overexpression model of the GTPase-activating protein RGS4 resulted in a reduction of hypertrophy with a corresponding increase in mortality [Citation102]. Brancaccio et al. have demonstrated the crucial role melusin – a protein involved in load sensing – plays in maintaining cardiac function during hypertrophic conditions [Citation103]. Interestingly, given the previously discussed use of calcineurin inhibitors to safely prevent hypertrophy, a separate study focusing on a separate, endogenous inhibitor Zaki-4β found a similar inhibition of hypertrophy, but significantly reduced function as evaluated by two parameters of LV diastolic function [Citation104].
These studies are far from conclusive and have not consistently evaluated long-term survival. Identifying a viable therapeutic strategy will require a detailed understanding of how these approaches to blocking hypertrophy exert their primary effect, and their impacts on linked processes.
7. Conclusion
Cardiac hypertrophy is an important pathophysiological response to chronic changes in metabolic demand. Hypertrophy assumes different structural and functional manifestations depending on a variety of environmental, genetic factors providing the backdrop for an ongoing debate about the viability of hypertrophy as a therapeutic target to prevent heart failure. Mounting studies provide examples of molecular perturbations that yield preserved hypertrophy without cardiac dysfunction raising the question of whether hypertrophy may be ‘redirected’ rather than inhibited for therapeutic benefit in some cases. Critical gaps remain in our ability to link-specific hypertrophic stress stimuli to cell and organ-level changes. Such knowledge may clarify the role of hypertrophy in pathophysiology and point the way to improved therapies.
8. Expert opinion – novel strategies for modulating hypertrophy for therapeutic benefit
Hypertrophy is a major independent risk factor for heart failure [Citation1], and has received much attention as a target for pharmacological intervention. Preclinical studies, including those discussed in previous sections, have provided important information on the complex web of signaling nodes and intermediates involved in the hypertrophic response to pathophysiological stress while providing proof of principle for the ability to modulate hypertrophy and cardiac function in disease. However, uncertainty remains regarding the best path forward in the search for new therapies [Citation3,Citation4,Citation6]. Therefore, a greater understanding is needed of the mechanistic determinants of the direction of cardiomyocyte growth and functional implications at the cell, organ, and organism level. Here, we provide an opinion on the major barriers and potential opportunities for the field moving forward.
Arguably, the greatest opportunities for progress in our understanding of hypertrophy and in the search for new therapeutic targets reside in the area of computational modeling. Cardiac hypertrophy is remarkably complex, with a myriad of interconnected signaling pathways operating across wide temporal and spatial scales. Thus, the challenge of defining a coherent paradigm that integrates existing knowledge and allows for testable predictions is tremendous. As the field attempts to reconcile conflicting data and identify more effective strategies for improving function in the setting of cardiac disease, computational modeling provides tremendous opportunity to understand complex interactions and emergent responses in complicated signaling processes. Many prior computational studies have focused on the biomechanics of the heart during hypertrophy, integrating changes in mechanical forces, hemodynamics, and myocardial growth [Citation105]. However, fewer studies have considered the multitude of different intracellular signaling pathways that are activated during hypertrophy. Recent work from the Saucerman lab has developed a large network computational model, integrating the interactions between over 100 molecules and pathways. Such an approach illustrated that Ras – as a nexus of multiple signaling pathways – was a critical modulator of hypertrophy [Citation106]. Additionally, this network model also was able to predict interactions between multiple perturbations [Citation107]. This approach has been expanded to investigate adrenergic stimulation during hypertrophy [Citation108], crosstalk between hypertrophy and apoptosis [Citation109], and a multiscale model of hypertrophy signaling and mechanics during TAC [Citation110]. While further studies of individual pathways are critical for our understanding of hypertrophy, it is also important to identify crosstalk between these pathways, to facilitate integrated approaches that can predict the effects of therapies aimed to control hypertrophy.
Computational models, of course, require experimental data for constraining model parameters and validation. Thus, it is fair to ask whether available data are sufficient to yield useful models or whether specific types of data/experiments are needed to improve models. As discussed throughout this article, one important gap for experimental data is related to details of how cardiomyocyte dimensions change in response to specific stimuli or molecular perturbation. These data become particularly important as we try to dissect different forms of hypertrophy and their functional consequences in the setting of disease. As the sarcomeric changes within these cells are the primary structural consequence of hypertrophy – and ultimately what we observe at the organ level – it is essential that we understand how these signaling modalities impact subcellular structural organization. Beyond cardiomyocyte structure, additional data on changes in energy metabolism and utilization with specific perturbations will be invaluable for developing a more solid theoretical framework. As the cell type with the highest mitochondrial content [Citation111], cardiomyocytes are uniquely sensitive to changes in mitochondrial function. Research has shown that the cardiac metabolism is altered in conditions of both physiological and pathological hypertrophy [Citation111–114], likely necessitating changes in force generation and contractile mechanics. As the cardiac myocyte continues to develop under these conditions of altered energy availability, the structural orientation optimal for maximal energetic efficiency will undoubtedly become altered as well. Further study will ultimately be necessary to determine if therapeutic alteration of energy availability may be a viable strategy for addressing these structural changes. When we consider upstream signals inducing molecular and cellular changes, additional information is needed on how exactly pathophysiological neurohumoral/biomechanical stimuli are integrated by the cardiomyocyte at the molecular level. For example, it could be valuable to better understand how the myocyte differentiates specific loading profiles and the downstream functional consequences when combined with different neurohumoral inputs. Finally, signaling within the cardiomyocyte is obviously important for the hypertrophic response but so is intercellular communication involving cardiomyocytes, fibroblasts, immune cells, and endothelial cells. Answers to these questions and more will help guide the field to a deeper theoretical understanding of hypertrophy that will facilitate development of new and improved therapies for cardiovascular disease patients.
Article highlights
Cardiac hypertrophy is an essential physiologic response to chronic changes in cardiac demand, and can be differentiated both by the conditions that lead to its development and the structural changes that result.
While the direct signal is unclear, we highlight several pathways that have high degrees of involvement in directing hypertrophic orientation and development.
Furthermore, key studies suggest the orientation of hypertrophic growth, and not just hypertrophy in general, can be a significant regulator of cardiac function.
The CaMKII/βIV spectrin/STAT3 axis and other signaling pathways are discussed for their role in developing hypertrophic states with preserved cardiac function, bringing attention to a gap in knowledge concerning the nature of adaptive versus maladaptive hypertrophy.
Computational approaches combined with experimental studies to more fully characterize hypertrophy at the cell and organ level will be necessary to identify effective therapeutic strategies.
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosure
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Additional information
Funding
References
- Levy D, Garrison RJ, Savage DD, et al. Prognostic implications of echocardiographically determined left ventricular mass in the framingham heart study. N Engl J Med. 1990; 322: 1561–1566.
- Frey N, Katus HA, Olson EN, et al. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589.
- Crozatier B, Ventura-Clapier R. Inhibition of hypertrophy, per se, may not be a good therapeutic strategy in ventricular pressure overload: other approaches could be more beneficial. Circulation. 2015; 131:1448–1457.
- Schiattarella GG, Hill JA. Inhibition of hypertrophy is a good therapeutic strategy in ventricular pressure overload. Circulation. 2015; 131:1435–1447.
- Pitoulis FG, Terracciano CM. Heart plasticity in response to pressure- and volume-overload: a review of findings in compensated and decompensated phenotypes. Front Physiol. 2020;11:92.
- Hill JA, Karimi M, Kutschke W, et al. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–2869.
- Meguro T, Hong C, Asai K, et al. Cyclosporine attenuates pressure-overload hypertrophy in mice while enhancing susceptibility to decompensation and heart failure. Circ Res. 1999;84:735–740.
- Kong Y, Tannous P, Lu G, et al. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation. 2006;113:2579–2588.
- Muraski JA, Fischer KM, Wu W, et al. Pim-1 kinase antagonizes aspects of myocardial hypertrophy and compensation to pathological pressure overload. Proc Natl Acad Sci U S A. 2008;105:13889–13894.
- Dorn GW 2nd. The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–970.
- Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380.
- Chaui-Berlinck JG, Monteiro LHA. Frank-Starling mechanism and short-term adjustment of cardiac flow. J Exp Biol. 2017;220:4391–4398.
- Shiels HA, White E. The Frank-Starling mechanism in vertebrate cardiac myocytes. J Exp Biol. 2008;211:2005–2013.
- Cingolani HE, Perez NG, Cingolani OH, et al. The Anrep effect: 100 years later. Am J Physiol Heart Circ Physiol. 2013;304:H175–82.
- Esler M. The sympathetic regulation of the heart. Eur Heart J. 2016;37:2808–2809.
- Brum G, Osterrieder W, Trautwein W. Beta-adrenergic increase in the calcium conductance of cardiac myocytes studied with the patch clamp. Pflugers Archiv. 1984;401:111–118.
- De Mello WC, Danser AH. Angiotensin II and the heart: on the intracrine renin-angiotensin system. Hypertension. 2000;35:1183–1188.
- Soonpaa MH, Kim KK, Pajak L, et al. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:H2183–9.
- Bernardo BC, Weeks KL, Pretorius L, et al. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010; 128: 191–227.
- McCain ML, Yuan H, Pasqualini FS, et al. Matrix elasticity regulates the optimal cardiac myocyte shape for contractility. Am J Physiol Heart Circ Physiol. 2014;306:H1525–39.
- Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018; 15:387–407.
- Fagard RH. Impact of different sports and training on cardiac structure and function. Cardiol Clin. 1997;15:397–412.
- Borghetti G, Von Lewinski D, Eaton DM, et al. Diabetic cardiomyopathy: current and future therapies. beyond glycemic control. Front Physiol. 2018;9:1514.
- Yildiz M, Oktay AA, Stewart MH, et al. Left ventricular hypertrophy and hypertension. Prog Cardiovasc Dis. 2020;63:10–21.
- Japp AG, Gulati A, Cook SA, et al. The diagnosis and evaluation of dilated cardiomyopathy. J Am Coll Cardiol. 2016;67:2996–3010.
- Rossi MA, Carillo SV. Cardiac hypertrophy due to pressure and volume overload: distinctly different biological phenomena? Int J Cardiol. 1991;31:133–141.
- Frey N, Olson EN. Cardiac hypertrophy: the good, the bad and the ugly. Annu Rev Physiol. 2003;65:45–79.
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600.
- Schiattarella GG, Hill TM, Hill JA. Is load-induced ventricular hypertrophy ever compensatory? Circulation. 2017; 136:1273–1275.
- Lim HW, Molkentin JD. Calcineurin and human heart failure. Nat Med. 1999;5:246–247.
- Lim HW, De Windt LJ, Steinberg L, et al. Calcineurin expression, activation, and function in cardiac pressure-overload hypertrophy. Circulation. 2000;101:2431–2437.
- Ding B, Price RL, Borg TK, et al. Pressure overload induces severe hypertrophy in mice treated with cyclosporine, an inhibitor of calcineurin. Circ Res. 1999;84:729–734.
- Martinez-Martinez S, Lozano-Vidal N, Lopez-Maderuelo MD, et al. Cardiomyocyte calcineurin is required for the onset and progression of cardiac hypertrophy and fibrosis in adult mice. FEBS J. 2019;286:46–65.
- Wilkins BJ, Dai YS, Bueno OF, et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118.
- Zou Y, Hiroi Y, Uozumi H, et al. Calcineurin plays a critical role in the development of pressure overload-induced cardiac hypertrophy. Circulation. 2001;104:97–101.
- Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun. 2004;322:1178–1191.
- MacDonnell SM, Weisser-Thomas J, Kubo H, et al. CaMKII negatively regulates calcineurin-NFAT signaling in cardiac myocytes. Circ Res. 2009;105:316–325.
- Lorenz K, Schmitt JP, Vidal M, et al. Cardiac hypertrophy: targeting Raf/MEK/ERK1/2-signaling. Int J Biochem Cell Biol. 2009;41:2351–2355.
- Purcell NH, Wilkins BJ, York A, et al. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc Natl Acad Sci U S A. 2007;104:14074–14079.
- Li XM, Ma YT, Yang YN, et al. Downregulation of survival signalling pathways and increased apoptosis in the transition of pressure overload-induced cardiac hypertrophy to heart failure. Clin Exp Pharmacol Physiol. 2009;36:1054–1061.
- Cipolletta E, Monaco S, Maione AS, et al. Calmodulin-dependent kinase II mediates vascular smooth muscle cell proliferation and is potentiated by extracellular signal regulated kinase. Endocrinology. 2010;151:2747–2759.
- Cipolletta E, Rusciano MR, Maione AS, et al. Targeting the CaMKII/ERK interaction in the heart prevents cardiac hypertrophy. PLoS One. 2015;10:e0130477.
- Bueno OF, De Windt LJ, Tymitz KM, et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350.
- Zhang J, Fan G, Zhao H, et al. Targeted inhibition of focal adhesion kinase attenuates cardiac fibrosis and preserves heart function in adverse cardiac remodeling. Sci Rep. 2017;7:43146.
- Peng X, Wu X, Druso JE, et al. Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proc Natl Acad Sci U S A. 2008;105:6638–6643.
- Tucci AR, Oliveira FOR Jr., Lechuga GC, et al. Role of FAK signaling in chagasic cardiac hypertrophy. Braz J Infect Dis. 2020;24:386–397.
- Cheng Z, Zhu Q, Dee R, et al. Focal adhesion kinase-mediated phosphorylation of beclin1 protein suppresses cardiomyocyte autophagy and initiates hypertrophic growth. J Biol Chem. 2017;292:2065–2079.
- Zheng D, Zhang M, Liu T, et al. Osteoprotegerin prompts cardiomyocyte hypertrophy via autophagy inhibition mediated by FAK/BECLIN1 pathway. Life Sci. 2021;264:118550.
- Clemente CF, Xavier-Neto J, Dalla Costa AP, et al. Focal adhesion kinase governs cardiac concentric hypertrophic growth by activating the AKT and mTOR pathways. J Mol Cell Cardiol. 2012;52:493–501.
- Peng X, Kraus MS, Wei H, et al. Inactivation of focal adhesion kinase in cardiomyocytes promotes eccentric cardiac hypertrophy and fibrosis in mice. J Clin Invest. 2006;116:217–227.
- Li J, Tan Y, Passariello CL, et al. Signalosome-regulated serum response factor phosphorylation determining myocyte growth in width versus length as a therapeutic target for heart failure. Circulation. 2020;142:2138–2154.
- Hullmann J, Traynham CJ, Coleman RC, et al. The expanding GRK interactome: implications in cardiovascular disease and potential for therapeutic development. Pharmacol Res. 2016;110:52–64.
- Gurevich VV, Gurevich EV. GPCR signaling regulation: the role of grks and arrestins. Front Pharmacol. 2019;10:125.
- Hauser AS, Attwood MM, Rask-Andersen M, et al. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16:829–842.
- Dorn GW. 2nd, GRK mythology: g-protein receptor kinases in cardiovascular disease. J Mol Med (Berl). 2009;87:455–463.
- DelPrincipe F, Egger M, Niggli E. Calcium signalling in cardiac muscle: refractoriness revealed by coherent activation. Nat Cell Biol. 1999;1:323–329.
- Sorriento D, Ciccarelli M, Cipolletta E, et al. “Freeze, don’t move”: how to arrest a suspect in heart failure - a review on available grk2 inhibitors. Front Cardiovasc Med. 2016;3:48.
- Bledzka KM, Manaserh IH, Grondolsky J, et al. A peptide of the amino-terminus of GRK2 induces hypertrophy and yet elicits cardioprotection after pressure overload. J Mol Cell Cardiol. 2021;154:137–153.
- Gold JI, Gao E, Shang X, et al. Determining the absolute requirement of G protein-coupled receptor kinase 5 for pathological cardiac hypertrophy: short communication. Circ Res. 2012;111:1048–1053.
- Coleman RC, Eguchi A, Lieu M, et al. A peptide of the N terminus of GRK5 attenuates pressure-overload hypertrophy and heart failure. Sci Signal. 2021;14. 10.1126/scisignal.abb5968.
- Sorriento D, Santulli G, Ciccarelli M, et al. The amino-terminal domain of grk5 inhibits cardiac hypertrophy through the regulation of calcium-calmodulin dependent transcription factors. Int J Mol Sci. 2018; 19:861.
- Lee JH, Seo HW, Ryu JY, et al. KR-39038, a novel grk5 inhibitor, attenuates cardiac hypertrophy and improves cardiac function in heart failure. Biomol Ther (Seoul). 2020;28:482–489.
- Jones WK, Brown M, Ren X, et al. NF-kappaB as an integrator of diverse signaling pathways: the heart of myocardial signaling? Cardiovasc Toxicol. 2003;3:229–254.
- Sorriento D, Santulli G, Franco A, et al. Integrating GRK2 and NFkappaB in the pathophysiology of cardiac hypertrophy. J Cardiovasc Transl Res. 2015;8:493–502.
- Unudurthi SD, Nassal D, Greer-Short A, et al. βIV-Spectrin regulates STAT3 targeting to tune cardiac response to pressure overload. J Clin Invest. 2018;128:5561–5572.
- Swaminathan PD, Purohit A, Hund TJ, et al. Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ Res. 2012;110:1661–1677.
- Westenbrink BD, Edwards AG, McCulloch AD, et al. The promise of CaMKII inhibition for heart disease: preventing heart failure and arrhythmias. Expert Opin Ther Targets. 2013;17:889–903.
- Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51(4):468–473.
- Mustroph J, Neef S, Maier LS. CaMKII as a target for arrhythmia suppression. Pharmacol Ther. 2017;176:22–31.
- Zhang T, Johnson EN, Gu Y, et al. The cardiac-specific nuclear B isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002;277:1261–1267.
- Zhang T, Kohlhaas M, Backs J, et al. CaMKII isoforms differentially affect calcium handling but similarly regulate hdac/mef2 transcriptional responses. J Biol Chem. 2007;282:35078–35087.
- Zhang T, Maier LS, Dalton ND, et al. The δCIsoform of CaMKII CaMKII is activated activated in cardiac cardiaC hypertrophy and induces induces dilated cardiomyopathy cardiomyopathy and heart failure. Circ Res. 2003;92:912–919.
- Ling H, Zhang T, Pereira L, et al. Requirement for Ca2+/calmodulin-dependent </sup>++/calmodulin–dependent kinase II in the transition from pressure overload-induced overload–induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–1240.
- Kreusser MM, Lehmann LH, Wolf N, et al. Inducible cardiomyocyte-specific deletion of CaM kinase II protects from pressure overload-induced heart failure. Basic Res Cardiol. 2016;111:65.
- Backs J, Backs T, Neef S, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009;106:2342–2347.
- Grimm M, Ling H, Brown JH. Crossing signals: relationships between beta-adrenergic stimulation and CaMKII activation. Heart Rhythm. 2011;8:1296–1298.
- Erickson JR, Joiner ML, Guan X, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474.
- He BJ, Joiner ML, Singh MV, et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med. 2011;17:1610–1618.
- Zhang R, Khoo MS, Wu Y, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417.
- Patel NJ, Nassal DM, Gratz D, et al. Emerging therapeutic targets for cardiac arrhythmias: role of STAT3 in regulating cardiac fibroblast function. Expert Opin Ther Targets. 2021;25:63–73.
- Kurdi M, Zgheib C, Booz GW. Recent developments on the crosstalk between stat3 and inflammation in heart function and disease. Front Immunol. 2018;9:3029.
- Hutchins AP, Diez D, Miranda-Saavedra D. Genomic and computational approaches to dissect the mechanisms of STAT3ʹs universal and cell type-specific functions. Jakstat. 2013;2:e25097.
- Zouein FA, Altara R, Chen Q, et al. Pivotal importance of stat3 in protecting the heart from acute and chronic stress: new advancement and unresolved issues. Front Cardiovasc Med. 2015;2:36.
- Negoro S, Kunisada K, Tone E, et al. Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc Res. 2000;47:797–805.
- Hilfiker-Kleiner D, Shukla P, Klein G, et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. 2010;122:145–155.
- Obana M, Maeda M, Takeda K, et al. Therapeutic activation of signal transducer and activator of transcription 3 by interleukin-11 ameliorates cardiac fibrosis after myocardial infarction. Circulation. 2010;121:684–691.
- Obana M, Miyamoto K, Murasawa S, et al. Therapeutic administration of IL-11 exhibits the postconditioning effects against ischemia-reperfusion injury via STAT3 in the heart. Am J Physiol Heart Circ Physiol. 2012;303:H569–77.
- Zou Y, Takano H, Mizukami M, et al. Leukemia inhibitory factor enhances survival of cardiomyocytes and induces regeneration of myocardium after myocardial infarction. Circulation. 2003;108:748–753.
- Brar BK, Stephanou A, Liao Z, et al. Cardiotrophin-1 can protect cardiac myocytes from injury when added both prior to simulated ischaemia and at reoxygenation. Cardiovasc Res. 2001;51:265–274.
- Mir SA, Chatterjee A, Mitra A, et al. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J Biol Chem. 2012;287:2666–2677.
- Zhao L, Cheng G, Jin R, et al. Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ Res. 2016;118:1918–1929.
- Unudurthi SD, Greer-Short A, Patel N, et al. Spectrin-based pathways underlying electrical and mechanical dysfunction in cardiac disease. Expert Rev Cardiovasc Ther. 2018;16:59–65.
- Nassal DM, Patel NJ, and Unudurthi SD, et al. Ca2+/calmodulin kinase II-dependent II–dependent regulation of βIV-spectrin modulates cardiac fibroblast gene expression, proliferation, and contractility. J Biol Chem. 2021; 297:100893.
- Patel NJ, Nassal DM, Greer-Short AD, et al. betaIV-Spectrin/STAT3 complex regulates fibroblast phenotype, fibrosis, and cardiac function. JCI Insight. 2019;4:e131046.
- Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure the jnc 7 report. JAMA. 2003;289:2560–2572.
- Kjeldsen SE, Dahlof B, Devereux RB, et al. Effects of losartan on cardiovascular morbidity and mortality in patients with isolated systolic hypertension and left ventricular hypertrophy: a losartan intervention for endpoint reduction (LIFE) substudy. JAMA. 2002;288:1491–1498.
- Cao DJ, Wang ZV, Battiprolu PK, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108:4123–4128.
- Berry JM, Cao DJ, Rothermel BA, et al. Histone deacetylase inhibition in the treatment of heart disease. Expert Opin Drug Saf. 2008;7:53–67.
- Sussman MA, Lim HW, Gude N, et al. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281:1690–1693.
- Akhter SA, Luttrell LM, Rockman HA, et al. Targeting the Receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science. 1998;280:574–577.
- Shimoyama M, Hayashi D, Takimoto E, et al. Calcineurin plays a critical role in pressure overload-induced cardiac hypertrophy. Circulation. 1999;100:2449–2454.
- Rogers JH, Tamirisa P, Kovacs A, et al. RGS4 causes increased mortality and reduced cardiac hypertrophy in response to pressure overload. J Clin Invest. 1999;104:567–576.
- Brancaccio M, Fratta L, Notte A, et al. Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat Med. 2003;9:68–75.
- Gelpi RJ, Gao S, Zhai P, et al. Genetic inhibition of calcineurin induces diastolic dysfunction in mice with chronic pressure overload. Am J Physiol Heart Circ Physiol. 2009;297:H1814–9.
- Yoshida K, Holmes JW. Computational models of cardiac hypertrophy. Prog Biophys Mol Biol. 2021;159:75–85.
- Ryall KA, Holland DO, Delaney KA, et al. Network reconstruction and systems analysis of cardiac myocyte hypertrophy signaling. J Biol Chem. 2012;287:42259–42268.
- Frank DU, Sutcliffe MD, Saucerman JJ. Network-based predictions of in vivo cardiac hypertrophy. J Mol Cell Cardiol. 2018;121:180–189.
- Khalilimeybodi A, Paap AM, Christiansen SLM, et al. Context-specific network modeling identifies new crosstalk in beta-adrenergic cardiac hypertrophy. PLoS Comput Biol. 2020;16:e1008490.
- Kang JH, Lee HS, Park D, et al. Context-independent essential regulatory interactions for apoptosis and hypertrophy in the cardiac signaling network. Sci Rep. 2017;7:34.
- Estrada AC, Yoshida K, Saucerman JJ, et al. A multiscale model of cardiac concentric hypertrophy incorporating both mechanical and hormonal drivers of growth. Biomech Model Mechanobiol. 2021;20:293–307.
- Kolwicz SC Jr., Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113:603–616.
- Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation. 2007;116:434–448.
- Xiang K, Qin Z, Zhang H, et al. Energy metabolism in exercise-induced physiologic cardiac hypertrophy. Front Pharmacol. 2020;11:1133.
- Tuomainen T, Tavi P. The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp Cell Res. 2017;360:12–18.