
ABSTRACT
Introduction
Renal ciliopathies represent a collection of genetic disorders characterized by deficiencies in the biogenesis, maintenance, or functioning of the ciliary complex. These disorders, which encompass autosomal dominant polycystic kidney disease (ADPKD), autosomal recessive polycystic kidney disease (ARPKD), and nephronophthisis (NPHP), typically result in cystic kidney disease, renal fibrosis, and a gradual deterioration of kidney function, culminating in kidney failure.
Areas covered
Here we review the advances in basic science and clinical research into renal ciliopathies which have yielded promising small compounds and drug targets, within both preclinical studies and clinical trials.
Expert opinion
Tolvaptan is currently the sole approved treatment option available for ADPKD patients, while no approved treatment alternatives exist for ARPKD or NPHP patients. Clinical trials are presently underway to evaluate additional medications in ADPKD and ARPKD patients. Based on preclinical models, other potential therapeutic targets for ADPKD, ARPKD, and NPHP look promising. These include molecules targeting fluid transport, cellular metabolism, ciliary signaling and cell-cycle regulation. There is a real and urgent clinical need for translational research to bring novel treatments to clinical use for all forms of renal ciliopathies to reduce kidney disease progression and prevent kidney failure.
1. Introduction
1.1. Overall aims
Within the last 20 years, research of ciliopathies has expanded exponentially. Ciliopathies, a group of genetic diseases that were once isolated or unknown, are now being characterized as a spectrum of pleiotropic inherited disorders with shared defects in the biogenesis, maintenance, or functioning of the ciliary complex. One subset of primary ciliopathy syndromes are renal ciliopathies, which all share a kidney defect, often leading to kidney failure (KF). Notably, there are currently no curative treatments for renal ciliopathies.
The increased understanding of the molecular composition, structure, and function of the primary cilium, as well as developments in ciliopathy disease models, have uncovered shared pathogenic mechanisms across the spectrum of renal ciliopathies. Together, these scientific advances have driven the identification of potential therapeutic targets. This review will explore promising small molecular compounds and drug targets within preclinical studies and clinical trials, discussing the prospects for potential and future treatments of renal ciliopathies.
1.2. An overview of the primary cilium
The primary cilium is a microtubule-based cellular appendage, found as a single organelle on nearly every mammalian cell, including renal tubular epithelial cells [Citation1]. It is a sensory organelle and functions as a signaling hub, which is vital in numerous biological processes such as left-right patterning in development, organogenesis, tubulogenesis, cell cycle regulation, and homeostasis.
During quiescent stages of the cell cycle (G0) the primary cilium is formed, which can also be mimicked by serum starvation in vitro. Upon initiation of ciliogenesis, the mature centriole, a cylindrical 9-fold triplet microtubule structure (150–200 nm diameter, 400–450 nm length) with distal and subdistal appendages, forms the basal body (BB) of the primary cilium [Citation2–4]. The BB anchors to the plasma membrane via distal appendage proteins (DAPs) that project radially from the BB, marking the transition zone region [Citation5,Citation6]. The core BB extends to form the 9-fold doublet microtubule-based axoneme, lacking a central axonemal pair (9+0), rendering it immotile. The primary cilium itself is encased in a ciliary membrane contiguous with the plasma membrane. A discrete ciliary compartment is formed using the transition zone (TZ) of the cilium which cross-links the ciliary membrane with the proximal axonemal microtubules, utilizing Y-links and ciliary necklace proteins and works with DAPs, septin ring barrier, nucleoporins, and other ciliary protein complexes to regulate selective targeting, sorting and localization of molecular cargo [Citation7–10]. Protein complexes integral to ciliary function include the BBSome, MKS complex, NPHP complex, Inversin, and CEP290 complexes [Citation6,Citation11–13]. Intraflagellar transport (IFT) is used to transport protein cargo within the ciliary compartment and is also required for formation of the central axoneme and numerous ciliary signaling pathways [Citation1]. Anterograde movement (up) and retrograde movement (down) is exchanged at the ciliary tip and at other points along the ciliary axoneme [Citation1,Citation14]. Upon entry into the cell cycle, due to the requirement of the mature centriole within the centrosome, the cilium is disassembled and resorbed [Citation15].
The primary cilium acts a vital transducer in numerous signaling pathways. Patched 1 (PTCH1), the Sonic hedgehog (Shh) receptor, localizes to the ciliary membrane and prevents entry of Smoothened (SMO) [Citation16]. Upon Shh binding to PTCH1, PTCH1 is trafficked out of the cilium allowing SMO to accumulate in the primary cilium, which activates Gli transcription factors. Shh signaling pathway is vital in processes such as cell determination and patterning, and is involved in proper kidney development and functioning, with defective signaling implicated in various renal ciliopathies [Citation16–19]. Components of the canonical Wnt signaling pathway, which is involved in cell proliferation, differentiation and survival, are located at the primary cilium [Citation20]. Components of the non-canonical Wnt planar cell polarity pathway (PCP), regulating cell morphology, migration, and orientated cell division, are also present at the primary cilium [Citation20]. PCP signaling is required for proper nephron tubular formation and maintenance, defects of which can lead to cystogenesis within the kidney tubules [Citation18,Citation21,Citation22]. There are other ciliary signaling pathways which are vital in cell cycle regulation and proliferation, that have been implicated in proper renal tubular formation and kidney function. These includes cyclic AMP (cAMP) signaling, mammalian target of rapamycin (mTOR) signaling, epidermal growth factor (EGF) signaling, AMP-activated protein kinase (AMPK) signaling and insulin-like growth factor (IGF) signaling, although this list is not exhaustive [Citation23,Citation24].
The primary cilia are also transducers of mechanical stimuli. In the renal tubules, primary cilia project from the apical surface of renal tubular epithelial cells into the lumen and are stimulated by urinary flow. Polycystin-1 (PC1) and polycystin-2 (PC2) act as mechanotransducers, which mediate numerous downstream signaling pathways such as PCP, mTOR, and Shh, which can be dysregulated in cystic kidney models [Citation25–28]. Primary cilia also sense morphogens, hormones, and odorants [Citation29]. Emerging studies have also indicated that primary cilia may have roles in releasing ‘ectosomes’ as signaling messengers [Citation23].
As proteins encoded by primary cilium-related genes are closely linked with the function of centrioles, it is logical that some ciliary genes, such as CEP290 and CEP164, have also been implicated in DNA damage response (DDR) pathways and cell cycle regulation, which effect renal cell proliferation, apoptosis and fibrosis [Citation23,Citation30,Citation31].
There are also other specialized primary ciliary structures, including the connecting cilium within the photoreceptor cells of the retina and olfactory sensory neurons [Citation32–34]. The other main subtype of cilia are motile cilia, which are found as groups of organelles on specialized cells such as the respiratory epithelium and reproductive tract [Citation35]. Sperm flagella is also a specialized motile cilium. Additionally, nodal cilia found at the embryonic node have overlapping structures with both primary and motile cilia and are vital for left-right patterning during development [Citation36]. However, the cilia type present on kidney epithelial cells are primary cilia, and therefore this will be focus of this renal ciliopathy review.
2. Renal ciliopathies
2.1. Renal ciliopathies overview
Primary ciliopathies are a group of rare genetic multi-system syndromes, caused by defects in the maintenance, biogenesis or functioning of the primary cilium. Primary ciliopathies are classified further into disease syndromes with defined clinical presentations, but they often have genetic and phenotypic overlaps. There are over 35 ciliopathy syndromes with an estimated grouped incidence of 1 in 1,000 people, with over 180 known primary ciliopathy genes, but approximately 50% of ciliopathy cases remain genetically unsolved [Citation1,Citation35,Citation37–39]. Also, primary ciliopathy genes have high pleiotropy, making it difficult for clear diagnosis, prognosis, and appropriate disease management based upon genotype alone. It might be that as the number of ciliary genes increases and syndromes overlap further, ciliopathies will become a continuum of disorders rather than the current system of discrete syndromes [Citation38].
Renal ciliopathy syndromes often manifest in the form of cystic kidney disease, but can also manifest as tubulointerstitial fibrosis, both can lead to KF () [Citation40]. The primary cilium is found on the apical surface of renal epithelial cells and projects into the lumen of the nephron, acting as both a chemical and mechanical signal transducer. Archetypal renal ciliopathy syndromes include autosomal dominant polycystic kidney disease (ADPKD), autosomal recessive polycystic kidney disease (ARPKD), and nephronophthisis (NPHP) [Citation40]. There are other renal primary ciliopathies with extra-renal manifestations affecting organs such as the retina, brain, skeletal system, metabolism, liver, auditory perception, fertility, and development. These include Bardet-Biedl syndrome (BBS), Joubert syndrome (JBTS), Senior-Løken syndrome (SLS), Jeune asphyxiating thoracic syndrome (JATD), and the perinatally lethal Meckel-Gruber syndrome (MKS). There are no curative treatments for renal ciliopathies, and renal manifestations are typically life limiting [Citation24,Citation41]. Current therapeutics involve symptom management and supportive treatments, some including dialysis, treatment of hypertension and anemia, and dietary restrictions including salt restriction, but they do not target the underlying cause of disease or prevent KF [Citation40].
Figure 1. Clinical phenotypes and molecular genetics of renal ciliopathies.
Note:Variability in the age of onset of KF and the median age of onset of clinical symptoms are shown as extended diamond bars. ADPKD mostly presents in adulthood (30-40 years of age) although a small proportion of cases reported in early life (1-10 years of age) due to biallelic or digenic or severe loss of function PKD1/PKD2 variants. The age of presentation for ARPKD varies with approximately one third of patients presenting before 1 year of age, one third between 1 and 20 years of age and one third after 20 years with mutations in PKHD1 being the most common cause. In juvenile NPHP, KF develops at a median age of 13 years whereas in adult form, which is clinically and histologically similar to the juvenile NPHP, KF develops after 15-20 years of age. Infantile NPHP is rare with KF appearing during first year of life. The current clinical trials with name of the drug, ClinicalTrials.gov identifier number and the age range of patients the study is recruiting is shown for ADPKD and ARPKD. There are currently no clinical trials in progress for NPHP. (Created using BioRender.com).
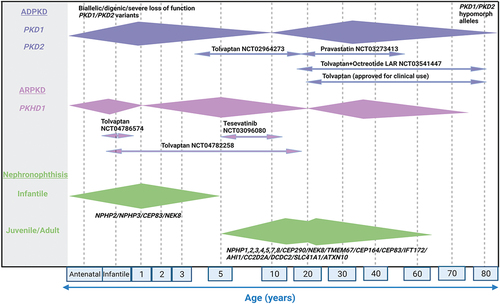
2.2. ADPKD
The most common inherited renal ciliopathy is ADPKD, frequently leading to KF, requiring renal replacement therapy [Citation38,Citation42]. ADPKD is estimated to effect 1 in 1000 people, and it typically presents in adulthood with hypertension and bilateral cystic kidneys, although age-dependent penetrance and disease variability means prevalence is likely underestimated [Citation43,Citation44]. Patients with typical ADPKD have numerous large fluid-filled bilateral kidney cysts, which cause kidney enlargement. The cysts themselves may arise from all nephron segments, but predominantly originate from the collecting duct [Citation24]. ADPKD is often characterized by misorientated hyperproliferation of renal epithelial cells, which causes the formation of renal tubular cysts [Citation45]. Subsequent cyst enlargement causes obstruction of neighboring nephrons, displacing renal parenchyma, and compressing microvasculature, causing urine drainage to be hindered, glomerular filtration to be gradually impaired, following which renal tubules become atrophic. The kidney injury causes inflammation and fibrosis pathways to be triggered [Citation45,Citation46]. ADPKD often results in KF.
Heterozygous mutations in PKD1 and PKD2, encoding polycystin-1 (PC1) and polycystin-2 (PC2) respectively, account for over 90% of ADPKD patients. PKD1 mutations account for 75–85% of identified mutations, and PKD2 mutations are less common, accounting for 15% of identified mutations and milder phenotypes with KF developing at a later age [Citation41,Citation47]. PC1 and PC2 are located in the primary cilium. PC1 acts as an ion channel and g-protein coupled receptor, and PC2 acts as an ion channel that forms a heteromeric complex with PC1 with fibrocystin, regulating PC1’s localization in kidney epithelia [Citation48,Citation49]. It has traditionally been proposed that the PC1/PC2 complex mediates Ca2+ signaling in the primary cilium, in response to fluid flow; however, more recently, this has been refuted, with the exact pathomechanism unclear [Citation41,Citation45,Citation50,Citation51]. Nevertheless, it is thought that loss or malfunction of PC1/PC2 has implications downstream for multiple pathways, including cAMP, mTOR, EGF, AMPK, and cMyc signaling, which effect orientated cell proliferation [Citation52].
The majority of patients present with ADPKD in adulthood (30–40 years old) with a typical radiological pattern of cystic kidney disease, with bilateral and diffuse distribution of kidney cysts and mild, moderate or severe replacement of kidney tissue by cysts, where all cysts contribute similarly to an increase in total kidney volume [Citation53]. There is a large variability in the age of presentation and disease progression can be measured using total kidney volume (TKV) following kidney magnetic resonance imaging to predict risk of KF [Citation54]. TKV has been used as a surrogate end point in interventional clinical trials [Citation55]. Pathogenic mutations in PKD1 and PKD2 can include frame-shift, nonsense, splice-site, missense and synonymous SNV changes, with more recent evidence for deep intronic alleles [Citation41]. In general patients with loss of function mutations in PKD1 and PKD2 present with a more severe, earlier-onset cystic kidney phenotype (). Prenatal and antenatal presentations of ADPKD are often associated with a PKD1 or PKD2 loss of function allele and the presence of an additional second allele, often hypomorphic, in PKD1 or PKD2 acting as a genetic modifier [Citation56]. Digenic heterozygous alleles in both PKD1 and PKD2 could also contribute to earlier onset ADPKD [Citation56]. Occasionally, severe early onset PKD phenotypes may also be caused by biallelic loss of function mutations in PKD1 or PKD2 (). The “somatic second hit” hypothesis of PKD1/PKD2 mutations also may affect phenotypic variability [Citation57]. On the other end of the spectrum, mild ADPKD phenotypes with preserved renal function may be due to single heterozygous hypomorphic mutations in PKD1 and PKD2 [Citation56].
Radiologically, there are other forms of atypical ADPKD, where there may be unilateral cystic kidney disease, segmental cystic kidney disease involving one pole of one or both kidneys, asymmetric cystic kidney disease and lopsided kidney disease, where less than or equal to 5 kidney cysts account for greater than or equal to 50% of total kidney volume [Citation53]. Such patients present with cystic kidney disease phenotypes, as well as the other renal and extra-renal phenotypes, such as polycystic liver disease. These are caused by mutations in genes encoding proteins not typically associated with primary cilia but are predicted to effect the PC1 or PC2 complex [Citation41]. An example is heterozygous mutations in the transcription factor HNF1B which causes atypical ADPKD with a wide range of renal phenotypes, such as congenital abnormalities of the kidney and urinary tract (CAKUT) or autosomal dominant tubulointerstitial kidney disease (ADTKD)-like presentations [Citation58]. Other genetic causes of atypical ADPKD include heterozygous variants in; IFT140, DNAJB11, GANAB, ALG8, ALG9, SEC61A1 and PRKCSH [Citation58–67]. Atypical ADPKD is characterized clinically with a milder disease progression phenotype with some mutations in genes such as GANAB and ALG8 having no instances of KF reported to date; however, some patients reach KF in later adulthood [Citation41,Citation61–64].
2.3. ARPKD
ARPKD is a rare renal ciliopathy than ADPKD, with an estimated incidence of 1 in 20,000 live births [Citation68]. Patients with ARPKD have numerous fluid-filled bilateral kidney cysts and poor corticomedullary differentiation, which causes kidney enlargement, subsequent kidney injury, and KF. ARPKD is also often associated with liver abnormalities. Unlike ADPKD, patients with ARPKD often present with symptoms in early childhood with 50% suffering with KF by 10 years of age. However, there are also later childhood and adult presentations of ARPKD which predominately have liver phenotypes including congenital hepatic fibrosis and ductal plate malformations, alongside milder cystic kidney disease () [Citation41,Citation69].
ARPKD is most commonly caused by mutations in PKHD1 that encodes fibrocystin, which interacts with PC1 and PC2 within the primary cilia [Citation50,Citation70–72]. Fibrocystin is a key regulator of cell proliferation, apoptosis and polarization [Citation73]. More recently, mutations in DZIP1L have been associated with ARPKD which develop KF later in life, although this accounts for less than 1% of individuals [Citation74]. DZIP1L is believed to be involved in proper localization of PC1 and PC2. Biallelic mutations in TULP3 have also been shown to give a phenotype resembling ARPKD, with progressive fibrocystic liver and kidney disease, as well as polycystic kidney disease [Citation75]. The encoded protein TULP3 acts as a critical adaptor protein for ciliary trafficking.
2.4. NPHP
NPHP patients present with a progressive decline in kidney function, often with a loss of urinary concentrating ability, resulting in polyuria and polydipsia. Histological features following renal biopsy reveal tubulointerstitial disease associated with progressive tubular interstitial fibrosis, inflammation, and tubular atrophy, and sometimes corticomedullary cysts [Citation19,Citation76]. NPHP is not typically associated with kidney enlargement other than in infantile cases [Citation23]. Notably, NPHP leads to a progressive loss of renal tubular function and represents the most common pediatric inherited cause of KF.
NPHP has a disease spectrum; there are traditionally three clinical subtypes of NPHP, infantile, juvenile, and adolescent [Citation41,Citation77]. Infantile NPHP has a severe phenotype, with a median age of onset of 1 year old and KF typically before 3 years old. Infantile NPHP presents with enlarged kidneys with cortical cysts, moderate fibrosis, and rapid disease progression, similar to ARPKD, and lacks the tubular basement membrane changes present in other NPHP subtypes [Citation23,Citation76]. Juvenile NPHP, the most common, has median age of onset of 13 years old with KF typically in the second decade of life, and patients present with small to normal sized kidneys with tubulointerstitial fibrosis, thickened tubular membranes and in less than 50% patients, corticomedullary cysts. There may also be adult-onset NPHP, which clinically presents as juvenile NPHP, albeit later [Citation41,Citation77]. NPHP gene mutations such as NPHP1 whole gene deletions may be detected in adult KF patients who have previously been labelled as KF of uncertain cause () [Citation78].
NPHP has an estimated incidence of 1 in 50,000–1 in 1,000,000 individuals [Citation76]. To date, there are over 20 NPHP causative genes identified, encoding protein products known as nephrocystins, inherited in an autosomal recessive pattern, accounting for less than 50% of NPHP ciliopathies [Citation40]. Mutations in the NPHP complex, a distinct module found in the transition zone of the primary cilium, causes NPHP. The NPHP complex is involved in transition zone and ciliary biogenesis, as well as cargo trafficking. The NPHP complex includes protein products from RPGRIP1L, NPHP4, NPHP1, NPHP5, NPHP3, and NEK8. Mutations in NPHP1, encoding nephrocystin-1, is the most common genetic cause of NPHP (approximately 20% of cases) [Citation77,Citation79]. Nephrocystin-1 is an adaptor protein that localizes to the primary cilium and apical membrane of kidney epithelial cells, with functions in cell signaling, adhesion and cell maintenance, interacting with PC1 and PC2 and nephrocystins [Citation80]. Other genetic causes of NPHP include genes encoding proteins which are localized to the primary cilium or centriole, including CEP290 and CEP164 [Citation41,Citation76,Citation81]. Specifically, infantile NPHP is commonly caused by mutations in INVS, NPHP3, NEK8 and CEP83 [Citation82–85]. Adults presenting with later onset NPHP have been shown to have disease causing variants in NPHP1 and NPHP3 [Citation79,Citation86,Citation87]. However, there are several NPHP genes that are not currently directly linked to primary ciliary function, including ZNF423, MAPKBP1 and GLIS2 [Citation38,Citation81,Citation88,Citation89].
2.5. Other renal ciliopathies with associated clinical manifestations
At least 15% of NPHP cases are associated with extra-renal manifestations [Citation23], which can be grouped together as other multisystem ciliopathy syndromes. If genes that cause syndromic NPHP-related ciliopathies are accounted for, there are over 90 implicated genes [Citation23,Citation90]. A well-described NPHP-related ciliopathy is JBTS, a primary ciliopathy syndrome characterized by brain axial magnetic resonance imaging (MRI) ‘molar tooth sign’, caused by cerebellar vermis aplasia (CVA), thickened and horizontalized cerebellar peduncles and deepened interpeduncular fossa. This neurological development disorder can lead to many problems beginning in early life including developmental delay, hypotonia leading to ataxia, breathing problems and intellectual disability [Citation35,Citation91]. JBTS may characterized by a neurological phenotype but can be a renal-retinal-cerebellar syndrome with the NPHP-associated kidney disease leading to KF, and retinal degeneration leading to blindness. Liver and skeletal phenotypes may also be present in patients with JBTS. It is estimated that 1 in 80,000–1 in 100,000 people have JBTS; however, the heterogenous nature of the disease means it is likely underdiagnosed [Citation92]. Mutations in over 35 genes have been identified as genetic causes of JBTS, most of which are autosomal recessive, apart from OFD1 which has X-linked recessive inheritance [Citation93]. Mutations in CEP290 are the most common cause of JBTS accounting for more than 38% of cases [Citation94].
BBS is an autosomal recessive ciliopathy where patients present with retinal degeneration usually leading to blindness as well as at least two of the following primary/major symptoms; renal dysfunction (which is frequent and often presents on prenatal ultrasound with renal and urinary tract malformation, such as cystic kidney, pelvic dilation, and ureter obstruction), intellectual disability, obesity, polydactyly, and hypogonadism [Citation95]. Patients can also present with secondary/minor symptoms, some of which include growth retardation, delayed development, speech disorders, thyroid problems, anosmia, and dental anomalies [Citation96]. BBS clinical diagnosis is based upon the presence of 4 primary symptoms, or 3 primary symptoms and at least two secondary symptoms (Beales criteria) [Citation97]. Renal biopsy in patients with chronic kidney disease (CKD) and BBS typically shows renal dysplasia and chronic tubulointerstitial nephropathy [Citation98,Citation99]. Overall BBS is estimated to occur in 1 in 100,000 live births, although there is a higher prevalence in non-European Union countries with high consanguinity, specifically Pakistan [Citation96,Citation100]. There are over 20 known BBS genes accounting for approximately 85% of cases. Many of the protein products of genes which cause BBS are found within the BBSome protein complex, which mediates the binding of IFT particles (IFT-A, IFT-B) to protein cargo at the base of the cilium and ciliary tip, enabling anterograde and retrograde ciliary transport [Citation11,Citation12]. The BBSome also interacts with both PC1, PC2 and NPHP-associated proteins [Citation101]. The BBSome complex contains; BBS2, BBS7, BBS9, BBS1, BBS4, BBS18, BBS8 and BBS5, which interacts with ADP ribosylation factor (ARF)-like GTPase ARL6/BBS3 required for membrane recruitment of the BBSome. The most prevalent causes of BBS are BBS1 mutations whose encoded protein is found within the BBSome and BBS10 mutations, whose encoded protein is found within the BBSome chaperonin-like complex to mediate BBSome assembly [Citation102,Citation103].
NPHP-like ciliopathies also include SLS where patients present with NPHP associated with retinitis pigmentosa. The most common cause of SLS is mutations in NPHP5 alias IQCB1 [Citation104,Citation105].
Meckel Gruber Syndrome (MKS) is a rare ciliopathy syndrome characterized by NPHP associated with skeletal defects and developmental abnormalities, often resulting in embryonic and postnatal lethality [Citation106]. Causative genes have been identified for 60% of MKS cases, mutations of which are present in the MKS complex located at the transition zone. It contains the core proteins; B9D1, B9D2, TCTN1, TCTN2, TCTN3, CC2D2A, TMEM216, TMEM67, MKS1, and TMEM237. The MKS complex is thought to be involved in transition zone biogenesis and regulation of trafficking in and out of the primary cilium [Citation6,Citation13,Citation107].
JATD is an autosomal recessive syndrome characterized by short-rib thoracic skeletal dystrophies, with short stature, narrow thorax with short ribs, short limbs, and polydactyly. JATD is also linked with cystic renal disease [Citation93]. Genes associated with JATD include CEP120, CSPP1, KIAA0586, and PIBF1. Oral-facial-digital syndromes (OFD) are ciliopathy syndromes with characteristic facial abnormalities, oral abnormalities, and polydactyly. The gene OFD1 causes X-linked dominant OFD with polycystic kidney disease [Citation108].
Exploration of all the known and potential genes underlying renal and multisystem primary ciliopathies is beyond the scope of this review; however, other reviews can be recommended [Citation40,Citation41].
3. Modelling renal ciliopathies
Conventionally, primary ciliopathies have been studied using 2D cell line cultures that can form primary cilia upon entry into G0, mimicked by serum starvation. Examples of immortalized renal cell lines include HEK293 (Human embryonic kidney), mIMCD-3 (murine kidney inner medullary collecting duct), MDCK (canine kidney distal tubule/collecting duct), LLC-PK1 (pig kidney proximal tubule) and A6 (frog kidney) [Citation109–114]. These cells can be utilized to study the normal functioning of renal primary cilia, be genetically modified to understand gene and variant-specific pathogenic mechanisms of disease and be used to examine the effects of potential therapeutics. Developed more recently, human urine-derived renal epithelial cells (hURECs) from healthy donors and patients can be used as a renal ‘liquid biopsy’, which is a powerful tool for understanding patient-specific molecular mechanisms of disease, working toward personalized therapies [Citation40,Citation115,Citation116].
More recently, 3D kidney tubuloids and organoids from hURECs and stem cells have been utilized to model kidney development and disease. Organoids have the advantage of containing different parts of the nephron and interstitial cells, which is especially useful for modelling the 3D renal tubule structure with cilia projecting into the lumen [Citation116–123]. Kidney tubuloids and organoids are also utilized for personalized medicine approaches, by culturing patient-derived stem cells or genetically engineering patient mutations [Citation23,Citation38,Citation109,Citation124]. Other more recent scientific advances include kidney-on-a-chip platforms which integrate living kidney cells and biophysical systems such as fluid flow, pressure, and chemical gradients, creating a model more accurate to the human in vivo system [Citation45,Citation125].
A large insight into renal ciliopathies has been gained by using animal models, typically zebrafish and rodents in which cilia form in the same tissues as humans, but Xenopus, Chick, and C. elegans have also been utilized [Citation109]. Zebrafish are useful tools for studying embryonic consequences of renal ciliopathies as they are easily genetically manipulated, the embryos and larva (24–72 hours post fertilization) are transparent allowing non-invasive imaging during development, and classic zebrafish ‘ciliopathy’ defects such as pericardiac edema, hydrocephalus, abnormal body curvature and pronephric cysts present rapidly [Citation45,Citation81,Citation126]. Additionally, zebrafish are low cost and have fast development, meaning they allow efficient functional studies and large-scale screening analysis [Citation23].
Rodents have been invaluable in ciliopathy research, especially as they can be utilized to study post-embryonic phenotypes including adult histology and fibrosis [Citation23]. Prior to the discovery of genetic causes of renal ciliopathies rodent strains which develop spontaneous renal cysts were utilized as disease models; an example is cpk mice as a murine model of ARPKD [Citation38,Citation127–129]. With improvement in gene sequencing and development of genetic modification techniques numerous transgenic animals with gene knockouts, knockins, and patient-specific point mutations have been designed. The use of inbred transgenic mouse models also helps to remove confounding influences such as oligogenecity and triallelism, to understand the exact pathogenic mechanisms driven by specific genetic mutations [Citation127]. Biological parameters such as kidney volume, kidney weight, and urine/plasma biomarkers [Citation130] can be measured, histopathology examined, and molecular pathways investigated by transcriptomics and proteomics, to uncover disease mechanisms and determine the efficacy of potential therapeutics.
4. Disease mechanisms of renal ciliopathies
Although renal ciliopathies can be classified into distinct syndromes, causative mutations in genes encoding proteins involved in the primary cilium or centrosome mean they may share overlapping mechanisms of disease, which may be amenable for therapeutic intervention (). Abnormal functioning of proteins involved in ciliogenesis, such as CEP164, can prevent proper cilia formation, which will effect a myriad of downstream ciliary signaling pathways. Additionally, mutations in genes encoding for proteins involved in cargo trafficking or regulation, such as CEP290, will have implications for signal pathway transduction, as well as mutations in components of signaling pathways themselves, such as PKD1. In regard to renal ciliopathies, abnormalities in signaling pathways such as cAMP, Shh, Wnt, mTOR, and AMPK, likely cause misoriented cellular divisions, increased proliferation, increased fluid secretion and subsequent cystogenesis, consequently leading to further kidney damage. Ciliary and centriolar proteins which have roles in DDR and cell cycle regulation may also be driving a renal cystogenesis phenotype alongside increased fibrosis and apoptosis. Increased inflammation and dysfunctional mitochondria are also by-products of dysregulated signaling pathways have been shown to contribute to the progression of renal ciliopathies. Extensive reviews of mechanisms of renal ciliopathy diseases have recently been performed [Citation23,Citation24]. Importantly, due to the wide range of cellular processes that primary cilia regulate, it is likely that in each syndrome there are multiple pathogenic drivers of disease. In some ways, this is advantageous as it offers many points for potential therapeutic targets. However, the cross talk between pathways and feedback loops introduces complications of changing one pathway without negatively affecting another. Further challenges arise with core biological pathways, such as Shh signaling, in which modification in vitro may be beneficial, but systemic treatment is unrealistic due to the expected severe side effects [Citation18,Citation24,Citation116].
Figure 2. Renal ciliopathies and potential drug targets and prospects for clinical trials.
Note: Summary diagram showing the molecular pathways and the therapeutic targets studied in cell based and rodent models (highlighted in green) and those studied in human clinical trials (highlighted in blue) in renal ciliopathies. The drug names are shown in red. (Created using BioRender.com).
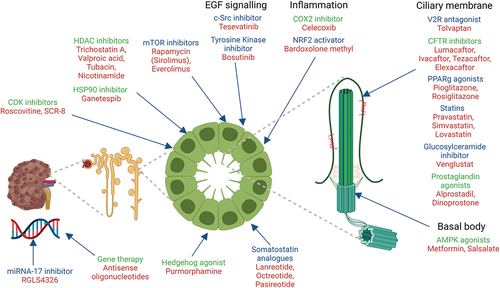
5. Preclinical drug targets and clinical trials in renal ciliopathies
Preclinical research on drug targets for renal ciliopathies has focused on understanding the molecular mechanisms underlying these disorders. This has led to ongoing clinical trials for renal ciliopathies, with several drugs in various stages of development (). Potential therapeutics are being evaluated for their ability to slow the progression of cystic kidney disease. Here we provide a brief overview of the current landscape.
Table 1. Molecular targets, drug compounds, preclinical trials, and clinical trials for renal ciliopathies.
5.1. Targeting transmembrane channels
ADPKD is currently the only renal ciliopathy where an approved treatment option called tolvaptan is available to patients which slows kidney disease progression. The arginine vasopressin receptor 2 (AV2R) is a G-protein coupled receptor (GPCR) that localizes to the basolateral membrane of renal tubular cells and the ciliary membrane, and is upregulated in ADPKD patients. AV2R upregulation causes increased cAMP and PKA activation, which in turn leads to aquaporin 2 (AQP2) mediated and chloride channel dependent fluid secretion, promoting cystogenesis [Citation221,Citation222]. Tolvaptan, is an arginine vasopressin type receptor (AVPR2) antagonist and has been used with success in selected ADPKD patients with progressive forms of the disease [Citation45,Citation131]. As tolvaptan is a very expensive drug, and the associated side effects of polyuria are significant, its use is restricted to only selected patients with rapidly progressive disease, as evidenced by declining kidney function and large kidney volumes. Reports of acute liver toxicity within clinical trials mean regular monitoring is necessary [Citation131,Citation223,Citation224]. A newer AV2R antagonist lixivaptan has been shown promise in in vitro preclinical trials, and shown to have less hepatotoxic effects in rat models, although in humans this drug was terminated early by the sponsor, due to reassessment of commercial potential of the drug (ClinicalTrials.gov, NCT04064346) [Citation136,Citation225,Citation226].
Although there are currently no approved treatment options for ARPKD, there are 2 clinical studies currently underway using tolvaptan in the treatment of ARPKD. The first study is a phase 3b multicenter, open label, non-randomized trial to evaluate the safety, tolerability, and efficacy of tolvaptan in infants and children 28 days to less than 18 years of age with ARPKD. The participants receiving the tolvaptan treatment will be evaluated for 18 months over the course of the trial (ClinicalTrials.gov, NCT04782258) [Citation132]. The second study is a phase 3 clinical trial currently recruiting patients from different locations across the globe. It will evaluate the effect of tolvaptan on the need for renal replacement therapy in pediatric patients 28 days to less than 12 weeks of age with ARPKD (ClinicalTrials.gov, NCT04786574) [Citation132].
In ADPKD, transepithelial chloride secretion through cystic fibrosis transmembrane conductance regulator (CFTR) and a non -CFTR activated chlorine channel, Transmembrane protein 16A (TMEM16A) are known to drive the cyst enlargement [Citation140]. These channels contribute to increased chloride transport into the lumen of the renal tubular cyst, which drives fluid secretion, promoting cyst growth [Citation227,Citation228]. In vivo studies have shown that small molecular CFTR modulator drugs can slow the cystic kidney phenotype and preserve kidney function in PKD1 murine models of ADPKD [Citation138]. Interestingly, repurposing of the currently approved cystic fibrosis drug VX-809 (lumacaftor), which acts as a CFTR misfolding corrector and potentiator, cause reduced cyst growth in PKD1 mouse models, alongside decreased intracellular calcium, cell proliferation and endoplasmic reticulum (ER) stress markers [Citation139]. The beneficial actions of lumacaftor and its analogues are due to the translocalization of CFTR to the basolateral membrane along with stimulation of apical sodium reabsorption via the epithelial sodium channel to reduce cyst growth [Citation229]. There are other approved CFTR corrector drug treatments including ivacaftor, tezacaftor, and elexacaftor. It would therefore be interesting to see whether these also have an effect on renal ciliopathy models; however, none of these are currently in clinical trials for PKD. Also, Tmem16a knockdown in mouse models of ADPKD using the FDA approved drugs niclosamide and benzbromarone, and also by using ani9, a specific small molecule inhibitor of Tmem16a [Citation140,Citation230,Citation231] have shown to strongly suppress the cystic phenotype in vivo, this is yet to make it to clinical trials. Additionally, peroxisome proliferator-activator receptor gamma (PPARγ) inhibits the synthesis of CFTR protein and inhibits ERK signaling, which can be exploited by agonists pioglitazone and rosiglitazone to ameliorate cystic kidney disease as shown in vitro and in vivo using PCK rat, Han:SPRD rat, and PKD1 mouse models [Citation141–145,Citation232]. However, high doses of rosiglitazone led to adverse effects including cardiac enlargement [Citation145]. Interestingly, pioglitazone in combination with tolvaptan showed improved renal survival and slowed cystic kidney disease progression in an adult-onset PKD mouse model [Citation146]. A recently concluded phase 2 clinical study showed using a low-dose pioglitazone slowed down the kidney cyst progression in humans with ADPKD (ClinicalTrials.gov, NCT02697617) [Citation146]. PPARγ agonists could also be explored for ARPKD and NPHP diseases.
5.2. Targeting lipid metabolism
More recently, statins have been explored as treatments of renal ciliopathies. Statins are hydroxy-3-methylglutarylCoA reductase inhibitors which modulate lipids, such as cholesterol, and effect AMPK activation [Citation233]. Cholesterol is also enriched in the ciliary membrane and has important roles in ciliary membrane signaling. Statins, such as lovastatin, have shown to reduce kidney weight and cyst volume in models of NPHP [Citation147]. Simvastatin has also showed promising results of ameliorating kidney function in phase 1 clinical trials in ADPKD patients, but there were disadvantages to the study, with too small patient numbers and a very short follow up time. Hence a bigger patient cohort with longer follow up duration is required to assess the effects of Simvastatin [Citation148]. Pravastatin is in phase 4 clinical trial for ADPKD designed to assess the efficacy and benefits of statin therapy in adults with ADPKD recruiting patients from 25 to 60 years of age. This clinical study evaluates parameters such as kidney volume and function, renal blood flow, plasma and urine protein markers and blood vessel stiffness at baseline and 2 years after pravastatin treatment in 150 patients with ADPKD (ClinicalTrials.gov, NCT03273413) [Citation149]. As statins have shown to improve renal function in the rat models of PKD, it would be interesting to see if statins have a beneficial effect on NPHP in humans [Citation23]. Glycosphingolipids also accumulate in PKD models which disrupts ciliary signaling. Treatments with glucosylceramide inhibitors in ADPKD and NPHP murine models have shown reduced cystogenesis [Citation150,Citation234]. Venglustat, a glucosylceramide synthase inhibitor is currently being used in clinical trials for ADPKD (ClinicalTrials.gov, NCT03523728) [Citation151].
5.3. Targeting inflammation
Inflammatory cells, such as macrophages, drives the cyst epithelial cell proliferation, growth, and kidney fibrosis in the early stages of ADPKD and ARPKD [Citation235]. Therefore, preventing inflammation could be another approach in treatment of PKD [Citation236]. Literature evidence shows that cyclooxygenase 2 (COX2), a prostaglandin metabolizer, is upregulated in cystic kidney diseases, including PKD and NPHP disease models, and hence could be considered a potential target for therapy [Citation152,Citation237,Citation238]. Celecoxib, a specific (COX2) inhibitor ameliorated the ADPKD disease progression and improved renal function in mouse model of ADPKD [Citation153], which has been replicated for other COX inhibitors in the Han:SPRD Cy rat model [Citation152,Citation154]. Diets enriched in flax-oil have also demonstrated reduced COX-derived oxylipin levels, with reduced kidney cyst progression and kidney fibrosis in Nphp3 mice models [Citation155]. Potentially treatment with COX inhibitors, or dietary supplementation may be useful for reducing inflammation in renal ciliopathy diseases. Another possible inflammatory target is the transcription factor nuclear factor erythroid 2-like 2 (NFR2), which increases the presence of antioxidants and detoxification proteins. Recent studies also indicated that NRF2 negatively regulated the primary ciliogenesis process and hedgehog signaling (Hh) [Citation239,Citation240]. Bardoxolone, a semisynthetic derivative of triterpenoids exerts antioxidant activity by promoting the activation of Nrf2 and the downregulation of the proinflammatory nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) signaling, and is considered a potential therapeutic option for patients with ADPKD [Citation156–158]. Bardoxolone methyl oral capsules used for treatment of patients with ADPKD is currently in phase 3 clinical trial to study the safety, tolerability and efficacy of the compound (ClinicalTrials.gov, NCT03918447) [Citation159]. Triptolide, an NF-kB inhibitor with anti-proliferative and pro-apoptotic properties was used in ADPKD mouse models to reduce cystic burden; however, there were adverse effects including infertility and immunosuppression [Citation160]. Inflammation could also be a driver of NPHP kidney phenotypes. NPHP1 regulates CCL2 chemokine expression alongside LKB1, with LKB1 loss causing increased CCL2 levels and subsequent macrophage activation, and thus CKD disease progression in vivo [Citation241]. Modulation of inflammatory pathways should be explored as a therapeutic option for NPHP.
5.4. Targeting cellular metabolism
One pathway thought to have a core role in renal ciliopathies is the mTOR pathway, which acts as a central regulator of cellular metabolism, protein translation, growth, cell cycle regulation and cell survival, integrating effectors from other pathways including IGF and AMPK signaling [Citation161]. Multiple in vitro and in vivo studies have demonstrated that the mTOR pathway is aberrantly activated in renal ciliopathy models and human cells, especially in the cystic renal epithelial cells, which stimulates uncontrolled cell growth and proliferation, driving cystogenesis [Citation26,Citation161]. It is thought that in a ‘normal’ environment, mTOR receptor 1 (mTORC1) is negatively regulated by ciliary flow sensing, with aberrant flow sensing leading to mTOR activation, which further downregulates PC1 [Citation26,Citation242,Citation243]. Rapamycin (alias sirolimus) and everolimus are mTOR complex 1 inhibitors. Rapamycin has shown to decrease cystogenesis and cell proliferation in models of renal ciliopathies, including NPHP zebrafish knockdown models and ADPKD, ARPKD, and NPHP murine models [Citation28,Citation161–167,Citation172,Citation244]. In murine models of ADPKD both rapamycin and mTOR kinase inhibitor (TORKi) caused a similar decreased cystic kidney burden and improved kidney function, associated with decrease mTOR and cell proliferation [Citation171]. In Nphp3 mouse models, rapamycin did not affect initial kidney cyst development, but reduced kidney cyst size and kidney fibrosis, indicating that it might be useful in later stages of NPHP [Citation27]. mTOR inhibitors have been studied in human PKD patients. ADPKD patients that have received a renal transplant, but retain the cystic kidney, are often given rapamycin as an immunosuppressant, to prevent transplant rejection. It was shown that rapamycin treatment showed a trend toward reduced kidney volumes of the remaining polycystic kidney compared to patients on other immunosuppressant treatments [Citation161,Citation168]. However, other clinical trials have not shown as much promise as disease models. Treatment of ADPKD patients with everolimus slowed total increase in kidney volume but not overall renal failure (ClinicalTrials.gov, NCT00414440), and treatment of ADPKD patients with sirolimus did not have an ameliorative effect (ClinicalTrials.gov, NCT00346918) [Citation169,Citation173]. A meta-analysis of 9 clinical trials from 2009–2016 of mTOR inhibitor treatment in ADPKD patients demonstrated no significant renal benefit with an increased risk of associated adverse effects [Citation245]. Use of folate-conjugated forms of rapamycin have been tested in preclinical models, to try and overcome the lack of efficiency in human studies [Citation170]. However, this has not been explored in human studies.
AMPK which localizes to the basal body of cilia functions as a sensor of low ATP levels and subsequently mitochondrion function, and is negatively regulated by CFTR and mTOR [Citation246]. Polycystic disease models have also shown impaired mitochondrial function and are more prone to ER stress and reactive oxygen species due to a shift in energy production from oxidative phosphorylation to glycolysis through inhibition of AMPK [Citation247–249]. Activation of AMPK by metformin reduced cyst growth in several models of disease, and investigated in clinical trials; however, there were concerns that the therapeutic AMPK activation in human kidneys might require a higher oral dose [Citation149,Citation192–196]. Hence, to overcome the non-sufficient efficacy of metformin alone, another drug salsalate, a pro drug dimer of acetyl salicylic acid was identified [Citation197,Citation250]. Salsalate was highly effective in attenuating the renal cysts in the PKD1 mouse models. It had a strong inhibitory effect on NF-κB, activated AMPK, and also down regulated cAMP, and stimulated mitochondrial biogenesis and function [Citation197]. With an excellent oral bioavailability and safety profile in humans, salsalate which has long been approved for use in treatment of arthritis is yet to be tested in clinical trials for ADPKD. Preclinical studies with AMPK agonists may also be useful for ARPKD and NPHP. Mitochondrial dysfunction itself could also be a potential target for treatment of cystic kidney disease. In vivo studies have shown that 2-deoxyglucose can inhibit aerobic glycolysis and subsequently reduce cystic epithelial proliferation, slowing cyst expansion and reducing kidney volume [Citation198]. Inhibition of Ca2+/Calmodulin-dependent protein kinase II (CaMKII) has also been shown to improve ER and oxidative stress, as well as mitochondrial function in jck mice [Citation199].
Setmelanotide, a hypothalamic melanocortin 4 receptor (MC4R) agonist showed potential in a phase 2 clinical study in decreasing the hunger and weight related issues in patients with Bardet-Biedl Syndrome (BBS). A phase 3 clinical trial is currently underway to test the long-term safety and efficacy of Setmelanotide for the treatment of obesity and hyperphagia in individuals with BBS (CliniclaTrials.gov, NCT03746522) [Citation251].
EGF signaling is upregulated in polycystic kidney disease, with EGF receptors (EGFR) overexpressed and mislocalized to the apical surface of renal epithelial cells [Citation252]. EGF activates ERK and RAF/MEK pathways, which drives cystic kidney disease. Tesevatinib, a multikinase inhibitor functions as an inhibitor of c-Src which attenuates the EGFR axis and cAMP pathways, as well as being a potent inhibitor of KDR which is a key mediator of cyst growth [Citation174]. Moreover, Tesevatinib ameliorated the renal cyst progression rodent models of ARPKD and significantly reduced the phosphorylation levels of c-Src, EGFR and ERK1/2 which led to a reduction of MAPK activity and the resultant cellular proliferation. Tesevatinib, is already in phase 2 clinical trial for ADPKD (ClinicalTrials.gov, NCT01559363). It is also currently in phase 1 clinical trial for ARPKD recruiting children aged 5–12 years whose kidney function is at least 50% normal (ClinicalTrials.gov, NCT03096080). Tesevatinib has been selected on the basis that both ADPKD and ARPKD patients share similar pathophysiological features and clinical manifestations in new-borns, children, and young adults. Another tyrosine kinase inhibitor, Bosutinib, has been utilized in clinical trials in ADPKD patients, showing some success in reducing kidney growth but glomerular filtration rate still declined, and gastrointestinal and liver toxicity was observed in patients recruited [Citation176]. Preclinical trials have shown the EGFR inhibitors reduced progression of renal cysts, cyst size and improved renal function in Bpk mouse model and Han:SPRD rat model of NPHP, but not in ADPKD PCK rat model [Citation174,Citation179]. Treatment was associated with increased cAMP, which might actually be beneficial in NPHP diseases, but might explain poor results with ADPKD models. Sorafenib is another multikinase inhibitor which can suppress cystogenesis in vitro in models of ADPKD; although it has not been tested in clinical trials for PKD [Citation180]. MAPK/ERK pathways can also be inhibited by PD184352, which has prevented cyst growth and protect kidney functions in pcy mice and Inv mice, which are models NPHP [Citation181,Citation182].
Somatostatin is a growth hormone inhibitor essential for regulation of growth factors, and inhibition of insulin and glucagon secretion, whilst lowering cAMP availability. Somatostatin G-coupled receptors are expressed in renal tubule cells and cholangiocytes [Citation253–257]. Somatostatin analogs such as lanreotide, octreotide, and pasireotide act in a similar pharmacokinetic action to somatostatin, and have been shown to slow cell proliferation in PKD models [Citation187,Citation258,Citation259]. In vitro spheroid models of NPHP disease treated with octreotide rescued ciliary defects and abnormal spheroid formation, they were also associated with a decrease in cAMP [Citation260]. Treatment of ADPKD patients with octreotide show some potential toward slowing disease progression, particularly in late-stage ADPKD; however, studies have been limited by small cohort size and patients show some adverse gastrointestinal effects [Citation183,Citation188] (ClinicalTrials.gov, NCT01377246, NCT00309283). Treatment with pasireotide in patients with severe polycystic liver disease, associated with polycystic kidney disease, slowed progression of liver and kidney growth, but did not improve glomerular filtration rate (GFR) decline and participants experienced increased frequency of adverse effects including diabetes and hyperglycemia (ClinicalTrials.gov, NCT01670110) [Citation191]. However, there is conflicting evidence in the effectiveness in using lanreotide treatment to slow declining kidney function and kidney enlargement in ADPKD (ClinicalTrials.gov, NCT01354405, NCT01616927) [Citation183–186]. Recently, a pilot clinical trial with tolvaptan and octreotide was shown to reduce GFR more effectively than octreotide alone, and significantly reduced cystic kidney volume and ameliorate the aquaretic effect of tolvaptan (ClinicalTrials.gov, NCT03541447) [Citation189].
Recently, prostaglandin receptor 2 (PGE2) agonists rescued the NPHP1-associated ciliary and kidney phenotype in vitro and in vivo, mediated by increased downstream cAMP signaling which is often reduced in NPHP ciliopathy models, and decreased fibrosis [Citation200]. This is an exciting potential therapeutic target for NPHP.
5.5. Targeting angiogenesis
Increased angiogenesis in early disease progression of ADPKD, with increased expression of vascular endothelial growth factor receptor (VEGFR) in endothelial cells is thought to drive cystogenesis [Citation261]. VEGFR mRNA inhibition in vivo models of NPHP led to reduced tubule cell proliferation and subsequently lower cystogenesis and recovered renal function [Citation201].
5.6. Targeting cilium disassembly
Histone deacetylase 6 (HDAC6) is an enzyme which mediates cilium disassembly, it is also associated with the heat shock protein 90 (HSP90) [Citation262–264]. In PC1 deficient cells, HDAC6 and HSP90 are upregulated; inhibition of HDAC6 restores the ciliary phenotype [Citation203]. Preclinical studies have shown that inhibition of HDAC6 in PKD models with ACY-1215 slows cyst growth and reduces cystic volume, associated with reduction in cAMP and CFTR activity. Other HDAC6 inhibitors including; Tubacin, Trichostatin A (TSA), Valproic acid (VPA) and Nicotinamide ameliorates cystic kidney disease, reduces cAMP expression and improve kidney function in ADPKD preclinical studies [Citation204–207,Citation265]. There have been small pilot studies of nicotinamide treatment in ADPKD patients, which did not show any difference between drug and placebo (ClinicalTrials.gov, NCT02558595). Another potential target is HSP90; inhibition of HSP90 with ganetespib or STA-2842 in ADPKD mouse models also slowed cyst growth, alongside reduced ciliation [Citation208,Citation209].
5.7. Targeting ciliary signaling
The primary cilium is a vital transducer of Hh signaling, which is required for proper kidney development and renal epithelial cell proliferation [Citation266,Citation267]. Hh signaling is also involved in proliferation of myofibroblasts which secrete ECM components, increasing fibrosis [Citation19]. Hh signaling is dysregulated in numerous ciliopathies [Citation268,Citation269], with lots of evidence pointing toward dysregulation in NPHP [Citation17,Citation18,Citation116]. CEP290 mutated spheroids show decreased Hh signaling; treatment with Purmorphamine, a Hh agonist, restores 3D spheroid structure [Citation116]. However, Pkd1, Nphp9 and Ttc21b/Ift39 murine models have increased Hh signaling, and inhibition of GLI and SMO with small molecular antagonists Gantb1 and Sant2 respectively, prevented renal cyst formation [Citation202]. It is likely that the direction of Hh dysregulation will be dependent on the genetic cause of disease, which will change the type of treatment required. However, as Hh signaling is a core developmental and homeostatic pathway, any manipulation of Hh signaling is thought to be likely to lead to the risk of serious side effects [Citation24].
There are other ciliary-mediated signaling pathways that are dysregulated in renal ciliopathies some including cAMP, calcium-sensing receptor activation, canonical Wnt and Hippo [Citation21,Citation270–285]. Direct modulation of these pathways in preclinical studies have shown promise at ameliorating the disease phenotype, but they have not yet progressed to clinical trials [Citation274,Citation277,Citation278,Citation284,Citation286,Citation287].
5.8. Targeting cell cycle regulation/DNA damage response
A major phenotype driving cystogenesis in renal ciliopathies is hyperproliferation of epithelial cells lining the cysts. Cyclin dependent kinases (CDK) regulate the cell cycle. Preclinical studies with murine models of ADPKD, ARPKD and NPHP, have used treatment with roscovitine, a reversible inhibitor of CDK1,2,5,7 and 9, to attenuate hyperproliferation by inducing G1 AND G2-M cell cycle arrest. This causes slower kidney functional decline and cystogenesis [Citation30,Citation210–213,Citation288]. Other CDK inhibitors have rescued ciliation in Cep290 KO cells [Citation214]. CDK inhibitors are currently being used in clinical trials for numerous cancer treatments, and other diseases such as cystic fibrosis, inflammatory bowel disease and ulcerative colitis, but have not been used in renal ciliopathy trials. Cell division cycle 25 A (Cdc25A) is overexpressed in PKD animal models; inhibition of Cdc25A with menadione (vitamin K3) slows cystogenesis in rodent models of PKD [Citation215].
DNA damage repair (DDR) pathways detect DNA damage and activates either repair mechanisms, senescence, or apoptosis. This is vital for removal of severely damaged cells and regulation of tissue repair. Multiple ciliary proteins have been implicated in DDR including CEP290, CEP164, ZNF243, NEK8, SDCCAG8 and MAPKBP1 knockout [Citation81,Citation88,Citation214,Citation288,Citation289]. Elevated DNA damage and replicative stress are found in models of renal ciliopathies. It is hypothesized that aberrant DDR leads to leads to increased DNA damage and impaired cell cycle regulation, driving increased proliferation and misregulated apoptosis [Citation81,Citation237]. Specifically, Cep290 murine models show primary mouse kidney cells with increased double-strand breaks, supernumerary centrioles and replication fork asymmetry, indicative of replicative stress; CDK inhibitors can rescue these phenotypes in vitro [Citation214]. Cep164 KD in HeLa and IMCD3 cells is also thought to cause increased DNA damage, genomic instability and replicative stress, although results have been conflicting with some studies showing no different in the cell cycle, checkpoint activation or DNA damage [Citation30,Citation31,Citation81,Citation290–292]. It is possible that ciliopathy proteins effect DDR in a cell specific manner. Abnormal DDR also contributes to increased fibrosis, which is a characteristic of NPHP. Cep164 KD in IMCD3 cells induces DNA damage, with s-phase block and anaphase lag, as well as increased epithelial-mesenchymal transition [Citation81]. More research is needed to understand the role of ciliopathy proteins in DDR, and how this may be exploited for therapeutic purposes.
5.9. Utilizing gene therapy
Aberrant activation of microRNAs, short non-coding RNAs, has been shown to promote the progression of ADPKD [Citation216,Citation293]. MicroRNA17 (miR-17) induces kidney cysts in PKD murine models via decreased PC1 and PC2 expression, which also has effects on mitochondrial function to promote cyst growth [Citation294,Citation295]. miR-17 is upregulated in ADPKD murine models, deletion of which reduces cyst growth, fibrosis, and inflammation, and improves mitochondrial function in a PKD mouse model [Citation216]. In a recent study Lee et al. reported that RGLS4326, a short oligonucleotide inhibitor of miR-17 significantly attenuated the cyst growth in vitro in cell culture models, and also in vivo in the PKD mouse models by upregulating Pkd1 and Pkd2 expression [Citation217]. A phase 1 clinical study of RGLS4326 in patients with ADPKD is currently under way (ClinicalTrials.gov, NCT04536688) [Citation218]. Other miRNAs are known to be involved in kidney cyst growth regulation, including miR-21, miR-193 and miR-214. The role of miRNA in ARPKD and NPHP is yet to be explored.
Another potential personalized therapeutic technique is to ‘correct’ the causative genetic defect. Antisense oligonucleotides (ASOs) can be used to bind to target mRNA to alter mRNA expression or splicing; ASOs have been shown to distribute to the kidney tubule [Citation24,Citation41]. ASOs have been utilized in models of Cep290 ciliopathies to skip over the effected in-frame exon, subsequently rescuing the ciliary and renal cystogenesis phenotype [Citation219]. This has also been completed for murine models of Alport nephropathy [Citation296,Citation297]. ASO therapies are currently showing promise in clinical trials for retinal ciliopathies, although they have not been utilized in clinical trials for renal ciliopathies [Citation298–301]. ASOs have also been utilized to regulate genetic modifiers of renal ciliopathies, such as BSND to slow the cystogenesis phenotype models of Cep290 ciliopathy disease [Citation220]. Modifier alleles are likely contribute to the heterogeneity of renal ciliopathies, and could offer a personalized treatment approach.
6. Expert opinion
Currently, for renal ciliopathies, the only approved treatment for slowing disease progression is tolvaptan for ADPKD, utilizing a very specific target, the AVPR2, expressed in the renal collecting duct. All other forms of treatment at present are merely supportive, aimed at treatment of anemia, hypertension, growth retardation, and other symptoms related to CKD. Currently kidney transplantation is the only potential long-term cure [Citation302]. There are, however, promising clinical trials for both adults and children with ADPKD and ARPKD including tolvaptan, pioglitazone, pravastatin, simvastatin, RGLS4326, bardoxolone methyl, metformin, lanreotide, octreotide, pasireotide, tesevatinib, and bosutinib. These drugs are clearly targeting many different aspects of the disease pathogenesis including cyst expansion and cell proliferation. The discovery of tolvaptan as an effective agent for ADPKD by using preclinical models including the pcy mouse (which has a Nphp3 mutation) gives hope that disease mechanisms and therefore therapeutic interventions may be shared across different ciliopathies, and from dominant to recessive forms of cystic kidney disease and nephronophthisis.
A common final pathway associated with many forms of CKD is renal fibrosis. Therefore, there remains the possibility of using drugs directed at this disease course, rather than a ciliopathy specific pathway/mechanism for the treatment of NPHP and other related renal ciliopathies. Recently, treatment of CKD related to diabetes has been revolutionized by two classes of drugs: the sodium-glucose transporter-2 (SGLT2) inhibitors [Citation303–305] and mineralocorticoid receptor antagonists (MRA) [Citation306].
SGLT2i drugs now have a proven track record in CKD patients with and without diabetes in reducing glomerular hypertension mediated through tubuloglomerular feedback independent of their effect on glycemic control. They remain the most exciting therapeutic advance in the treatment of kidney disease and as nephroprotective agents since the discovery of angiotensin converting enzyme (ACE) inhibitors. They remain, however, untested in ADPKD patients as such patients were excluded from most clinical trials exploring kidney protection [Citation307]. Intriguingly, SGLT2i may have anti-inflammatory role as well as impacting autophagy and improving mitochondrial dysfunction [Citation308,Citation309]. We propose that establishing the potential benefit of SGLT2i in patients with ADPKD is a research priority.
MRA drugs such as spironolactone have been available as antihypertensive agents for many years. However, recently attention has been focused on finerenone. This is relevant as finerenone has been studied for its potential to reduce fibrosis, particularly in the context of CKD. As a selective MRA, finerenone works by blocking the activity of the mineralocorticoid receptor, which is involved in regulating salt and water balance as well as promoting inflammation and fibrosis. By inhibiting this receptor, finerenone may reduce the harmful effects associated with excessive fibrosis in CKD. Clinical studies, such as the FIDELIO-DKD trials, have evaluated the effects of finerenone on kidney outcomes in patients with CKD and type 2 diabetes [Citation310]. This trial showed that finerenone treatment reduced the risk of kidney failure, including the need for dialysis or transplantation, in these patients. Additionally, it demonstrated a decrease in the progression of albuminuria and a reduction in cardiovascular events. While the precise mechanisms through which finerenone reduces fibrosis are not yet fully understood, it is believed that by targeting the mineralocorticoid receptor, the drug can mitigate the inflammation and fibrotic processes in the kidneys, potentially slowing down the progression of fibrosis and preserving kidney function. It therefore becomes a very attractive drug to slow down the tubulointerstitial fibrosis associated with renal ciliopathies, independent of the ciliary defect. Further work in relevant pre-clinical models is required as well as designing future clinical trials for testing this drug in relevant phenotypic groups. The use of biomarkers for renal fibrosis may aid the assessment of these agents [Citation130].
We have already discussed briefly the use of anti-miR-17 oligonucleotides for the treatment of PKD in preclinical studies. Again, following the theme of agents to prevent renal fibrosis, anti-miR-21 oligonucleotide therapies have been applied to animal models of renal fibrosis, specifically the Col4a3-/- Alport syndrome mice [Citation311]. Use of anti-miR-21 drugs provided enough evidence for a clinical trial in Alport syndrome patients (called HERA) to assess the efficacy of lademirsen (an anti miR-21 agent) in reducing the decline in renal function [Citation312]. This clinical study was unfortunately terminated early, after 24 weeks, as it did not lead to a meaningful improvement in kidney function when compared with placebo. Targeting miR-21 may still represent a promising therapy for other kidney diseases, aside from Alport syndrome, but pharmaceutical companies may be wary of this agent given the disappointment of the HERA trial. Targeting kidney fibrosis remains a promising approach [Citation313]. But, any future drug targeting fibrosis of the kidneys will now need to demonstrate added benefit to a standard of care that combines renin-angiotensin system with MRA or SGLT2 inhibitors.
Notably, there are currently no clinical trials underway for NPHP using small molecules, such as rapamycin in spite of the in vitro and in vivo evidence. This could be explained by the fact that there is severe phenotypic and genotypic heterogeneity in NPHP patients and relatively shorter therapeutic window compared to ADPKD [Citation23]. There is a need, however, to define better disease progression in this group of patients so that clinical trial ready cohorts can be identified. For NPHP, targeting cilia-related signaling pathways seems to be the most promising approach. Manipulating ciliary signaling and downstream pathways may represent druggable targets for the treatment of renal ciliopathies.
Overall, the development of new treatments for renal ciliopathies is an active area of research, and advances in gene therapy, stem cell therapy, and drug development hold promise for improving outcomes for patients with these disorders. There is a recognized need for the development of longitudinal cohorts of well-phenotyped and genotyped renal ciliopathy patients with enriched clinical and genomic datasets to underpin future clinical trials of precision therapies and personalized medicine approaches. This will allow cohorts of patients to be well characterized and to be “trial-ready” for novel therapies and other disease interventions. As an example, in the UK, longitudinal data is already captured by the well-established UK Renal Registry and National Registry of Rare Kidney Diseases (RaDaR) platform. RaDaR currently has 242 participants registered with NPHP or ARPKD and 8006 with ADPKD, constituting among the largest cohorts in these disorders reported anywhere in the world. We also believe that by the renal ciliopathy research community interacting with well-organized patient networks this will empower patients, utilize patient and public involvement into design of clinical trials (industry), and allow patient focused research to become truly translational.
7. Conclusions
In conclusion, renal ciliopathies pose a significant challenge to healthcare professionals, as current treatments are largely supportive, and a long-term cure is only possible through kidney transplantation. However, recent clinical trials have shown promising results for drugs such as tolvaptan, pioglitazone, and bardoxolone methyl and clinical indications for use of these agents will hopefully broaden to pediatric disease settings for ADPKD and ARPKD. Although there are no ongoing trials for NPHP, there is recognition that patient cohorts need to be identified and characterized so that they will be trial ready. Across the spectrum of renal ciliopathies, preclinical models have provided insight into the understanding the function of PKD- and NPHP-related proteins, highlighting their ciliary and extraciliary functions. Dissecting further their role in cellular biology is a prerequisite to developing novel specific therapies for these complex disorders.
Article highlights
Renal ciliopathies include a range of inherited kidney diseases, including polycystic kidney disease and nephronophthisis, which lead to progressive chronic kidney disease and kidney failure.
There is a need for improved treatments for all renal ciliopathies.
Targeting kidney cyst growth has proved to be useful in autosomal dominant polycystic kidney disease in adults.
New treatments for childhood onset diseases including autosomal recessive polycystic kidney disease and nephronophthisis are urgently needed as there are no current therapies.
Pre-clinical models of renal ciliopathies provide a diverse set of disease mechanisms to target and these insights need to be urgently translated into clinical care.
Harmonizing diagnostic, investigative, and management approaches for renal ciliopathy patients will allow patient cohorts to become clinical trial ready and allow new therapeutic agents to enter clinical practice.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants, or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017 Sep;18(9):533–547.
- Sonnen KF, Schermelleh L, Leonhardt H, et al. 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol Open. 2012 Oct 15;1(10):965–976. DOI:10.1242/bio.20122337
- Andersen JS, Wilkinson CJ, Mayor T, et al. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003 Dec 04;426(6966):570–574. DOI:10.1038/nature02166
- Cajanek L, Nigg EA. Cep164 triggers ciliogenesis by recruiting Tau tubulin kinase 2 to the mother centriole. Proc Natl Acad Sci U S A. 2014 Jul 15;111(28):E2841–50. DOI:10.1073/pnas.1401777111
- Yang, Yang TT, Chong WM, et al. Super-resolution architecture of mammalian centriole distal appendages reveals distinct blade and matrix functional components. Nat Commun. 2018;9(1). DOI:10.1038/s41467-018-04469-1
- Shi X, Garcia G 3rd, Van De Weghe JC, et al. Super-resolution microscopy reveals that disruption of ciliary transition-zone architecture causes Joubert syndrome. Nat Cell Biol. 2017 Oct;19(10):1178–1188.
- Reiter JF, Blacque OE, Leroux MR. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012 Jun 29;13(7):608–618. DOI:10.1038/embor.2012.73
- Hu Q, Milenkovic L, Jin H, et al. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 2010 Jul;329(5990):436–439.
- Kee HL, Dishinger JF, Blasius TL, et al. A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat Cell Biol. 2012 Mar;14(4):431–437.
- Yang TT, Su J, Wang WJ, et al. Superresolution pattern recognition reveals the architectural map of the ciliary transition zone. Sci Rep. 2015 Sep 14;5(1):14096. DOI:10.1038/srep14096
- Nachury MV, Loktev AV, Zhang Q, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007 Jun 15;129(6):1201–1213. DOI:10.1016/j.cell.2007.03.053
- Nakayama K, Katoh Y. Ciliary protein trafficking mediated by IFT and BBSome complexes with the aid of kinesin-2 and dynein-2 motors. J Biochem. 2018 Mar 1;163(3):155–164. DOI:10.1093/jb/mvx087
- Czarnecki PG, Shah JV. The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol. 2012 Apr;22(4):201–210.
- Alkanderi S, Molinari E, Shaheen R, et al. ARL3 mutations cause Joubert syndrome by disrupting ciliary protein composition. Am J Hum Genet. 2018 Oct 4;103(4):612–620. DOI:10.1016/j.ajhg.2018.08.015
- Smith CEL, Lake AVR, Johnson CA. Primary cilia, ciliogenesis and the actin cytoskeleton: a little less resorption, a little more actin please. Front Cell Dev Biol. 2020;8:622822.
- Gigante ED, Caspary T. Signaling in the primary cilium through the lens of the Hedgehog pathway. Wiley Interdiscip Rev Dev Biol. 2020 Nov;9(6):e377.
- Breslow DK, Hoogendoorn S, Kopp AR, et al. A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat Genet. 2018 Mar;50(3):460–471. DOI:10.1038/s41588-018-0054-7
- Hynes AM, Giles RH, Srivastava S, et al. Murine Joubert syndrome reveals Hedgehog signaling defects as a potential therapeutic target for nephronophthisis. P Natl Acad Sci USA. 2014 Jul 8;111(27):9893–9898. DOI:10.1073/pnas.1322373111
- Attanasio M, Uhlenhaut NH, Sousa VH, et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet. 2007 Aug;39(8):1018–1024.
- Anvarian Z, Mykytyn K, Mukhopadhyay S, et al. Cellular signalling by primary cilia in development, organ function and disease. Nat rev Nephrol. 2019 Apr;15(4):199–219.
- Fischer E, Legue E, Doyen A, et al. Defective planar cell polarity in polycystic kidney disease. Nat Genet. 2006 Jan;38(1):21–23.
- Lancaster MA, Louie CM, Silhavy JL, et al. Impaired Wnt-beta-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat Med. 2009 Sep;15(9):1046–1054.
- Stokman MF, Saunier S, Benmerah A. Renal ciliopathies: sorting out therapeutic approaches for nephronophthisis. Front Cell Dev Biol. 2021;9:653138.
- Molinari E, Sayer JA. Emerging treatments and personalised medicine for ciliopathies associated with cystic kidney disease. Expert Opin Orphan Drugs. 2017;5(10):785–798.
- Lienkamp S, Ganner A, Walz G. Inversin, Wnt signaling and primary cilia. Differentiation. 2012 Feb;83(2):S49–55.
- Boehlke C, Kotsis F, Patel V, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol. 2010 Nov;12(11):1115–1122.
- Gattone VH, Sinders RM, Hornberger TA, et al. Late progression of renal pathology and cyst enlargement is reduced by rapamycin in a mouse model of nephronophthisis. Kidney Int. 2009 Jul;76(2):178–182.
- Wahl PR, Serra AL, Le Hir M, et al. Inhibition of mTOR with sirolimus slows disease progression in Han : SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transpl. 2006 Mar;21(3):598–604.
- Ishikawa H, Marshall WF. Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol. 2011 Apr;12(4):222–234.
- Slaats GG, Ghosh AK, Falke LL, et al. Nephronophthisis-associated CEP164 regulates cell cycle progression, apoptosis and epithelial-to-mesenchymal transition. PLoS Genet. 2014 Oct;10(10):e1004594.
- Sivasubramaniam S, Sun X, Pan YR, et al. Cep164 is a mediator protein required for the maintenance of genomic stability through modulation of MDC1, RPA, and CHK1. Genes Dev. 2008 Mar 01;22(5):587–600. DOI:10.1101/gad.1627708
- Bachmann-Gagescu R, Neuhauss SC. The photoreceptor cilium and its diseases. Curr Opin Genet Dev. 2019 Jun;56:22–33.
- May-Simera H. The sensory antennae in the eye. Prog Retin Eye Res. 2017;60:144–180. DOI:10.1016/j.preteyeres.2017.05.001
- Jenkins PM, McEwen DP, Martens JR. Olfactory cilia: linking sensory cilia function and human disease. Chem Senses. 2009 Jun;34(5):451–464.
- Mitchison HM, Valente EM. Motile and non-motile cilia in human pathology: from function to phenotypes. J Pathol. 2017 Jan;241(2):294–309.
- Little RB, Norris DP. Right, left and cilia: How asymmetry is established. Semin Cell Dev Biol. 2021 Feb;110:11–18.
- Wheway G, Schmidts M, Mans DA, et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat Cell Biol. 2015 Aug;17(8):1074–1087.
- McConnachie DJ, Stow JL, Mallett AJ. Ciliopathies and the Kidney: A Review. Am J Kidney Dis. 2021 Mar;77(3):410–419.
- Ciliopathy Alliance. 2020. Accessed 01 05 2023. Available from: https://ciliopathyalliance.org/
- Devlin LA, Sayer JA. Renal ciliopathies. Curr Opin Genet Dev. 2019 Jun;56:49–60.
- Barroso-Gil M, Olinger E, Sayer JA. Molecular genetics of renal ciliopathies. Biochem Soc Trans. 2021 Jun 30;49(3):1205–1220. DOI:10.1042/BST20200791
- Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919–935. DOI:10.1016/S0140-6736(18)32782-X
- Lanktree MB, Haghighi A, Guiard E, et al. Prevalence estimates of polycystic kidney and liver disease by population sequencing. J Am Soc Nephrol. 2018 Oct;29(10):2593–2600.
- Lanktree MB, Haghighi A, di Bari I, et al. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol. 2021 May 8;16(5):790–799. DOI:10.2215/CJN.02320220
- Duong Phu M, Bross S, Burkhalter MD, et al. Limitations and opportunities in the pharmacotherapy of ciliopathies. Pharmacol Ther. 2021 Sep;225:107841.
- Chapin HC, Caplan MJ. The cell biology of polycystic kidney disease. J Cell Bio. 2010 Nov 15;191(4):701–710. DOI:10.1083/jcb.201006173
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007 Jul;18(7):2143–2160.
- Su Q, Hu F, Ge X, et al. Structure of the human PKD1-PKD2 complex. Science. 2018 Sep 7;361(6406). DOI:10.1126/science.aat9819
- Ta CM, Vien TN, Ng LCT, et al. Structure and function of polycystin channels in primary cilia. Cell Signal. 2020 Aug;72:109626.
- Wang SX, Zhang JJ, Nauli SM, et al. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol. 2007 Apr;27(8):3241–3252.
- Delling M, Indzhykulian AA, Liu X, et al. Primary cilia are not calcium-responsive mechanosensors. Nature. 2016 Mar 31;531(7596):656–660. DOI:10.1038/nature17426
- Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50. DOI:10.1038/s41572-018-0047-y
- Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015 Jan;26(1):160–172.
- McEwan P, Bennett Wilton H, Ong ACM, et al. A model to predict disease progression in patients with autosomal dominant polycystic kidney disease (ADPKD): the ADPKD outcomes model. BMC Nephrol. 2018 Feb 13;19(1):37. DOI:10.1186/s12882-017-0804-2
- Edwards ME, Blais JD, Czerwiec FS, et al. Standardizing total kidney volume measurements for clinical trials of autosomal dominant polycystic kidney disease. Clin Kidney J. 2019 Feb;12(1):71–77.
- Rossetti S, Kubly VJ, Consugar MB, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009 Apr;75(8):848–855.
- Qian F, Germino GG. “Mistakes Happen”: Somatic mutation and disease. Am J Hum Genet. 1997 Nov;61(5):1000–1005.
- Faguer S, Decramer S, Chassaing N, et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int. 2011 Oct;80(7):768–776.
- Cornec-Le Gall E, Olson RJ, Besse W, et al. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet. 2018 May 3;102(5):832–844. DOI:10.1016/j.ajhg.2018.03.013
- Huynh VT, Audrezet MP, Sayer JA, et al. Clinical spectrum, prognosis and estimated prevalence of DNAJB11-kidney disease. Kidney Int. 2020 Aug;98(2):476–487.
- Porath B, Gainullin VG, Cornec-Le Gall E, et al. Mutations in GANAB , encoding the glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet. 2016 Jun 2;98(6):1193–1207. DOI:10.1016/j.ajhg.2016.05.004
- Besse W, Choi J, Ahram D, et al. A noncoding variant in GANAB explains isolated polycystic liver disease (PCLD) in a large family. Hum Mutat. 2018 Mar;39(3):378–382.
- Besse W, Dong K, Choi J, et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest. 2017 Sep 1;127(9):3558. DOI:10.1172/JCI96729
- Besse W, Chang AR, Luo JZ, et al. ALG9 mutation carriers develop kidney and liver cysts. J Am Soc Nephrol. 2019 Nov;30(11):2091–2102.
- Li A, Davila S, Furu L, et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003 Mar;72(3):691–703.
- Bolar NA, Golzio C, Zivna M, et al. Heterozygous loss-of-function SEC61A1 mutations cause autosomal-dominant tubulo-interstitial and glomerulocystic kidney disease with anemia. Am J Hum Genet. 2016 Jul 7;99(1):174–187. DOI:10.1016/j.ajhg.2016.05.028
- Senum SR, Li Y(M, Benson KA, et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am J Hum Genet. 2022 Jan 6;109(1):136–156. DOI:10.1016/j.ajhg.2021.11.016
- Devuyst O, Knoers NV, Remuzzi G, et al. Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet. 2014 May 24;383(9931):1844–1859. DOI:10.1016/S0140-6736(14)60659-0
- Burgmaier K, Kilian S, Bammens B, et al. Clinical courses and complications of young adults with Autosomal Recessive Polycystic Kidney Disease (ARPKD). Sci Rep. 2019 May 28;9(1):7919. DOI:10.1038/s41598-019-43488-w
- Goetz SC, Bangs F, Barrington CL, et al. The Meckel syndrome- associated protein MKS1 functionally interacts with components of the BBSome and IFT complexes to mediate ciliary trafficking and hedgehog signaling. PLoS ONE. 2017;12(3):e0173399. DOI:10.1371/journal.pone.0173399
- Molinari E, Srivastava S, Dewhurst RM, et al. Use of patient derived urine renal epithelial cells to confirm pathogenicity of PKHD1 alleles. BMC Nephrol. 2020 Oct 15;21(1):435. DOI:10.1186/s12882-020-02094-z
- Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002 Mar;30(3):259–269.
- Sweeney WE Jr., Avner ED. Molecular and cellular pathophysiology of autosomal recessive polycystic kidney disease (ARPKD). Cell Tissue Res. 2006 Dec;326(3):671–685.
- Lu H, Galeano MCR, Ott E, et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat Genet. 2017 Jul;49(7):1025–1034.
- Devane J, Ott E, Olinger EG, et al. Progressive liver, kidney, and heart degeneration in children and adults affected by TULP3 mutations. Am J Hum Genet. 2022 May 5;109(5):928–943. DOI:10.1016/j.ajhg.2022.03.015
- Srivastava S, Molinari E, Raman S, et al. Many genes-one disease? Genetics of Nephronophthisis (NPHP) and NPHP-associated disorders. Front Pediatr. 2017 ;5:287.
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009 Jan;20(1):23–35.
- Snoek R, van Setten J, Keating BJ, et al. NPHP1 (Nephrocystin-1) Gene deletions cause adult-onset ESRD. J Am Soc Nephrol. 2018 Jun;29(6):1772–1779.
- Hoefele J, Nayir A, Chaki M, et al. Pseudodominant inheritance of nephronophthisis caused by a homozygous NPHP1 deletion. Pediatr Nephrol. 2011 Jun;26(6):967–971.
- Salomon R, Saunier S, Niaudet PN. Nephronophthisis. Pediatr Nephrol. 2009 Dec;24(12):2333–2344.
- Chaki M, Airik R, Ghosh AK, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012 Aug 3;150(3):533–548. DOI:10.1016/j.cell.2012.06.028
- Otto EA, Schermer B, Obara T, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003 Aug;34(4):413–420.
- Tory K, Rousset-Rouviere C, Gubler MC, et al. Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int. 2009 Apr;75(8):839–847.
- Failler M, Gee HY, Krug P, et al. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am J Hum Genet. 2014 Jun 5;94(6):905–914. DOI:10.1016/j.ajhg.2014.05.002
- Otto EA, Trapp ML, Schultheiss UT, et al. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol. 2008 Mar;19(3):587–592.
- Olbrich H, Fliegauf M, Hoefele J, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet. 2003 Aug;34(4):455–459.
- Georges B, Cosyns JP, Dahan K, et al. Late-onset renal failure in Senior-Loken syndrome. Am J Kidney Dis. 2000 Dec;36(6):1271–1275.
- Macia MS, Halbritter J, Delous M, et al. Mutations in MAPKBP1 cause juvenile or late-onset cilia-independent nephronophthisis. Am J Hum Genet. 2017 Feb 2;100(2):372. DOI:10.1016/j.ajhg.2017.01.025
- Shamseldin HE, Shaheen R, Ewida N, et al. The morbid genome of ciliopathies: an update. Genet Med. 2020 Jun;22(6):1051–1060.
- Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol. 2017 Mar 1;9(3):a028191. DOI:10.1101/cshperspect.a028191
- Parisi M, Glass I. Joubert syndrome. In: Adam M, Mirzaa GM, and Pagon RA, editors. GeneReviews(R). Seattle (WA): University of Washington, Seattle; 1993-2023. https://www.ncbi.nlm.nih.gov/books/NBK1325/
- Brancati F, Dallapiccola B, Valente EM. Joubert syndrome and related disorders. Orphanet J Rare Dis. 2010 Jul;5:20.
- Parisi MA. The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity. Trans Sci Rare Dis. 2019 Jul 4;4(1–2):25–49.
- Radha Rama Devi A, Naushad SM, Lingappa L. Clinical and molecular diagnosis of Joubert syndrome and related disorders. Pediatr Neurol. 2020 May;106:43–49.
- Forsythe E, Kenny J, Bacchelli C, et al. Managing Bardet-Biedl syndrome-now and in the future. Front Pediatr. 2018 ;6:23.
- Maria M, Lamers IJ, Schmidts M, et al. Genetic and clinical characterization of Pakistani families with Bardet-Biedl syndrome extends the genetic and phenotypic spectrum. Sci Rep. 2016 Oct 06;6:34764.
- Beales PL, Elcioglu N, Woolf AS, et al. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999 Jun;36(6):437–446.
- Putoux A, Attie-Bitach T, Martinovic J, et al. Phenotypic variability of Bardet-Biedl syndrome: focusing on the kidney. Pediatr Nephrol. 2012 Jan;27(1):7–15.
- Forsythe E, Sparks K, Best S, et al. Risk factors for severe renal disease in bardet-biedl syndrome. J Am Soc Nephrol. 2017 Mar;28(3):963–970.
- Alliance C. Ciliopathy alliance: bardet-biedl syndrome. 2020 [01 Mar 2023] Available from: https://ciliopathyalliance.org/ciliopathies
- Barbelanne M, Hossain D, Chan DP, et al. Nephrocystin proteins NPHP5 and Cep290 regulate BBSome integrity, ciliary trafficking and cargo delivery. Hum Mol Genet. 2015 Apr 15;24(8):2185–2200.
- UK B What is bardet biedl syndrome. 2020 [01 Mar 2023] Available from: https://bbsuk.org.uk/about-bbs-uk/bardet-biedl-syndrome/
- Seo S, Baye LM, Schulz NP, et al. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc Natl Acad Sci U S A. 2010 Jan 26;107(4):1488–1493. DOI:10.1073/pnas.0910268107
- Otto EA, Loeys B, Khanna H, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005 Mar;37(3):282–288.
- Luo F, Tao YH. Nephronophthisis: A review of genotype-phenotype correlation. Nephrology (Carlton). 2018 Oct;23(10):904–911.
- Hartill V, Szymanska K, Sharif SM, et al. Meckel-Gruber Syndrome: An Update on Diagnosis, Clinical Management, and Research Advances. Front Pediatr. 2017 ;5:244.
- Humbert MC, Weihbrecht K, Searby CC, et al. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci U S A. 2012 Nov 27;109(48):19691–19696.
- Field M, Scheffer IE, Gill D, et al. Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations. Eur J Hum Genet. 2012 Jul;20(7):806–809.
- Wheway G, Mitchison HM, Genomics England Research C. Opportunities and Challenges for Molecular Understanding of Ciliopathies-The 100,000 Genomes Project. Front Genet. 2019;10:127.
- Rafferty KA Jr., Sherwin RW. The length of secondary chromosomal constrictions in normal individuals and in a nucleolar mutant of Xenopus laevis. Cytogenetics. 1969;8(6):427–438.
- Graham FL, Smiley J, Russell WC, et al. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977 Jul;36(1):59–74.
- Perantoni A, Berman JJ. Properties of Wilms’ tumor line (TuWi) and pig kidney line (LLC-PK1) typical of normal kidney tubular epithelium. In Vitro. 1979 Jun;15(6):446–454.
- Rauchman MI, Nigam SK, Delpire E, et al. An osmotically tolerant inner medullary collecting duct cell line from an SV40 transgenic mouse. Am J physiol. 1993 Sep;265(3 Pt 2):F416–24.
- Gaush CR, Hard WL, Smith TF. Characterization of an established line of canine kidney cells (MDCK). Proc Soc Exp Biol Med. 1966 Jul;122(3):931–935.
- Molinari E, Decker E, Mabillard H, et al. Human urine-derived renal epithelial cells provide insights into kidney-specific alternate splicing variants. Eur J Hum Genet. 2018 Dec;26(12):1791–1796.
- Srivastava S, Ramsbottom SA, Molinari E, et al. A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies. Hum Mol Genet. 2017;26(23):4657–4667.
- Forbes TA, Howden SE, Lawlor K, et al. Patient-iPSC-derived kidney organoids show functional validation of a ciliopathic renal phenotype and reveal underlying pathogenetic mechanisms. Am J Hum Genet. 2018 May 3;102(5):816–831.
- Schutgens F, Rookmaaker MB, Margaritis T, et al. Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat Biotechnol. 2019 Mar;37(3):303–313.
- Kuraoka S, Tanigawa S, Taguchi A, et al. PKD1-dependent renal cystogenesis in human induced pluripotent stem cell-derived ureteric bud/collecting duct organoids. J Am Soc Nephrol. 2020 Oct;31(10):2355–2371.
- Yousef Yengej FA, Jansen J, Rookmaaker MB, et al. Kidney organoids and tubuloids. Cells. 2020 May 26;9(6):1326.
- Uchimura K, Wu H, Yoshimura Y, et al. Human pluripotent stem cell-derived kidney organoids with improved collecting duct maturation and injury modeling. Cell Rep. 2020 Dec 15;33(11):108514. DOI:10.1016/j.celrep.2020.108514
- Cruz NM, Song X, Czerniecki SM, et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat Mater. 2017 Nov;16(11):1112–1119.
- Low JH, Li P, Chew EGY, et al. Generation of human PSC-derived kidney organoids with patterned nephron segments and a de novo vascular network. Cell Stem Cell. 2019 Sep 5;25(3):373–387 e9. DOI:10.1016/j.stem.2019.06.009
- Steichen C, Giraud S, Hauet T. Combining kidney organoids and genome editing technologies for a better understanding of physiopathological mechanisms of renal diseases: state of the art. Front Med. 2020;7:10.
- Ashammakhi N, Wesseling-Perry K, Hasan A, et al. Kidney-on-a-chip: untapped opportunities. Kidney Int. 2018 Dec;94(6):1073–1086.
- Molinari E, Ramsbottom SA, Sammut V, et al. Using zebrafish to study the function of nephronophthisis and related ciliopathy genes. F1000Res. 2018;7:1133.
- Nagao S, Yamaguchi T. Review of the use of animal models of human polycystic kidney disease for the evaluation of experimental therapeutic modalities. J Clin Med. 2023 Jan 14;12(2):668.
- Nagao S, Kugita M, Yoshihara D, et al. Animal models for human polycystic kidney disease. Exp Anim. 2012;61(5):477–488.
- Richards T, Modarage K, Malik SA, et al. The cellular pathways and potential therapeutics of Polycystic kidney disease. Biochem Soc Trans. 2021 Jun 30;49(3):1171–1188.
- Mansour SG, Puthumana J, Coca SG, et al. Biomarkers for the detection of renal fibrosis and prediction of renal outcomes: a systematic review. BMC Nephrol. 2017 Feb 20;18(1):72.
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012 Dec 20;367(25):2407–2418.
- Liebau MC, Hartung EA, Perrone RD. Perspectives on drug development in autosomal recessive polycystic kidney disease. Clin J Am Soc Nephrol. 2022 Oct;17(10):1551–1554.
- Wang X, Gattone V 2nd, Harris PC, et al. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. 2005 Apr;16(4):846–851.
- Aihara M, Fujiki H, Mizuguchi H, et al. Tolvaptan delays the onset of end-stage renal disease in a polycystic kidney disease model by suppressing increases in kidney volume and renal injury. J Pharmacol Exp Ther. 2014 May;349(2):258–267.
- Hopp K, Hommerding CJ, Wang X, et al. Tolvaptan plus pasireotide shows enhanced efficacy in a PKD1 model. J Am Soc Nephrol. 2015 Jan;26(1):39–47.
- Di Mise A, Wang X, Ye H, et al. Pre-clinical evaluation of dual targeting of the GPCRs CaSR and V2R as therapeutic strategy for autosomal dominant polycystic kidney disease. FASEB J. 2021 Oct;35(10):e21874.
- Wang Y, Chen F, Wang J, et al. Two novel homozygous mutations in NPHP1 lead to late onset end-stage renal disease: a case report of an adult nephronophthisis in a Chinese intermarriage family. BMC Nephrol. 2019 May 16;20(1):173.
- Yang BX, Sonawane ND, Zhao D, et al. Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2008 Jul;19(7):1300–1310.
- Yanda MK, Cha B, Cebotaru CV, et al. Pharmacological reversal of renal cysts from secretion to absorption suggests a potential therapeutic strategy for managing autosomal dominant polycystic kidney disease. J Biol Chem. 2019 Nov 8;294(45):17090–17104.
- Cabrita I, Kraus A, Scholz JK, et al. Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. Nat Commun. 2020 Aug 28;11(1):4320.
- Blazer-Yost BL, Haydon J, Eggleston-Gulyas T, et al. Pioglitazone attenuates cystic burden in the PCK rodent model of polycystic kidney disease. PPAR Res. 2010;2010:274376.
- Nofziger C, Brown KK, Smith CD, et al. PPARgamma agonists inhibit vasopressin-mediated anion transport in the MDCK-C7 cell line. Am J Physiol Renal Physiol. 2009 Jul;297(1):F55–62.
- Yoshihara D, Kurahashi H, Morita M, et al. PPAR-gamma agonist ameliorates kidney and liver disease in an orthologous rat model of human autosomal recessive polycystic kidney disease. Am J Physiol Renal Physiol. 2011 Feb;300(2):F465–74.
- Muto S, Aiba A, Saito Y, et al. Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum Mol Genet. 2002 Jul 15;11(15):1731–1742.
- Dai B, Liu YW, Mei CL, et al. Rosiglitazone attenuates development of polycystic kidney disease and prolongs survival in Han:SPRD rats. Clin sci. 2010 Oct;119(7–8):323–333.
- Kanhai AA, Bange H, Verburg L, et al. Renal cyst growth is attenuated by a combination treatment of tolvaptan and pioglitazone, while pioglitazone treatment alone is not effective. Sci Rep-UK. 2020 Feb 3;10(1):1672.
- Klawitter J, Zafar I, Klawitter J, et al. Effects of lovastatin treatment on the metabolic distributions in the Han:SPRD rat model of polycystic kidney disease. BMC Nephrol. 2013 Jul 31;14:165.
- van Dijk MA, Kamper AM, van Veen S, et al. Effect of simvastatin on renal function in autosomal dominant polycystic kidney disease. Nephrol Dial Transpl. 2001 Nov;16(11):2152–2157.
- Capuano I, Riccio E, Caccavallo S, et al. ADPKD and metformin: from bench to bedside. Clin Exp Nephrol. 2019 Nov;23(11):1341–1342.
- Natoli TA, Smith LA, Rogers KA, et al. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat Med. 2010 Jul;16(7):788–792.
- Perrone RD, Hariri A, Minini P, et al. The STAGED-PKD 2-stage adaptive study with a patient enrichment strategy and treatment effect modeling for improved study design efficiency in patients with ADPKD. Kidney Med. 2022 Oct;4(10):100538.
- Sankaran D, Bankovic-Calic N, Ogborn MR, et al. Selective COX-2 inhibition markedly slows disease progression and attenuates altered prostanoid production in Han: SPRD-cy rats with inherited kidney disease. Am J Physiol-Renal. 2007 Sep;293(3):F821–F830.
- Monirujjaman M, Aukema HM. Cyclooxygenase 2 inhibition slows disease progression and improves the altered renal lipid mediator profile in the Pkd2(WS25/-) mouse model of autosomal dominant polycystic kidney disease. J Nephrol. 2019 Jun;32(3):401–409.
- Ibrahim NH, Gregoire M, Devassy JG, et al. Cyclooxygenase product inhibition with acetylsalicylic acid slows disease progression in the Han:SPRD-Cy rat model of polycystic kidney disease. Prostaglandins Other Lipid Mediat. 2015 Jan-Mar;116-117:19–25.
- Yamaguchi T, Devassy JG, Gabbs M, et al. Dietary flax oil rich in alpha-linolenic acid reduces renal disease and oxylipin abnormalities, including formation of docosahexaenoic acid derived oxylipins in the CD1-pcy/pcy mouse model of nephronophthisis. Prostaglandins Leukot Essent Fatty Acids. 2015 Mar;94:83–89.
- Celentano S, Capolongo G, Pollastro RM. Bardoxolone: a new potential therapeutic agent in the treatment of autosomal dominant polycystic kidney disease?. G Ital Nefrol. 2019 Sep 24;36(5):2019-vol5. Italian. PMID: 31580543.
- Rossing P, Block GA, Chin MP, et al. Effect of bardoxolone methyl on the urine albumin-to-creatinine ratio in patients with type 2 diabetes and stage 4 chronic kidney disease. Kidney Int. 2019 Oct;96(4):1030–1036.
- de Zeeuw D, Akizawa T, Audhya P, et al. Bardoxolone methyl in Type 2 diabetes and Stage 4 chronic kidney disease. New Engl J Med. 2013 Dec 26;369(26):2492–2503.
- Ito M, Tanaka T, Nangaku M. Nuclear factor erythroid 2-related factor 2 as a treatment target of kidney diseases. Curr Opin Nephrol Hy. 2020 Jan;29(1):128–135.
- Leuenroth SJ, Bencivenga N, Igarashi P, et al. Triptolide reduces cystogenesis in a model of ADPKD. J Am Soc Nephrol. 2008 Sep;19(9):1659–1662.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006 Apr 4;103(14):5466–5471.
- Tao YX, Kim J, Schrier RW, et al. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol. 2005 Jan;16(1):46–51.
- Li A, Fan S, Xu YC, et al. Rapamycin treatment dose-dependently improves the cystic kidney in a new ADPKD mouse model via the mTORC1 and cell-cycle-associated CDK1/cyclin axis. J Cell Mol Med. 2017 Aug;21(8):1619–1635.
- Tobin JL, Beales PL. Restoration of renal function in zebrafish models of ciliopathies. Pediatr Nephrol. 2008 Nov;23(11):2095–2099.
- Zafar I, Ravichandran K, Belibi FA, et al. Sirolimus attenuates disease progression in an orthologous mouse model of human autosomal dominant polycystic kidney disease. Kidney Int. 2010 Oct;78(8):754–761.
- Shillingford JM, Piontek KB, Germino GG, et al. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010 Mar;21(3):489–497.
- e Zeeuw D, Akizawa T, Audhya P, et al. BEACON Trial Investigators. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013 Dec 26;369(26):2492–503.
- Qian Q, Du H, King BF, et al. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol. 2008 Mar;19(3):631–638.
- Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010 Aug 26;363(9):820–829.
- Shillingford JM, Leamon CP, Vlahov IR, et al. Folate-conjugated rapamycin slows progression of polycystic kidney disease. J Am Soc Nephrol. 2012 Oct;23(10):1674–1681.
- Holditch SJ, Brown CN, Atwood DJ, et al. A study of sirolimus and mTOR kinase inhibitor in a hypomorphic Pkd1 mouse model of autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol. 2019 Jul 1;317(1):F187–F196.
- Wu M, Wahl PR, Le Hir M, et al. Everolimus retards cyst growth and preserves kidney function in a rodent model for polycystic kidney disease. Kidney Blood Press R. 2007;30(4):253–259.
- Walz G, Budde K, Mannaa M, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010 Aug 26;363(9):830–840.
- Sweeney WE, Chen YG, Nakanishi K, et al. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 2000 Jan;57(1):33–40.
- Chang MY, A CMO. Targeting new cellular disease pathways in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2018 Aug 1;33(8):1310–1316.
- Tesar V, Ciechanowski K, Pei Y, et al. Bosutinib versus placebo for autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2017 Nov;28(11):3404–3413.
- Sweeney WE Jr., von Vigier RO, Frost P, et al. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 2008 Jul;19(7):1331–1341.
- Elliott J, Zheleznova NN, Wilson PD. c-Src inactivation reduces renal epithelial cell-matrix adhesion, proliferation, and cyst formation. Am J Physiol-Cell Ph. 2011 Aug;301(2):C522–C529.
- Torres VE, Sweeney WE, Wang XF, et al. EGF receptor tyrosine kinase inhibition attenuates the development of PKD in Han : SPRD rats. Kidney Int. 2003 Nov;64(5):1573–1579.
- Yamaguchi T, Reif GA, Calvet JP, et al. Sorafenib inhibits cAMP-dependent ERK activation, cell proliferation, and in vitro cyst growth of human ADPKD cyst epithelial cells. Am J Physiol-Renal. 2010 Nov;299(5):F944–F951.
- Calvet JP. MEK inhibition holds promise for polycystic kidney disease. J Am Soc Nephrol. 2006 Jun;17(6):1498–1500.
- Okumura Y, Sugiyama N, Tanimura S, et al. ERK regulates renal cell proliferation and renal cyst expansion in inv mutant mice. Acta Histochem Cytochem. 2009 Apr 28;42(2):39–45.
- Caroli A, Perico N, Perna A, et al. Effect of longacting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): a randomised, placebo-controlled, multicentre trial. Lancet. 2013 Nov 2;382(9903):1485–1495.
- Meijer E, Visser FW, van Aerts RMM, et al. Effect of lanreotide on kidney function in patients with autosomal dominant polycystic kidney disease: The DIPAK 1 randomized clinical trial. JAMA. 2018 Nov 20;320(19):2010–2019.
- Treille S, Bailly JM, Van Cauter J, et al. The use of lanreotide in polycystic kidney disease: a single-centre experience. Case Rep Nephrol Urol. 2014 Jan;4(1):18–24.
- Gevers TJ, Hol JC, Monshouwer R, et al. Effect of lanreotide on polycystic liver and kidneys in autosomal dominant polycystic kidney disease: an observational trial. Liver Int. 2015 May;35(5):1607–1614.
- Masyuk TV, Radtke BN, Stroope AJ, et al. Pasireotide is more effective than octreotide in reducing hepatorenal cystogenesis in rodents with polycystic kidney and liver diseases. Hepatology. 2013 Jul;58(1):409–421.
- Perico N, Ruggenenti P, Perna A, et al. Octreotide-LAR in later-stage autosomal dominant polycystic kidney disease (ALADIN 2): A randomized, double-blind, placebo-controlled, multicenter trial. PLOS Med. 2019 Apr;16(4):e1002777.
- Trillini M, Caroli A, Perico N, et al. Effects of octreotide-long-acting release added-on tolvaptan in patients with autosomal dominant polycystic kidney disease: pilot, randomized, placebo-controlled, cross-over trial. Clin J Am Soc Nephrol. 2023 Feb 1;18(2):223–233.
- Ghosh AK, Hurd T, Hildebrandt F. 3D spheroid defects in NPHP knockdown cells are rescued by the somatostatin receptor agonist octreotide. Am J Physiol-Renal. 2012 Oct;303(8):F1225–F1229.
- Hogan MC, Chamberlin JA, Vaughan LE, et al. Pansomatostatin agonist pasireotide long-acting release for patients with autosomal dominant polycystic kidney or liver disease with severe liver involvement: a randomized clinical trial. Clin J Am Soc Nephrol. 2020 Sep 7;15(9):1267–1278.
- Takiar V, Nishio S, Seo-Mayer P, et al. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci U S A. 2011 Feb 8;108(6):2462–2467.
- Chang MY, Ma TL, Hung CC, et al. Metformin inhibits cyst formation in a zebrafish model of polycystin-2 deficiency. Sci Rep-UK. 2017 Aug 2;7(1):7161.
- Seliger SL, Abebe KZ, Hallows KR, et al. A randomized clinical trial of metformin to treat autosomal dominant polycystic kidney disease. Am J Nephrol. 2018;47(5):352–360.
- Sorohan BM, Ismail G, Andronesi A, et al. A single-arm pilot study of metformin in patients with autosomal dominant polycystic kidney disease. BMC Nephrol. 2019 Jul 23;20(1):276.
- Pisani A, Riccio E, Bruzzese D, et al. Metformin in autosomal dominant polycystic kidney disease: experimental hypothesis or clinical fact?. BMC Nephrol. 2018 Oct 22;19(1):282.
- Leonhard WN, Song XW, Kanhai AA, et al. Salsalate, but not metformin or canagliflozin, slows kidney cyst growth in an adult-onset mouse model of polycystic kidney disease. EBioMedicine. 2019 Sep;47:436–445.
- Rowe I, Chiaravalli M, Mannella V, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. 2013 Apr;19(4):488–493.
- Bracken C, Beauverger P, Duclos O, et al. CaMKII as a pathological mediator of ER stress, oxidative stress, and mitochondrial dysfunction in a murine model of nephronophthisis. Am J Physiol-Renal. 2016 Jun 1;310(11):F1414–F1422.
- Garcia H, Serafin AS, Silbermann F, et al. Agonists of prostaglandin E(2) receptors as potential first in class treatment for nephronophthisis and related ciliopathies. Proc Natl Acad Sci U S A. 2022 May 3;119(18):e2115960119.
- Tao Y, Kim J, Yin Y, et al. VEGF receptor inhibition slows the progression of polycystic kidney disease. Kidney Int. 2007 Dec;72(11):1358–1366.
- Tran PV, Talbott GC, Turbe-Doan A, et al. Downregulating hedgehog signaling reduces renal cystogenic potential of mouse models. J Am Soc Nephrol. 2014 Oct;25(10):2201–2212.
- Yanda MK, Liu Q, Cebotaru V, et al. Histone deacetylase 6 inhibition reduces cysts by decreasing cAMP and Ca(2+) in knock-out mouse models of polycystic kidney disease. J Biol Chem. 2017 Oct 27;292(43):17897–17908.
- Cebotaru L, Liu QG, Yanda MK, et al. Inhibition of histone deacetylase 6 activity reduces cyst growth in polycystic kidney disease. Kidney Int. 2016 Jul;90(1):90–99.
- Cao Y, Semanchik N, Lee SH, et al. Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. P Natl Acad Sci USA. 2009 Dec 22;106(51):21819–21824.
- Fan LX, Li XJ, Magenheimer B, et al. Inhibition of histone deacetylases targets the transcription regulator Id2 to attenuate cystic epithelial cell proliferation. Kidney Int. 2012 Jan;81(1):76–85.
- Zhou X, Fan LX, Sweeney WE, et al. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J Clin Investig. 2013 Jul;123(7):3084–3098.
- Nikonova AS, Deneka AY, Kiseleva AA, et al. Ganetespib limits ciliation and cystogenesis in autosomal-dominant polycystic kidney disease (ADPKD). FASEB J. 2018 May;32(5):2735–2746.
- Seeger-Nukpezah T, Proia DA, Egleston BL, et al. Inhibiting the HSP90 chaperone slows cyst growth in a mouse model of autosomal dominant polycystic kidney disease. Proc Natl Acad Sci U S A. 2013 Jul 30;110(31):12786–12791.
- Bukanov NO, Smith LA, Klinger KW, et al. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006 Dec 14;444(7121):949–952.
- Bukanov NO, Moreno SE, Natoli TA, et al. CDK inhibitors R-roscovitine and S-CR8 effectively block renal and hepatic cystogenesis in an orthologous model of ADPKD. Cell Cycle. 2012 Nov 1;11(21):4040–4046.
- Husson H, Moreno S, Smith LA, et al. Reduction of ciliary length through pharmacologic or genetic inhibition of CDK5 attenuates polycystic kidney disease in a model of nephronophthisis. Hum Mol Genet. 2016 Jun 1;25(11):2245–2255.
- Airik R, Airik M, Schueler M, et al. Roscovitine blocks collecting duct cyst growth in Cep164-deficient kidneys. Kidney Int. 2019 Aug;96(2):320–326.
- Slaats GG, Saldivar JC, Bacal J, et al. DNA replication stress underlies renal phenotypes in CEP290-associated Joubert syndrome. J Clin Investig. 2015 Sep;125(9):3657–3666.
- Masyuk TV, Radtke BN, Stroope AJ, et al. Inhibition of Cdc25A suppresses hepato-renal cystogenesis in rodent models of polycystic kidney and liver disease. Gastroenterology. 2012 Mar;142(3):622–633 e4.
- Yheskel M, Lakhia R, Cobo-Stark P, et al. Anti-microRNA screen uncovers miR-17 family within miR-17 similar to 92 cluster as the primary driver of kidney cyst growth. Sci Rep-UK. 2019 Feb 13;9:1920.
- Lee EC, Valencia T, Allerson C, et al. Discovery and preclinical evaluation of anti-miR-17 oligonucleotide RGLS4326 for the treatment of polycystic kidney disease. Nat Commun. 2019 Sep 12;10(1):4148.
- Zhou JX, Li X. Non-Coding RNAs in Hereditary Kidney Disorders. Int J Mol Sci. 2021 Mar 16;22(6):3014.
- Ramsbottom SA, Molinari E, Srivastava S, et al. Targeted exon skipping of a CEP290 mutation rescues Joubert syndrome phenotypes in vitro and in a murine model. Proc Natl Acad Sci U S A. 2018 Dec 4;115(49):12489–12494.
- Ramsbottom SA, Thelwall PE, Wood KM, et al. Mouse genetics reveals Barttin as a genetic modifier of Joubert syndrome. Proc Natl Acad Sci U S A. 2020 Jan 14;117(2):1113–1118.
- Torres VE. Vasopressin antagonists in polycystic kidney disease. Kidney Int. 2005 Nov;68(5):2405–2418.
- Gattone VH 2nd, Wang X, Harris PC, et al. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003 Oct;9(10):1323–1326.
- Endo M, Katayama K, Matsuo H, et al. Role of liver transplantation in tolvaptan-associated acute liver failure. Kidney Int Rep. 2019 Nov;4(11):1653–1657.
- Sans-Atxer L, Joly D. Tolvaptan in the treatment of autosomal dominant polycystic kidney disease: patient selection and special considerations. Int J Nephrol Renov. 2018;11:41–51.
- Di Mise A, Venneri M, Ranieri M, et al. Lixivaptan, a new generation diuretic, counteracts vasopressin-induced Aquaporin-2 trafficking and function in renal collecting duct cells. Int J Mol Sci. 2019 Dec 26;21(1):183.
- Wang X, Constans MM, Chebib FT, et al. Effect of a Vasopressin V2 receptor antagonist on polycystic kidney disease development in a rat model. Am J Nephrol. 2019;49(6):487–493.
- Hanaoka K, Devuyst O, Schwiebert EM, et al. A role for CFTR in human autosomal dominant polycystic kidney disease. Am J physiol. 1996 Jan;270(1 Pt 1):C389–99.
- Ye M, Grantham JJ. The Secretion of fluid by renal cysts from patients with autosomal-dominant polycystic kidney-disease. New Engl J Med. 1993 Jul 29;329(5):310–313.
- Yanda MK, Cebotaru L. VX-809 mitigates disease in a mouse model of autosomal dominant polycystic kidney disease bearing the R3277C human mutation. FASEB J. 2021 Nov;35(11):e21987.
- Seo Y, Kim J, Chang J, et al. Synthesis and biological evaluation of novel Ani9 derivatives as potent and selective ANO1 inhibitors. Eur J Med Chem. 2018 Dec 5;160:245–255.
- Miner K, Labitzke K, Liu BX, et al. Drug repurposing: the anthelmintics niclosamide and nitazoxanide are potent TMEM16A antagonists that fully bronchodilate airways. Front Pharmacol. 2019 Feb 14;10:51.
- Lakhia R. The role of PPAR alpha in autosomal dominant polycystic kidney disease. Curr Opin Nephrol Hy. 2020 Jul;29(4):432–438.
- Sun W, Lee TS, Zhu M, et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006 Dec 12;114(24):2655–2662.
- Natoli TA, Modur V, Ibraghimov-Beskrovnaya O. Glycosphingolipid metabolism and polycystic kidney disease. Cell Signal. 2020 May;69:109526.
- Swenson-Fields KI, Vivian CJ, Salah SM, et al. Macrophages promote polycystic kidney disease progression. Kidney Int. 2013 May;83(5):855–864.
- Marra AN, Adeeb BD, Chambers BE, et al. Prostaglandin signaling regulates renal multiciliated cell specification and maturation. Proc Natl Acad Sci U S A. 2019 Apr 23;116(17):8409–8418.
- Zhang JQJ, Saravanabavan S, Munt A, et al. The role of DNA damage as a therapeutic target in autosomal dominant polycystic kidney disease. Expert Rev Mol Med. 2019 Nov 26;21:e6.
- Yamaguchi T, Lysecki C, Reid A, et al. Renal cyclooxygenase products are higher and lipoxygenase products are lower in early disease in the pcy mouse model of adolescent nephronophthisis. Lipids. 2014 Jan;49(1):39–47.
- Martin-Hurtado A, Martin-Morales R, Robledinos-Anton N, et al. NRF2-dependent gene expression promotes ciliogenesis and Hedgehog signaling. Sci Rep. 2019 Sep 25;9(1):13896.
- Liu P, Dodson M, Fang D, et al. NRF2 negatively regulates primary ciliogenesis and hedgehog signaling. PLoS Biol. 2020 Feb;18(2):e3000620.
- Viau A, Bienaime F, Lukas K, et al. Cilia-localized LKB1 regulates chemokine signaling, macrophage recruitment, and tissue homeostasis in the kidney. EMBO J. 2018 Aug 1;37(15):e98615.
- Pema M, Drusian L, Chiaravalli M, et al. mTORC1-mediated inhibition of polycystin-1 expression drives renal cyst formation in tuberous sclerosis complex. Nat Commun. 2016 Mar 2;7:10786.
- Lai YD, Jiang Y. Reciprocal regulation between primary cilia and mTORC1. Genes-Basel. 2020 Jun 26;11(6):711.
- Shao L, El-Jouni W, Kong F, et al. Genetic reduction of cilium length by targeting intraflagellar transport 88 protein impedes kidney and liver cyst formation in mouse models of autosomal polycystic kidney disease. Kidney Int. 2020 Nov;98(5):1225–1241.
- Lin CH, Chao CT, Wu MY, et al. Use of mammalian target of rapamycin inhibitors in patient with autosomal dominant polycystic kidney disease: an updated meta-analysis. Int Urol Nephrol. 2019 Nov;51(11):2015–2025.
- Herzig S, Shaw, RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018 Feb;19(2):121–135.
- Padovano V, Podrini C, Boletta A, et al. Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat rev Nephrol. 2018 Nov;14(11):678–687.
- Podrini C, Cassina L, Boletta A. Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell Signal. 2020 Mar;67:109495.
- Ishimoto Y, Inagi R, Yoshihara D, et al. Mitochondrial abnormality facilitates cyst formation in autosomal dominant polycystic kidney disease. Mol Cell Biol. 2017 Dec 15;37(24):e0033717.
- Anderson K, Wherle L, Park M, et al. Salsalate, an old, inexpensive drug with potential new indications: a review of the evidence from 3 recent studies. Am Health Drug Benefits. 2014 Jun;7(4):231–235.
- Haws RM, Gordon G, Han JC, et al. The efficacy and safety of setmelanotide in individuals with Bardet-Biedl syndrome or Alstrom syndrome: Phase 3 trial design. Contemp Clin Trials Commun. 2021 Jun;22:100780.
- Nakamura T, Ebihara I, Nagaoka I, et al. Growth factor gene expression in kidney of murine polycystic kidney disease. J Am Soc Nephrol. 1993 Jan;3(7):1378–1386.
- Gunther T, Tulipano G, Dournaud P, et al. International Union of Basic and Clinical Pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature. Pharmacol Rev. 2018 Oct;70(4):763–835.
- Law SF, Manning D, Reisine T. Identification of the subunits of GTP-binding proteins coupled to somatostatin receptors. J Biol Chem. 1991 Sep 25;266(27):17885–17897.
- Dasgupta P. Somatostatin analogues: multiple roles in cellular proliferation, neoplasia, and angiogenesis. Pharmacol Ther. 2004 Apr;102(1):61–85.
- Ferjoux G, Bousquet C, Cordelier P, et al. Signal transduction of somatostatin receptors negatively controlling cell proliferation. J Physiol Paris. 2000 May-Aug;94(3–4):205–210.
- Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014 Jan;25(1):18–32.
- Sun L, Yu CY, Mackey LV, et al. Lanreotide and its potential applications in polycystic kidney and liver diseases. Curr Top Med Chem. 2015;16(2):133–140.
- Patel V, Chowdhury R, Igarashi P. Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens. 2009 Mar;18(2):99–106.
- Ghosh AK, Hurd T, Hildebrandt F. 3D spheroid defects in NPHP knockdown cells are rescued by the somatostatin receptor agonist octreotide. Am J Physiol Renal Physiol. 2012 Oct 15;303(8):F1225–9.
- Reed BY, Masoumi A, Elhassan E, et al. Angiogenic growth factors correlate with disease severity in young patients with autosomal dominant polycystic kidney disease. Kidney Int. 2011 Jan;79(1):128–134.
- Yu F, Ran J, Zhou J. Ciliopathies: does HDAC6 represent a new therapeutic target?. Trends Pharmacol Sci. 2016 Feb;37(2):114–119.
- Kovacs JJ, Murphy PJM, Gaillard S, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005 May 27;18(5):601–607.
- Prodromou NV, Thompson CL, Osborn DP, et al. Heat shock induces rapid resorption of primary cilia. J Cell Sci. 2012 Sep 15;125(Pt 18):4297–4305.
- Chun P. Therapeutic effects of histone deacetylase inhibitors on kidney disease. Arch Pharm Res. 2018 Feb;41(2):162–183.
- Li B, Rauhauser AA, Dai J, et al. Increased hedgehog signaling in postnatal kidney results in aberrant activation of nephron developmental programs. Hum Mol Genet. 2011 Nov 1;20(21):4155–4166.
- Kopinke D, Norris AM, Mukhopadhyay S. Developmental and regenerative paradigms of cilia regulated hedgehog signaling. Semin Cell Dev Biol. 2021 Feb;110:89–103.
- Ruiz-Perez VL, Blair HJ, Rodriguez-Andres ME, et al. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development. 2007 Aug 15;134(16):2903–2912.
- Aguilar A, Meunier A, Strehl L, et al. Analysis of human samples reveals impaired SHH-dependent cerebellar development in Joubert syndrome/Meckel syndrome. P Natl Acad Sci USA. 2012 Oct 16;109(42):16951–16956.
- Yamaguchi T, Nagao S, Kasahara M, et al. Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am J Kidney Dis. 1997 Nov;30(5):703–709.
- Smith LA, Bukanov NO, Husson H, et al. Development of polycystic kidney disease in juvenile cystic kidney mice: Insights into pathogenesis, ciliary abnormalities, and common features with human disease. J Am Soc Nephrol. 2006 Oct;17(10):2821–2831.
- Wallace DP. Cyclic AMP-mediated cyst expansion. BBA-Mol Basis Dis. 2011 Oct;1812(10):1291–1300.
- Calvet JP. The Role of Calcium and Cyclic AMP in PKD. In: Li X, editor. Polycystic kidney disease. Brisbane (AU): Codon Publications; 2015. Nov. Chapter 8.
- Chen NX, Moe SM, Eggleston-Gulyas T, et al. Calcimimetics inhibit renal pathology in rodent nephronophthisis. Kidney Int. 2011 Sep;80(6):612–619.
- Simons M, Gloy J, Ganner A, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005 May;37(5):537–543.
- Bergmann C, Fliegauf M, Bruchle NO, et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet. 2008 Apr;82(4):959–970.
- Wang Q, Cobo-Stark P, Patel V, et al. Adenylyl cyclase 5 deficiency reduces renal cyclic AMP and cyst growth in an orthologous mouse model of polycystic kidney disease. Kidney Int. 2018 Feb;93(2):403–415.
- Schueler M, Braun DA, Chandrasekar G, et al. DCDC2 mutations cause a renal-hepatic ciliopathy by disrupting Wnt signaling. Am J Hum Genet. 2015 Jan 8;96(1):81–92.
- Habbig S, Bartram MP, Muller RU, et al. NPHP4, a cilia-associated protein, negatively regulates the hippo pathway. J Cell Bio. 2011 May 16;193(4):633–642.
- Habbig S, Bartram MP, Sagmuller JG, et al. The ciliopathy disease protein NPHP9 promotes nuclear delivery and activation of the oncogenic transcriptional regulator TAZ. Hum Mol Genet. 2012 Dec 15;21(26):5528–5538.
- Frank V, Habbig S, Bartram MP, et al. Mutations in NEK8 link multiple organ dysplasia with altered Hippo signalling and increased c-MYC expression. Hum Mol Genet. 2013 Jun 1;22(11):2177–2185.
- Grampa V, Delous M, Zaidan M, et al. Novel NEK8 mutations cause severe syndromic renal cystic dysplasia through YAP dysregulation. PLoS Genet. 2016 Mar;12(3):e1005894.
- Muller RU, Schermer B. Hippo signaling-a central player in cystic kidney disease?. Pediatr Nephrol. 2020 Jul;35(7):1143–1152.
- Xu C, Wang L, Zhang Y, et al. Tubule-specific Mst1/2 deficiency induces CKD via YAP and Non-YAP mechanisms. J Am Soc Nephrol. 2020 May;31(5):946–961.
- Szeto SG, Narimatsu M, Lu ML, et al. YAP/TAZ are mechanoregulators of TGF-beta-smad signaling and renal fibrogenesis. J Am Soc Nephrol. 2016 Oct;27(10):3117–3128.
- Rees S, Kittikulsuth W, Roos K, et al. Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. J Am Soc Nephrol. 2014 Feb;25(2):232–237.
- Omar F, Findlay JE, Carfray G, et al. Small-molecule allosteric activators of PDE4 long form cyclic AMP phosphodiesterases. Proc Natl Acad Sci U S A. 2019 Jul 2;116(27):13320–13329.
- Choi HJ, Lin JR, Vannier JB, et al. NEK8 links the ATR-regulated replication stress response and S phase CDK activity to renal ciliopathies. Mol Cell. 2013 Aug 22;51(4):423–439.
- Airik R, Slaats GG, Guo Z, et al. Renal-retinal ciliopathy gene Sdccag8 regulates DNA damage response signaling. J Am Soc Nephrol. 2014 Nov;25(11):2573–2583.
- Pan YR, Lee EY. UV-dependent interaction between Cep164 and XPA mediates localization of Cep164 at sites of DNA damage and UV sensitivity. Cell Cycle. 2009 Feb 15;8(4):655–664.
- Hoff S, Epting D, Falk N, et al. The nucleoside-diphosphate kinase NME3 associates with nephronophthisis proteins and is required for ciliary function during renal development. J Biol Chem. 2018 Sep 28;293(39):15243–15255.
- Daly OM, Gaboriau D, Karakaya K, et al. CEP164-null cells generated by genome editing show a ciliation defect with intact DNA repair capacity. J Cell Sci. 2016 May 1;129(9):1769–1774.
- Ramalingam H, Yheskel M, Patel V. Modulation of polycystic kidney disease by non-coding RNAs. Cell Signal. 2020 Jul;71:109548. DOI:10.1016/j.cellsig.2020.109548
- Hajarnis S, Lakhia R, Yheskel M, et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun. 2017 Feb 16;8:14395.
- Kuo IY, Brill AL, Lemos FO, et al. Polycystin 2 regulates mitochondrial Ca(2+) signaling, bioenergetics, and dynamics through mitofusin 2. Sci Signal. 2019 May 7;12(580):eaat7397.
- Gomez IG, MacKenna DA, Johnson BG, et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Investig. 2015 Jan;125(1):141–156.
- Yamamura T, Horinouchi T, Adachi T, et al. Development of an exon skipping therapy for X-linked Alport syndrome with truncating variants in COL4A5. Nat Commun. 2020 Jun 2;11(1):2777.
- Xue K, MacLaren RE. Antisense oligonucleotide therapeutics in clinical trials for the treatment of inherited retinal diseases. Expert Opin Investig Drugs. 2020 Oct;29(10):1163–1170.
- Duijkers L, van den Born LI, Neidhardt J, et al. Antisense oligonucleotide-based splicing correction in individuals with Leber congenital amaurosis due to compound heterozygosity for the c.2991+1655A>G mutation in CEP290. Int J Mol Sci. 2018 Mar 7;19(3):753.
- Barny I, Perrault I, Michel C, et al. Basal exon skipping and nonsense-associated altered splicing allows bypassing complete CEP290 loss-of-function in individuals with unusually mild retinal disease. Hum Mol Genet. 2018 Aug 1;27(15):2689–2702.
- Barny I, Perrault I, Michel C, et al. AON-mediated exon skipping to bypass protein truncation in retinal dystrophies due to the recurrent CEP290 c.4723A > T mutation fact or fiction?. Genes-Basel. 2019 May;10(5):368.
- Tayfur AC, Besbas N, Bilginer Y, et al. Follow-up of patients with juvenile nephronophthisis after renal transplantation: a single center experience. Transplant Proc. 2011 Apr;43(3):847–849.
- Nespoux J, Vallon V. Renal effects of SGLT2 inhibitors: an update. Curr Opin Nephrol Hypertens. 2020 Mar;29(2):190–198.
- Verma S, McMurray JJV. SGLT2 inhibitors and mechanisms of cardiovascular benefit: a state-of-the-art review. Diabetologia. 2018 Oct;61(10):2108–2117.
- Dhillon S. Dapagliflozin: a review in Type 2 diabetes. Drugs. 2019 Jul;79(10):1135–1146.
- Marcath LA. Finerenone. Clin Diabetes. 2021 Jul;39(3):331–332.
- Afsar B, Afsar RE, Demiray A, et al. Sodium-glucose cotransporter inhibition in polycystic kidney disease: fact or fiction. Clin Kidney J. 2022 Jul;15(7):1275–1283.
- Afsar B, Hornum M, Afsar RE, et al. Mitochondrion-driven nephroprotective mechanisms of novel glucose lowering medications. Mitochondrion. 2021 May;58:72–82.
- Sen T, Heerspink HJL. A kidney perspective on the mechanism of action of sodium glucose co-transporter 2 inhibitors. Cell Metab. 2021 Apr 6;33(4):732–739.
- Bakris GL, Agarwal R, Anker SD, et al. Effect of finerenone on chronic kidney disease outcomes in Type 2 diabetes. N Engl J Med. 2020 Dec 3;383(23):2219–2229.
- Rubel D, Boulanger J, Craciun F, et al. Anti-microRNA-21 therapy on top of ace inhibition delays renal failure in Alport syndrome mouse models. Cells. 2022 Feb 9;11(4):594.
- Chavez E, Rodriguez J, Drexler Y, et al. Novel therapies for Alport syndrome. Front Med. 2022;9:848389.
- Ramos AM, Gonzalez-Guerrero C, Sanz A, et al. Designing drugs that combat kidney damage. Expert Opin Drug Dis. 2015 May;10(5):541–556.