
ABSTRACT
Introduction
Microbial infections and resultant sepsis are leading causes of death in hospitals, representing approximately 20% of total deaths worldwide. Despite the difficulties in translating experimental insights into effective therapies for often heterogenous patient populations, an improved understanding of the pathogenic mechanisms underlying experimental sepsis is still urgently needed. Sepsis is partly attributable to dysregulated innate immune responses manifested by hyperinflammation and immunosuppression at different stages of microbial infections.
Areas covered
Here we review our recent progress in searching for late-acting mediators of experimental sepsis and propose high mobility group box 1 (HMGB1) and procathepsin-L (pCTS-L) as potential therapeutic targets for improving outcomes of lethal sepsis and other infectious diseases.
Expert opinion
It will be important to evaluate the efficacy of HMGB1- or pCTS-L-targeting agents for the clinical management of human sepsis and other infectious diseases in future studies.
1. Introduction
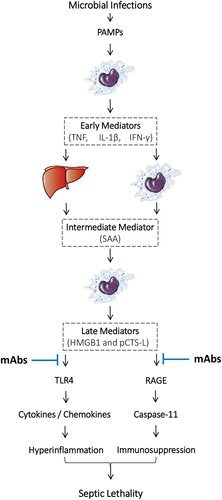
Microbial infections and resultant sepsis are leading causes of death in hospitals, representing approximately 20% of total deaths worldwide [Citation1]. The pathogenesis of sepsis is complex but partly attributable to dysregulated innate immune responses to lethal infections as manifested by hyperinflammation and/or immunosuppression at different stages of disease progression [Citation2]. To mount initial inflammatory response to infections, innate immune cells rely on various pattern recognition receptors [PRRs, e.g. the toll-like receptor 4 (TLR4) or TLR9] [Citation3,Citation4] to recognize distinct classes of molecules shared by different microbes that are termed as ‘pathogen-associated molecular patterns’ [PAMPs, e.g. bacterial lipopolysaccharide (LPS) or CpG-DNA] (). For example, upon detecting small amounts of LPS by an LPS-binding protein (LBP) [Citation5], a co-receptor cluster of differentiation 14 (CD14) [Citation6] delivers it to TLR4 [Citation3], which triggers an immediate release of ‘early’ proinflammatory cytokines such as tumor necrosis factor (TNF) [Citation7], interleukin-1β (IL-1β) [Citation8], and interferon-γ (IFN-γ) [Citation9] (). When overproduced, these early cytokines also induce some intermediate pro-inflammatory mediators such as serum amyloid A (SAA, ) [Citation10], which employs TLR4 [Citation11] and the receptor for advanced glycation end product (RAGE) [Citation12] to induce the expression of macrophage hemichannels (e.g. connexin 43 and pannexin 1) [Citation13,Citation14] and secretory phospholipase A2 (e.g. sPLA2-IIE/V) [Citation15], as well as the release of late-acting mediators such as high mobility group box 1 (HMGB1) [Citation16] and procathepsin-L (pCTS-L) [Citation17] (). Here we review experimental evidence to support HMGB1 and pCTS-L as potential therapeutic targets for improving survival in sepsis.
Figure 1. Overall summary of pathogenic roles of late-acting mediators in dysregulated innate immune responses to lethal infections. Note: Microbe-derived pathogen-associated molecular patterns (PAMPs such as LPS) rely on cell surface pattern recognition receptors (PRRs such as TLR4) to activate innate immune cells to immediately release ‘early’ proinflammatory mediators (such as TNF, IL-1β, and IFN-γ), which then stimulate hepatocytes and innate immune cells to synthesize and secrete an ‘intermediate’ mediator, SAA. SAA then activates innate immune cells to release late-acting proinflammatory mediators such HMGB1 and pCTS-L, which bind cell surface PRRs such as TLR4 and RAGE to induce: (i) the expression of cytokines/chemokines to trigger hyperinflammation; and (ii) the expression of pro-Casp-11 to activate inflammasome and pyroptosis. The HMGB1- and pCTS-L-mediated hyperinflammation and pyroptosis-associated immunosuppression may adversely contribute to the pathogenesis of lethal sepsis. A panel of HMGB1- and pCTS-L-neutralizing mAbs may interrupt their interactions with TLR4 and RAGE, thereby impairing HMGB1- or pCTS-L-mediated dysregulated inflammation or immunosuppression to confer protection against lethal sepsis.
2. HMGB1 as a late-acting damage-associated molecular pattern (DAMP) mediator of lethal sepsis
More than 50 years ago, a group of nuclear proteins were co-purified along with histones from animal tissues and termed as ‘high mobility group’ (HMG) proteins based on their relative high mobility on SDS-PAGE gels [Citation18]. Subsequently, these HMG proteins are sub-divided into HMGB1/2, HMGN1/2, and HMGA1 families in light of their size, sequence similarities, and DNA binding properties [Citation19]. Among them, HMGB1 is the most ubiquitous () and evolutionarily conserved protein that exhibit almost complete amino acid sequence homology between rodents and humans. As a chromosomal protein, nuclear HMGB1 has been suggested in diverse cellular functions that include the determination of nucleosomal structure and stability, as well as the modulation of transcription factor interaction with cognate DNA sequences to regulate gene expression () [Citation20].
Table 1. Comparison of HMGB1 and pCTS-L expression and secretion pathways.
2.1. HMGB1 is actively secreted by innate immune cells and passively released by pyroptotic cells
In 1999, we first reported that crude bacterial endotoxins (containing trace amounts of bacterial proteins, lipids and nucleic acids) and early proinflammatory cytokines (e.g. TNF) could induce HMGB1 release from innate immune cells, thereby serving as a late mediator of lethal endotoxemia () [Citation21]. Lacking a signal peptide sequence (), HMGB1 could not be actively secreted through classical endoplasmic reticulum (ER) – Golgi exocytotic pathways () [Citation21]. Instead, various inflammatory stimuli induced post-translational acetylation [Citation22], lactylation [Citation22] or phosphorylation [Citation23,Citation24] of the nuclear localization sequence (NLS) of HMGB1, enabling translocation of nuclear HMGB1 to cytoplasmic vesicles that could be secreted extracellularly via non-classical exosomal secretory pathways () [Citation22,Citation25–28]. Alternatively, cytoplasmic HMGB1 could also be passively released through pyroptosis () [Citation29], a programmed necrotic cell death pathway mediated by Caspase-1-dependent canonical and Caspase-11/4/5-dependent non-canonical inflammasome activation pathways [Citation30].
The ‘canonical’ inflammasome activation can be triggered by the oligomerization of ‘nucleotide-binding oligomerization domain (NOD)-like receptors’ (NLRs, e.g. NLRP1, NLRP3, or NLRC4) with adaptor molecules such as the ‘apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain’ (ASC) adaptor, which recruits and activates pro-caspase-1 to catalyze GSDMD cleavage and formation of cytoplasmic GSDMD membrane pores that propel cell membrane disruption and pyroptosis [Citation31]. The optimal activation of canonical inflammasome relies on a priming process orchestrated by PAMPs (e.g. LPS) to up-regulate NLRP3 expression, as well as some damage-associated molecular pattern (DAMP)-mediated oligomerization of NLRP3 with ASC adaptor and pro-casp-1 enzyme. It may partly explains why ultra-pure LPS (completely free from any contaminating bacterial lipids, proteins and nucleic acids) completely fails to induce pyroptosis and HMGB1 release unless the LPS priming is accompanied with a secondary stimulation with DAMPs (e.g. ATP) [Citation31,Citation32].
Alternatively, if LPS is delivered to cytoplasmic Casp-11/4/5 via CD14/TLR4 receptor-mediated endocytosis or bacteria-derived outer membrane vesicles (OMV) [Citation33], it directly activates Casp-11/4/5-dependent non-canonical inflammasome pathway of pyroptosis [Citation30]. Similarly, some LPS-inducible cytokines (e.g. interferons such as IFN-α/β and IFN-γ) can prime innate immune cells to up-regulate Casp-11/4/5 expression [Citation34,Citation35], and similarly induce HMGB1 release [Citation25,Citation36,Citation37]. Likewise, some acute phase proteins (e.g. serum amyloid A, SAA) can stimulate NLRP3 inflammasome activation [Citation38,Citation39] and HMGB1 release [Citation16] possibly through inducing the expression of hemichannel molecules (e.g. Pannexin 1 and Connexin 43) [Citation13,Citation14] and enzymes [e.g. secretory phospholipase A2 (sPLA2-IIE/V) and the double-stranded RNA-dependent protein kinase R (PKR)] implicated in the regulation of inflammasome activation and HMGB1 release [Citation13–15,Citation26,Citation32]. Thus, it is possible that cytoplasmic HMGB1 could be secreted extracellularly through Casp-1- or Casp-11/4/5-mediated inflammasome activation and pyroptosis, especially after the formation of GSDMD membrane pores and rupture of cytoplasmic membranes [Citation29,Citation40].
2.2. Extracellular HMGB1 contributes to dysregulated innate immune responses
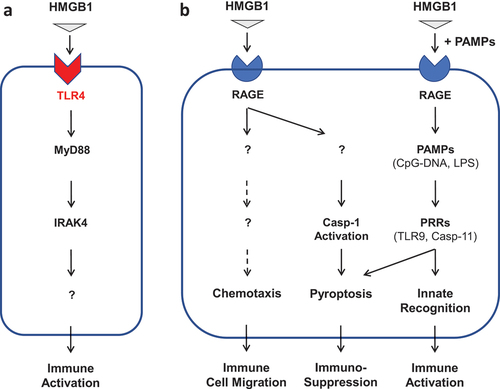
When HMGB1 is secreted by innate immune cells in relatively low levels, it binds high-affinity TLR4 receptor (KD = 22.0 nM) [Citation41–44] to augment inflammatory responses during an early stage of sepsis (; ) [Citation44]. Consequently, it can directly activate innate immune cells such as macrophages [Citation45], neutrophils [Citation46], and endothelial cells [Citation47] to produce more proinflammatory cytokines and chemokines. However, if HMGB1 is passively released by pyroptotic cells at relatively higher levels, it might also bind microbial PAMPs (e.g. CpG-DNA or LPS) and RAGE [Citation48,Citation49] and promoted the RAGE-receptor-mediated endocytosis of these PAMPs () [Citation50]. Consequently, HMGB1 facilitates the innate recognition of various PAMPs (e.g. CpG-DNA or LPS) by respective cytoplasmic PRRs such as TLR9 [Citation51–53] or Casp-11 [Citation50] to trigger hyperinflammation or pyroptosis-associated immunosuppression () [Citation50,Citation54]. By itself, extracellular HMGB1 can also bind RAGE to induce chemotaxis of dendritic cells [Citation55,Citation56] and neutrophils [Citation57], thereby facilitating the recruitment of innate immune cells to sites of the infection and injury to orchestrate hyperinflammatory responses (). On the other hand, extracellular HMGB1 could also induce immune tolerance [Citation58], pyroptosis [Citation50,Citation54], and immunosuppression [Citation59] that might adversely compromise the host’s ability to eradicate microbial infections [Citation60]. Therefore, extracellular HMGB1 serves as a late-acting DAMP and pathogenic mediator of lethal sepsis partly by triggering dysregulated hyperinflammation or immunosuppression (; ) [Citation21,Citation44,Citation61].
Figure 2. Divergent roles of TLR4 and RAGE in the regulation of HMGB1-mediated hyperinflammation and immunosuppression. Note: Extracellular HMGB1 can differentially bind distinct PRRs (such as TLR4 and RAGE) with different affinities. Consequently, HMGB1 may induce divergent inflammatory responses that include immune cell migration (chemotaxis), immune activation (hyperinflammation), or pyroptosis-associated immunosuppression.
Notably, extracellular HMGB1 has also been implicated in the pathogenesis of the dys-regulated coagulopathy [Citation62], because plasma HMGB1 levels positively correlate with the development of disseminated intravascular coagulation in septic patients [Citation63]. In animal models of trauma/hemorrhagic shock [Citation64] and venous thrombosis [Citation65], the platelet-derived HMGB1 has been suggested as a key mediator of the platelet aggregation and thrombosis [Citation66]. Consistently, HMGB1 induces tissue factor expression [Citation67] and platelet aggregation [Citation68], which may trigger thrombin activation and conversion of soluble fibrinogens into insoluble fibrin, a three-dimensional structural network capable of trapping blood cells to form thrombosis [Citation69].
2.3. HMGB1 as a late mediator of lethal sepsis
In murine model of lethal endotoxemia induced by intraperitoneal administration of bacterial endotoxins, HMGB1 was first detected in the blood within 8 h, and subsequently reached plateau levels from 16 to 32 h [Citation21]. Similarly, in animal model of sepsis induced by a surgical procedure termed as ‘cecal ligation and puncture’ (CLP), HMGB1 was also accumulated in the circulation in a delayed fashion, beginning around 18 h and peaking for at least 18–36 h [Citation61]. This late appearance of systemic HMGB1 paralleled with the onset of animal death from experimental endotoxemia and sepsis, and distinguished itself from other early proinflammatory cytokines [Citation70]. In animal models of lethal endotoxemia or sepsis, HMGB1-neutralizing polyclonal or monoclonal antibodies conferred a dose-dependent protection [Citation21,Citation61,Citation71], even when the first dose of antibodies was given in a delayed fashion – at 24 h post the onset of sepsis. These findings have established HMGB1 as a relatively ‘late’ mediator of experimental sepsis with relatively wider therapeutic windows than that offered by early cytokines [Citation44,Citation70,Citation72–74].
2.4. Endogenous and pharmacological modulators of HMGB1 release or actions
To counter-regulate hyperinflammatory responses, mammals have evolutionally developed multiple negative regulatory mechanisms to counter-regulate HMGB1 release or actions. For example, emerging evidence has supported some important roles of several endogenous proteins such as thrombomodulin (TM) [Citation75], haptoglobin (HP) [Citation76], complement component factor 1q (C1q) [Citation77], and tetranectin (TN) [Citation78] in the regulation HMGB1 release or extracellular activities (). For instance, TM could directly bind HMGB1 to prevent its interactions with macrophage receptors [Citation79], and consequently prevent HMGB1-induced inflammation [Citation75,Citation85]. Similarly, an acute phase protein, HP, could also capture HMGB1 to trigger the CD163-dependent endocytosis of HMGB1/HP complex, and consequently induce the production of anti-inflammatory enzymes (heme oxygenase-1) and cytokines (e.g. IL-10) [Citation76,Citation86]. In addition, a complement component factor of the classical complement pathway [Citation58], C1q, could similarly interact with HMGB1 (KD = 200 nM) and induce the production of anti-inflammatory cytokines (e.g. IL-10) () [Citation77,Citation87].
Table 2. Pharmacological and endogenous inhibitors of HMGB1 release or actions.
More recently, we discovered that another circulating protein, TN, could capture extracellular HMGB1 and facilitate the endocytosis of TN/HMGB1 complexes to enhance HMGB1-induced pyroptosis () [Citation78]. On one hand, pyroptosis allows excessive release of HMGB1 that may adversely drive a dysregulated inflammatory response to lethal infections. On the other hand, pyroptosis also leads to immune cell depletion and associated immunosuppression that may have compromised the host innate immunity against invading pathogens (; ). Accordingly, we have generated a panel of TN-specific mAbs that prevented harmful HMGB1/TN interaction, and consequently rescued mice from lethal sepsis [Citation78]. This finding has pointed to an exciting possibility to develop therapeutic antibodies against seemingly harmless proteins that collude with pathogenic sepsis mediators [Citation88–90].
Our seminal discovery of HMGB1 as a late mediator of lethal sepsis has also stimulated substantial interest in search for HMGB1-targeting small molecule inhibitors. A major component of licorice, glycyrrhizin (GZA), could directly bind HMGB1 [Citation80] to disrupt its engagement with RAGE receptor [Citation81] and suppress HMGB1-mediated inflammatory responses () [Citation91]. A chemical derivative of the GZA, carbenoxolone (CBX), however, effectively inhibited LPS-induced HMGB1 secretion [Citation82] by inhibiting the LPS-induced activation of hemichannels [Citation13,Citation14] or PKR [Citation32,Citation82] that are possibly involved in inflammasome activation and pyroptosis (). Furthermore, a red sage (Danshen) ingredient derivative, tanshinone IIA sodium sulfonate (TSN-SS), selectively inhibited LPS-induced HMGB1 release [Citation83] by enhancing endocytosis of extracellular HMGB1 and eventually subjecting it to degradation via a lysosome-dependent pathway () [Citation84].
3. Procathepsin-L (pCTS-L) as an inducible late-acting mediator of lethal sepsis
Cathepsins (CTS) refer to a family of intracellular peptide hydrolases that include cysteine proteases of the papain family (Cathepsin B, C, F, H, L, K, O, S, V, W, and X), serine carboxypeptidases (Cathepsin G) and aspartic proteases (Cathepsin D and E) [Citation92]. As a primary lysosomal protease with >85% amino acid sequence homology between humans and rodents () [Citation93,Citation94], Cathepsin L (CTS-L) is mainly responsible for degrading endocytosed proteins to generate immunogenic antigens for adaptive immunity () [Citation95,Citation96]. Like many other cathepsins, CTS-L is also first synthesized as an inactive proenzyme (procathepsin L, pCTS-L), and can be further processed to become mature and active CTS-L enzyme (). Unlike other papain enzymes, however, CTS-L is up-regulated in some malignantly transformed tumor cells by various growth factors such as the platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF), and the pro-region-containing procathepsin L (pCTS-L) can be secreted extracellularly () [Citation97] to facilitate tumor invasion and metastasis [Citation109]. Even in non-transformed cells, many inflammatory (e.g. LPS or IFN-γ) [Citation98–100] and noxious stimuli (e.g. alcohol consumption, cigarette smoking, and UV irradiation) [Citation101–103] can similarly induce the expression and secretion of pCTS-L in both innate immune cells [Citation98,Citation102] and nonimmune cells that include hepatocytes [Citation101], dermal fibroblasts [Citation103], and synovial fibroblasts [Citation99] ().
Figure 3. Structural domains and amino acid sequence of murine and human precathepsin L (preCTS-L), procathepsin L (pCTS-L), and cathepsin L (CTS-L). Note: A nascent human and murine pre-cathepsin L (preCTS-L) are synthesized as a 333 or 334 amino acid precursor with an N-terminal 16- or 17-residue signal sequence (shown in blue). The signal sequence can direct the ribosome to the rough endoplasmic reticulum (ER) membrane to initiate the translocation of the growing peptide chain across the ER membrane, during which the signal peptide is cleaved off by a signal peptidase to release the pro-enzyme, procathepsin L (pCTS-L). Consequently, pCTS-L can be secreted extracellularly through the classical ER – Golgi exocytotic pathway, or alternatively cleaved in the endosome to release the cathepsin L (CTS-L), which is further proteolytically processed to produce the active enzyme consisting of a heavy and light chain. The epitope sequence (P13, residue 194–214) for a panel of protective anti-pCTS-L monoclonal antibodies (mAb2, mAb20, and mAb26) was underlined.
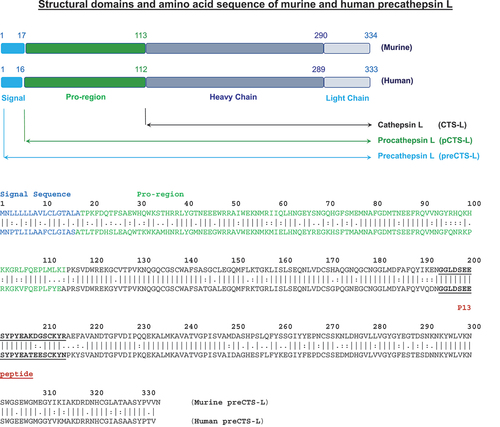
Table 3. Stimuli of pCTS-L release and its extracellular roles in diseases.
3.1. Identification of pCTS-L as an SAA-inducible and secretory protein
To search for other late-acting mediators of sepsis, we characterized SAA-inducible proteins in murine macrophage-conditioned culture medium, and identified a 40-kDa secretory protein as pCTS-L by mass spectrometry and immunoblotting assays [Citation17]. In human primary peripheral blood mononuclear cells (PBMCs), both LPS and SAA increased the intra- and extra-cellular levels of pCTS-L, suggesting that microbial PAMPs and host inflammatory cytokines induced pCTS-L expression and secretion [Citation17] (). Unlike HMGB1, a nascent precursor protein (pre-protein), precathepsin-L (preCTS-L), has a signal peptide sequence (, ), which directs the ribosome to the rough endoplasmic reticulum (ER) membrane and initiates the translocation of the growing peptide chain across the ER membrane, during which the signal peptide is cleaved off by a signal peptidase. Consequently, the resultant proenzyme (pCTS-L) might be actively secreted extracellularly through the classical ER – Golgi exocytotic pathway ().
In murine model of experimental sepsis, the expression of Ctsl mRNA was similarly elevated in the heart, intestine, kidney, liver, lung, and spleen [Citation17] (). Consistently, circulating pCTS-L protein levels were time-dependently elevated in septic animals, approaching plateau levels around 24–32 h post CLP, a time period when some septic animals started to die, suggesting a relatively late and systemic pCTS-L accumulation in a pre-clinical model of sepsis [Citation17]. Although pCTS-L was not detected in the plasma of normal healthy subjects, it was significantly elevated in septic patients in parallel with the systemic accumulation of the late-acting DAMP, HMGB1 [Citation17]. Furthermore, the plasma pCTS-L levels also positively correlated with corresponding sequential organ failure assessment (SOFA) scores [Citation17], as well as blood levels of several cytokines (e.g. IL-6) and chemokines (e.g. GRO, IL-8, and MCP-1) previously known as surrogate markers of experimental [Citation110,Citation111] and clinical sepsis [Citation112]. Collectively, these findings have suggested a sustainably late and systemic accumulation of pCTS-L in both pre-clinical and clinical settings.
3.2. Extracellular pCTS-L contributes to dysregulated inflammation and tissue injury via TLR4 and RAGE receptors
In vitro, recombinant pCTS-L dose-dependently induced several cytokines (e.g. IL-6 and TNF) and chemokines (e.g. RANTES, MCP-1, ENA-78/LIX, IL-8, GRO-α/KC, and GRO-α/β/γ) in human PBMCs [Citation17], suggesting that pCTS-L may promote dysregulated inflammation when it is excessively produced under pathological conditions (). These inflammatory activities were not likely attributable to some contaminating bacterial endotoxins, because endotoxin-free recombinant pCTS-L that was generated in eukaryotic (HEK293 kidney) cells similarly induced these cytokines/chemokines in human PBMCs [Citation17].
Mechanistically, pCTS-L elicits dysregulated inflammation through several PRR receptors such as TLR4 and RAGE (), because surface plasmon resonance (SPR) analysis revealed high affinities for pCTS-L interaction with both TLR4 (KD = 20.2 nM) and RAGE (KD = 3.5 nM) [Citation17]. In contrast, genetic disruption of both TLR4 and RAGE completely abrogated pCTS-L-induced release of all cytokines (IL-6 and IL-12) and chemokines (e.g. RANTES, MCP-1, MIP-1γ, LIX/ENA-78, KC/GRO-α, and MIP-2/GRO-β). Similarly, genetic knockout of TLR4 and RAGE rendered animals resistant to pCTS-L-induced inflammation, and pharmacological inhibition of pCTS-L interaction with TLR4 and RAGE with neutralizing mAbs simultaneously attenuated the pCTS-L-induced production of both TLR4-dependent (e.g. ENA-78/LIX, IL-8, RANTES, and MCP-1) and RAGE-dependent chemokines (e.g. GRO-α/KC and GRO-β/MIP-2) by human PBMCs [Citation17].
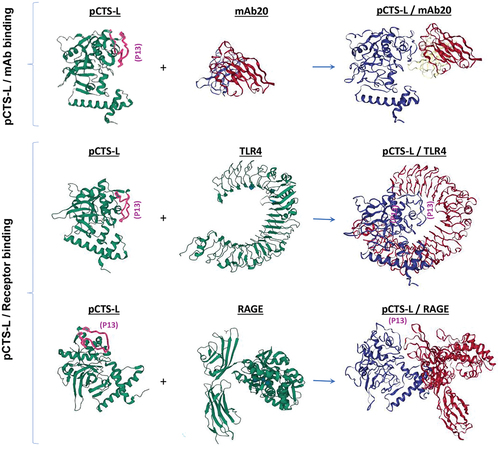
Figure 4. Schematic models of pCTS-L interaction with a neutralizing mAb20 and two pattern recognition receptors (TLR4 and RAGE). Note: We used the Machine Learning based antibody modeling (ABodyBuilder-ML) to predict the three-dimensional structure (IMGT model) of a protective anti-pCTS-L mAb20, and then employed the ClusPro Web Server to predict possible structures of pCTS-L/mAb or pCTS-L/receptor complexes that exhibited the least Gibbs free energy. The epitope sequence for a panel of protective mAbs was marked on pCTS-L (as P13 in purple) to illustrate its relative position within pCTS-L/mAb or the pCTS-L/PRR complexes. Note the epitope sequence (P13) for protective mAbs could be docked onto the CDR loops of a protective mAb20 (Top Panel). However, this P13 epitope sequence could be sequestered into the hydrophobic crevices of TLR4 (Middle Panel) and positioned sideways in close proximity to the V-domain of RAGE (Bottom Panel). Thus, the engagement of the P13 epitope sequence by a protective mAb may competitively prevent the co-current engagement of pCTS-L with its PRRs such as TLR4 and RAGE.
As aforementioned, excessive inflammation is often associated with a parallel up-regulation and activation of Casp-11/4/5, which can trigger dysregulated inflammasome activation, pyroptosis, and passive DAMP release [Citation113]. Indeed, we found that recombinant pCTS-L dose-dependently induced pro-Casp-11 expression and Casp-11 maturation in primary murine peritoneal macrophages derived from wildtype but not from TLR4/RAGE-deficient mice () [Citation17]. Taken together, these findings have suggested some important roles for TLR4 and RAGE in the regulation of pCTS-L-mediated hyperinflammation as well as Casp-11-associated pyroptosis and immunosuppression ().
3.3. Pathogenic role of pCTS-L in lethal sepsis
To understand the role of pCTS-L in sepsis, we intraperitoneally injected mice with recombinant pCTS-L at suprapathological doses and examined its effects on gene expression and tissue injuries. Administration of pCTS-L markedly upregulated several hepatic genes such as fibrinogen-α (Fga), fibrinogen-β (Fgb), and fibrinogen-γ (Fgg), which encode substrates for thrombin-catalyzed production of fibrin, a three-dimensional structural network capable of trapping blood cells to form thrombus [Citation114]. Consistently, intraperitoneal administration of pCTS-L at suprapathological concentrations caused marked injuries to the liver, as judged by an obvious increase in hepatocellular necrosis, cytoplasmic vacuolization, and associated sinusoidal congestion (thrombosis) [Citation17]. Similarly, the pCTS-L-induced liver injury was much more rigorous in wildtype C57BL/6 mice than that in mutant C57BL/6 mice deficient in both TLR4 and RAGE, further supporting some important roles for TLR4 and RAGE in the regulation of pCTS-L-mediated tissue injuries.
To evaluate the role of pCTS-L in sepsis, we first employed the genetic gene-knockout approach using a few breeding pairs of heterozygous Ctsl+/- KO mice, because female Ctsl−/− KO were infertile due to the failure to release mature oocytes during ovulation [Citation115]. In agreement with previous reports that genetic deletion of Ctsl led to a significant attenuation of pancreatitis [Citation107], atherosclerosis [Citation104], vascular intimal hyperplasia [Citation108], arthritis [Citation105], and colitis [Citation106] (), we observed that genetic disruption of pCTS-L lessened the CLP-induced liver injury [Citation17], further expanding a pathogenic role of pCTS-L in lethal sepsis (). This newly un-covered and novel role of extracellular pCTS-L in other inflammatory diseases that now include sepsis.
Even though gene knockout strategies are widely used to elucidate potential roles of various signaling molecules in many diseases, cautions should always be exercised when using these genetic approaches to evaluate extracellular roles of various inflammatory mediators. For example, despite the aforementioned pathogenic role of HMGB1 in lethal sepsis [Citation44], genetic disruption of HMGB1 expression unexpectedly renders animals more susceptible to both infections [Citation116] and injuries [Citation117], suggesting distinct roles of intracellular and extracellular HMGB1 in health and disease conditions [Citation118]. Accordingly, we generated polyclonal antibodies against murine pCTS-L and used these pharmacological agents to further characterize the extracellular role of pCTS-L in experimental sepsis (). Anti-murine pCTS-L total IgGs (pAbs) conferred dose-dependent and significant protections against lethal sepsis when the first dose was given at 2 h post CLP () [Citation17]. Consistently, pCTS-L antigen affinity-purified IgGs of protective pAbs effectively abrogated the pCTS-L-induced production of both TLR4-dependent cytokines (e.g. IL-6) and chemokines (e.g. MIP-1γ, LIX, RANTES, and MCP-1), as well as the RAGE-dependent neutrophilic chemokines (such as KC/GRO-α and MIP-2/GRO-β). These findings have suggested that anti-pCTS-L pAbs conferred protection against lethal sepsis possibly by attenuating pCTS-L-induced hyperinflammation likely orchestrated by both TLR4 and RAGE receptors ().
3.4. Generation of monoclonal antibodies (mAbs) against murine and human pCTS-L
To develop potential therapeutic antibodies, we strategically immunized mice with either murine or human pCTS-L and produced mAbs that could bind both murine and human pCTS-L for dual purposes. First, we could only test mAbs capable of binding murine pCTS-L for potential therapeutic efficacies using murine model of experimental sepsis. Moreover, we can only develop mAbs cross-reacting with bot murine and human pCTS-L as potential therapies for future sepsis trials. The therapeutic efficacy of each mAb was tested in a delayed regimen – starting at 24 h after CLP, a time point when circulating pCTS-L approached plateau levels and some septic animals started to succumb to death. All the mAbs cross-reacted with an epitope sequence corresponding to residue 175–193 (P12) of murine and human pCTS-L did not significantly affect animal survival when given at a wide range of doses (0.5–2.0 mg/kg). In contrast, three mAbs recognizing an epitope sequence corresponding to residue 194–214 (P13; ) of murine and human pCTS-L effectively rescued animals from lethal sepsis even when the 1st dose was given at 24 h post the onset of sepsis () [Citation17]. Consistently, the protective mAb significantly attenuated sepsis-induced systemic accumulation of several surrogate markers (e.g. IL-6, MIP-2/GRO-β and KC/GRO-α) of experimental sepsis. In vitro, the pCTS-L-induced production of several cytokines (e.g. IL-6) and chemokines (e.g. RANTES, MCP-1, ENA-78/LIX, IL-8, GRO-α/KC, and GRO-α/β/γ) in human PBMCs was effectively suppressed by the co-addition of protective anti-pCTS-L mAbs, suggesting that anti-pCTS-L mAbs confer protection against sepsis possibly by neutralizing its proinflammatory activities () [Citation17].
To further understand the neutralizing mechanism of these protective anti-pCTS-L mAbs, we evaluated their impact on pCTS-L interaction with TLR4 and RAGE receptors using SPR techniques. When an irrelevant control monoclonal antibody (‘c-mAb’) was injected onto the pCTS-L – conjugated sensor at an extremely high concentration (e.g.1200 nM), it did not affect subsequent pCTS-L binding to either TLR4 or RAGE [Citation17]. In a sharp contrast, when a pCTS-L-neutralizing IgG (mAb20) was first injected onto the pCTS-L – conjugated sensor, it markedly reduced pCTS-L interaction with TLR4 or RAGE, as manifested by an almost 55-fold (from 20.3 ± 2.3 nM to 1144.3 ± 173.6 nM) and 10-fold (from 3.1 ± 0.4 nM to 30.4 ± 9.8 nM) increase in the disassociation equilibrium constant KD for TLR4 and RAGE, respectively [Citation17]. Protein-protein docking analysis revealed that the epitope sequence (P13) of pCTS-L for protective mAbs was sequestered into the hydrophobic pocket of TLR4, but positioned sideways to the V-domain of RAGE () [Citation17]. It is thus possible that the engagement of P13-epitope sequence of pCTS-L by these P13-reactive mAbs poses different degree of physical hindrance to its co-current binding to different PRRs such as TLR4 and RAGE (). Therefore, these findings have suggested that some protective mAbs (e.g. mAb2, mAb20, and mAb26) may attenuate pCTS-L-induced inflammation by inhibiting its interaction with some putative PRR receptors (; ), and consequently rescue mice from lethal sepsis even when given in a delayed fashion.
To search for small molecule pCTS-L inhibitors, we developed a semi-high-throughput macrophage cell-based bioassay to screen a NatProduct Collection of 800 compounds, and identified a lipophilic sterol, lanosterol (LAN), as a selective inhibitor of pCTS-L-induced production of cytokines (e.g. TNF and IL-6) and chemokines (e.g. MCP-1 and ENA-78) in innate immune cells [Citation119]. To improve its bioavailability, we employed phospholipids with long and saturated acyl chains (such as the 1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine, DPPC) to generate liposomes nanoparticles carrying lanosterol at equal molar ratio. These LAN-containing liposomes similarly inhibited pCTS-L-induced production of several chemokines (e.g. MCP-1, RANTES, and MIP-2) in human PBMCs, and effectively rescued mice from lethal sepsis even when given at 24 h post CLP [Citation119]. This protection was associated with a significant attenuation of sepsis-induced systemic accumulation of serval surrogate biomarkers (e.g. IL-6, KC, and sTNF-RI), supporting an exciting possibility to develop liposome nanoparticles carrying anti-inflammatory sterols as potential therapies for human sepsis and other inflammatory diseases.
4. Conclusions
HMGB1 is actively secreted by innate immune cells via non-classical secretory pathways [Citation22,Citation25–28] or passively released through Casp-1 or Casp-11/4/5-mediated canonical or non-canonical inflammasome-dependent pyroptosis [Citation29,Citation40].
Extracellular HMGB1 can bind various microbial PAMPs (e.g. CpG-DNA or LPS) and RAGE [Citation48,Citation49] to promote RAGE-receptor-mediated endocytosis [Citation50] and innate recognition of these DAMPs by respective cytoplasmic PRRs such as TLR9 [Citation51–53] or Casp-11 [Citation50] to trigger hyperinflammation or pyroptosis-associated immunosuppression [Citation50,Citation54].
Extracellular HMGB1 can also directly bind TLR4 and RAGE [Citation41–44] to induce chemotaxis and activation of immune cells that may lead to hyperinflammation [Citation45,Citation46,Citation55–57], immune tolerance [Citation58], pyroptosis [Citation50,Citation54] and immunosuppression [Citation59] that adversely compromise the host’s ability to eradicate invading pathogens [Citation60].
Extracellular HMGB1 also contributes to dysregulated coagulopathy [Citation62–66], because it directly induces tissue factor expression [Citation67] and platelet aggregation [Citation68], thereby triggering thrombin activation and conversion of soluble fibrinogens into insoluble fibrin, a three-dimensional structural network capable of trapping blood cells to form thrombosis [Citation69].
HMGB1 is systematically accumulated in a delayed fashion, and its neutralizing antibodies confer protection against lethal sepsis [Citation21,Citation61,Citation71], even when the first dose of antibodies is given in a delayed fashion, establishing HMGB1 as a ‘late’ mediator of lethal sepsis with a relatively wider therapeutic window [Citation44,Citation70,Citation72–74].
A number of endogenous proteins such as thrombomodulin (TM) [Citation75], haptoglobin (HP) [Citation76], complement component factor 1q (C1q) [Citation77] and tetranectin (TN) [Citation78] can bind HMGB1 to counter-regulate its extracellular proinflammatory activities.
A number of medicinal ingredients such as glycyrrhizin (GZA) [Citation80,Citation81,Citation91] and its derivative (carbenoxolone, CBX) [Citation82] and tanshinone IIA sodium sulfonate (TSN-SS) [Citation83,Citation84] confer protection against lethal sepsis by inhibiting LPS-induced HMGB1 release through distinct mechanisms.
Cathepsin L is first synthesized as an inactive proenzyme (procathepsin L, pCTS-L), and pCTS-L can be secreted extracellularly by malignantly transformed tumor cells [Citation97] to facilitate tumor invasion and metastasis [Citation109].
Inflammatory (e.g. LPS, IFN-γ or SAA) [Citation17,Citation98–100] and noxious stimuli (e.g. alcohol, cigarette smoking, and UV irradiation) [Citation101–103] stimulate the expression and secretion of pCTS-L in both innate immune cells [Citation17,Citation98,Citation102] and nonimmune cells such as hepatocytes [Citation101], dermal fibroblasts [Citation103] and synovial fibroblasts [Citation99].
In murine model of experimental sepsis, the expression of Ctsl mRNA is elevated in the heart, intestine, kidney, liver, lung, and spleen, and its protein levels are elevated in the circulation in an delayed fashion [Citation17].
Blood pCTS-L levels are similarly elevated in septic patients in parallel with HMGB1 [Citation17], and positively correlate with blood levels of several cytokines (IL-6) and chemokines (GRO, IL-8, and MCP-1) known as surrogate markers of experimental [Citation110,Citation111] and clinical sepsis [Citation112].
Extracellular pCTS-L binds some PRR receptors such as TLR4 and RAGE and induces several cytokines (e.g. IL-6 and TNF) and chemokines (e.g. RANTES, MCP-1, ENA-78/LIX, IL-8, GRO-α/KC, and GRO-α/β/γ) in human PBMCs [Citation17].
Extracellular pCTS-L also dose-dependently induces pro-Casp-11 expression and Casp-11 maturation in peritoneal macrophages derived from wildtype but not from TLR4/RAGE-deficient mice, suggesting a possible role for pCTS-L in Casp-11-associated pyroptosis and immunosuppression [Citation17].
Intraperitoneal administration of recombinant pCTS-L at suprapathological doses upregulated several hepatic genes (e.g. fibrinogen-α/β/γ, Fga/b/g) that encode substrates for thrombin-catalyzed production of fibrin, and cause marked sinusoidal congestion and thrombosis in the liver [Citation17].
Genetic disruption of Ctsl expression confers protection against experimental pancreatitis [Citation107], atherosclerosis [Citation104], vascular intimal hyperplasia [Citation108], arthritis [Citation105], colitis [Citation106] and sepsis [Citation17].
pCTS-L-neutralizing monoclonal antibodies confer a dose-dependent and significant protection against lethal sepsis even when the first dose is given at 24 h post CLP [Citation17].
pCTS-L-specific polyclonal and monoclonal antibodies effectively attenuate pCTS-L-induced production of both TLR4-dependent IL-6, MIP-1γ, LIX, RANTES and MCP-1, as well as the RAGE-dependent KC/GRO-α and MIP-2/GRO-β [Citation17].
pCTS-L-neutralizing and protective mAbs may attenuate pCTS-L-induced inflammation possibly by inhibiting its interaction with some putative PRR receptors such as TLR4 and RAGE [Citation17].
Lanosterol-containing liposomes selectively inhibited pCTS-L-induced inflammation and rescued mice from lethal sepsis when given at 24 h post disease onset [Citation119].
5. Expert opinion
Microbial infections and sepsis cause approximately 20% of total deaths worldwide [Citation1], and annually costs more than $62 billion in the U.S. alone [Citation120]. Despite an improved understanding of the underlying pathophysiology, many anti-inflammatory agents [Citation121] or neutralizing antibodies against endotoxins or early cytokines (e.g. TNF or IL-1β) failed in clinical trials [Citation2,Citation122–127]. These failures partly reflected the complexity of underlying pathogenesis of clinical sepsis, as well as the heterogeneity of septic patient populations [Citation128]. In a sharp contrast to the relatively unified characteristics (e.g. age, body weight, and genetic background) of experimental animals in pre-clinical settings, patients recruited to previous clinical trials typically exhibited vast genetic and epigenetic heterogeneities associated with divergent comorbidities that may dramatically complicated the pathogenesis and progression of clinical sepsis. Therefore, future preclinical studies of HMGB1- and pCTS-L-targeting therapies might need to be further expanded to experimental models of sepsis with divergent underlying comorbidities (e.g. cancer and diabetes) in order to better simulate clinical sepsis syndromes that are often associated with high heterogeneities and divergent comorbidities [Citation129].
In many of the earlier sepsis trials, however, there was little consideration of patient stratification into biologically homogeneous subgroups due to the lack of biomarkers available to accurately diagnose sepsis [Citation129], which might have failed to detect certain beneficial effects merely unique to a subgroup of patients. Indeed, re-analysis of a prior Phase III randomized IL-1β receptor antagonist sepsis trial revealed a significant improvement in the survival of a subgroup of septic patients with concurrent hepatobiliary dysfunction/disseminated intravascular coagulation [Citation130]. Furthermore, there was little consideration of the appropriate timing of sepsis treatments in previous trials, partly because the kinetics of ‘early’ and ‘late’ mediators were not characterized in any recruited septic patients. Some proinflammatory cytokines such as TNF and IL-1β are produced relatively early during experimental and clinical sepsis and may still be needed for potentially beneficial and defensive inflammatory responses against invading pathogens. In contrast, HMGB1 and pCTS-L are released relatively late in experimental sepsis and better positioned to cause dysregulated inflammation and immunosuppression (). Although it is often difficult or impossible to precisely pinpoint the time course of clinical sepsis, HMGB1- or pCTS-L-targeting therapies could be given to patients presenting signs of sepsis as well as elevated blood levels of these late-acting mediators. On one hand, these late-acting mediators may offer a relatively wider therapeutic window for potential sepsis therapies. On the other hand, the pharmacological inhibition of HMGB1 and/or pCTS-L with specific antibodies or inhibitors may also preserve some potentially early and beneficial inflammatory responses orchestrated by TNF, IL-1β or other cytokines.
5.1. Therapeutic potential of HMGB1 and pCTS-L inhibitors in sepsis
Currently, there is still no effective therapy [Citation89] for septic patients other than adjunctive use of antibiotics, fluid resuscitation [Citation131], and supportive care [Citation120]. The phenomenal failure in sepsis trials during the last 30–40 years [Citation2,Citation122–127] has dramatically diminished the enthusiasm both for preclinical investigation of the sepsis pathophysiology and clinical development of innovative sepsis therapies. Consequently, the lingering fear from previous failures has also negatively shadowed the business decision process of HMGB1-targeting clinical trials. Nevertheless, previous experimental investigations of pathogenic early cytokines (such as TNF) in pre-clinical animal models have led to successful development of anti-TNF therapies for patients with other inflammatory diseases, such as rheumatoid arthritis [Citation132]. It is thus still important to further explore the therapeutic efficacy of several promising HMGB1- and pCTS-L-specific mAbs or inhibitors in clinical settings. For instance, a selective HMGB1 inhibitor, TSN-SS, has been widely used in China as a drug for patients with cardiovascular disorders. The dual effects of TSN-SS in attenuating HMGB1-mdiated inflammation and cardiovascular dysfunctions make it a promising candidate for sepsis clinical trials [Citation133]. Similarly, it would also be important to test the therapeutic efficacy of TN-reactive mAbs that effectively disrupted harmful interaction of TN with a pathogenic sepsis mediator, HMGB1 [Citation78]. The discovery of mAbs capable of disrupting TN/HMGB1 interaction and rescuing animals from lethal sepsis may provide clues for future development of novel therapies for human sepsis [Citation88]. Finally, our recent discovery of a panel of pCTS-L-neutralizing mAbs that can disrupt the harmful interaction of pCTS-L with TLR4 and RAGE to attenuate dysregulated inflammation or immunosuppression has suggested another exciting possibility of developing antibody strategies for human sepsis. It will thus be important and exciting to translate these pre-clinical findings into clinical applications through generating humanized HMGB1- or pCTS-L-neutralizing mAbs for further testing in future sepsis clinical trials.
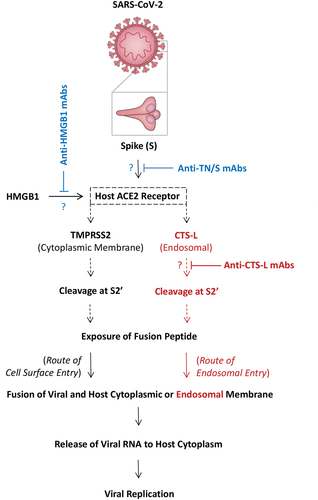
Recently, emerging evidence has suggested extracellular HMGB1 as a potential biomarker [Citation134] and therapeutic target for severe COVID-19 [Citation135–138], because HMGB1 contributes to dysregulated inflammatory responses to SARS-CoV-2 infections [Citation136], and upregulates the expression of host ACE2 needed for SARS-CoV-2 S binding and viral entry () [Citation137,Citation138]. Furthermore, we discovered that some anti-tetranectin (TN) mAbs capable of rescuing mice from lethal sepsis also cross-reacted with the ACE2 receptor binding motif (RBM) of SARS-CoV-2 spike protein (KD = 17.4–62.8 nM) [Citation139]. These TN/RBM-reactive mAbs not only competitively inhibited RBM-ACE2 interaction () [Citation139], but also selectively impaired the RBM-induced secretion of the granulocyte macrophage colony-stimulating factor (GM-CSF) [Citation139]. Finally, recent studies have revealed a possible role of CTS-L up-regulation in the regulation of SARS-CoV-2 entry into host cells [Citation140], as well as elevated circulating CTS-L levels in patients with COVID-19 [Citation141]. Specifically, CTS-L might be needed in the acidic endosomes for the route of endosomal entry of SARS-CoV-2 () [Citation141], and mAbs capable of attenuating the proinflammatory activities of pCTS-L might also inhibit the enzymatic activities of CTS-L to restrict CTS-L-mediated viral entry (). Therefore, it will be important to test the therapeutic efficacy of HMGB1-, TN/RBM- and/or pCTS-L-reactive mAbs for the treatment human sepsis, COVID-19 and other lethal infectious diseases. After all, it will be truly meaningful to invest time and resource to test the relevance of current experimental findings in future clinical trials.
Figure 5. Potential effects of neutralizing monoclonal antibodies on the HMGB1-induced ACE2 expression, the physical interaction of SARS-CoV-2 S protein with host ACE2 receptor, or the CTS-L-mediated cleavage of SARS-CoV-2 S protein. Note: Upon ACE2 engagement, SARS-CoV-2 spike (S) protein undergoes conformational changes in a target cell through proteolytic cleavage at the S2’ site either by the TMPSS2 on host cytoplasmic membrane or the CTS-L in the acidic endosomes. This cleavage of the spike protein at the S2’ site by either enzyme similarly leads to exposure of the fusion peptide to cytoplasmic or endosomal membranes, initiating the fusion between viral and host cellular membranes to form fusion pore and consequent release of viral RNA to host cell cytosol for viral replication. It is important to test whether mAbs capable of inhibiting the proinflammatory activities of HMGB1 and pCTS-L or harmful interactions between viral S protein and host ACE2 receptor could limit viral infection in preclinical and clinical settings.
Article highlights
HMGB1 is actively secreted by innate immune cells via non-classical secretory pathways and passively released through Casp-1 or Casp-11/4/5-mediated canonical or non-canonical inflammasome-dependent pyroptosis.
Extracellular HMGB1 can bind various microbial pathogen-associated molecular pattern molecules (PAMPs such as CpG-DNA or LPS) to promote their RAGE-receptor-mediated endocytosis and innate recognition by respective cytoplasmic pattern recognition receptors (PRRs such as TLR9 or Casp-11) to trigger hyperinflammation or pyroptosis-associated immunosuppression.
HMGB1 is systematically accumulated in a delayed fashion in experimental sepsis, and its neutralizing monoclonal antibodies rescued animals from lethal sepsis even when the first dose was given at 24 h post disease onset.
In murine model of experimental sepsis, the blood levels of procathepsin L (pCTS-L) are similarly elevated in a delayed fashion, and its neutralizing monoclonal antibodies rescued animals from lethal sepsis even when the first dose was given at 24 h post disease onset.
Blood pCTS-L levels are similarly elevated in septic patients in parallel with the systemic HMGB1 accumulation, and positively correlate with blood levels of several surrogate makers such as IL-6, IL-8, and MCP-1.
List of abbreviations used in the manuscript
| ACE2 | = | angiotensin-converting enzyme 2 |
| ASC | = | apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain |
| C1q | = | complement component factor 1q |
| CBX | = | carbenoxolone |
| CD14 | = | co-receptor cluster of differentiation 14 |
| CLP | = | cecal ligation and puncture |
| COVID-19 | = | coronavirus disease 2019 |
| CTS-L | = | cathepsin L |
| DAMP | = | damage-associated molecular pattern |
| ER | = | endoplasmic reticulum |
| Fg | = | fibrinogen |
| GM-CSF | = | granulocyte macrophage colony-stimulating factor |
| GSDMD | = | gasdermin D |
| GZA | = | glycyrrhizin |
| HMGB1 | = | high mobility group box 1 |
| HP | = | haptoglobin |
| IFN | = | interferon |
| IL | = | interleukin |
| KD | = | equilibrium dissociation constant |
| KO | = | knock-out |
| LBP | = | LPS-binding protein |
| LPS | = | lipopolysaccharide |
| mAb | = | monoclonal antibodies |
| NLR | = | nucleotide-binding oligomerization domain (NOD)-like receptors |
| PAMP | = | pathogen-associated molecular patterns |
| PBMCs | = | peripheral blood mononuclear cells |
| ia_version=’0’>pCTS-L | = | procathepsin-L |
| PKR | = | double-stranded RNA-dependent protein kinase R |
| preCTS-L | = | precathepsin L |
| PRR | = | pattern recognition receptor |
| RAGE | = | receptor for advanced glycation end product |
| RBM | = | the ACE2 receptor binding motif (RBM) of SARS-CoV-2 spike protein |
| S | = | spike protein |
| SAA | = | serum amyloid A |
| SARS-CoV-2 | = | severe acute respiratory syndrome (SARS)-like coronavirus 2 |
| SDS-PAGE | = | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SOFA | = | sequential organ failure assessment |
| sPLA2 | = | secretory phospholipase A2 |
| TLR | = | toll-like receptor |
| TM | = | thrombomodulin |
| TMPRSS2 | = | the transmembrane protease serine 2 |
| TN | = | tetranectin |
| TNF | = | tumor necrosis factor |
| TSN-SS | = | tanshinone IIA sodium sulfonate |
Declaration of interests
H. Wang is a co-inventor of two U.S. patents entitled ‘Antagonists of HMG-1 for treating inflammatory conditions’ (US Patent 8,053,206) and ‘Inhibition of inflammatory cytokine production with tanshinones’ (US Patent 8,513,227B2); and three patent applications entitled ‘Tetranectin-targeting monoclonal antibodies to fight against lethal sepsis and other pathologies,’ “Use of SARS-CoV-2 receptor binding motif (RBM) reactive monoclonal antibodies to treat COVID-19’,” and ‘Use of procathepsin L-neutralizing monoclonal antibodies to treat sepsis and other inflammatory diseases,’ as well as a provisional patent application entitled “Use of procathepsin L (pCTS-L)-inhibiting lanosterol-carrying liposome nanoparticles to treat lethal sepsis.”
A. Li is a co-inventor of three patent applications entitled ‘Tetranectin-targeting monoclonal antibodies to fight against lethal sepsis and other pathologies,’ ‘Use of SARS-CoV-2 receptor binding motif (RBM) reactive monoclonal antibodies to treat COVID-19,’ and ‘Use of procathepsin L-neutralizing monoclonal antibodies to treat sepsis and other inflammatory diseases,’ as well as a provisional patent application entitled ‘Use of procathepsin L (pCTS-L)-inhibiting lanosterol-carrying liposome nanoparticles to treat lethal sepsis.’
W. Chen is a co-inventor of a patent application entitled ‘Tetranectin-targeting monoclonal antibodies to fight against lethal sepsis and other pathologies.’
X. Qiang is a co-inventor of a patent application entitled ‘Use of SARS-CoV-2 receptor binding motif (RBM) reactive monoclonal antibodies to treat COVID-19.’
C.S. Zhu. is a co-inventor of two patent applications entitled ‘Use of SARS-CoV-2 receptor binding motif (RBM) reactive monoclonal antibodies to treat COVID-19’ and ‘Use of procathepsin L-neutralizing monoclonal antibodies to treat sepsis and other inflammatory diseases.’
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Additional information
Funding
References
- Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–211. doi: 10.1016/S0140-6736(19)32989-7
- Tindal EW, Armstead BE, Monaghan SF, et al. Emerging therapeutic targets for sepsis. Expert Opin Ther Targets. 2021;25(3):175–189. doi: 10.1080/14728222.2021.1897107
- Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085
- Ohto U, Ishida H, Shibata T, et al. Toll-like receptor 9 contains two DNA binding sites that function cooperatively to promote receptor dimerization and activation. Immunity. 2018;48(4):649–658.e4. doi: 10.1016/j.immuni.2018.03.013
- Hailman E, Lichenstein HS, Wurfel MM, et al. Lipopolysaccharide (LPS)-binding protein accelerates the binding of LPS to CD14. J Exp Med. 1994;179(1):269–277. doi: 10.1084/jem.179.1.269
- Wright SD, Ramos RA, Tobias PS, et al. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249(4975):1431–1433. doi: 10.1126/science.1698311
- Tracey KJ, Fong Y, Hesse DG, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330(6149):662–664. doi: 10.1038/330662a0
- Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87(6):2095–2147. doi: 10.1182/blood.V87.6.2095.bloodjournal8762095
- Heinzel FP. The role of IFN-gamma in the pathology of experimental endotoxemia. J Immunol. 1990;145(9):2920–2924. doi: 10.4049/jimmunol.145.9.2920
- Ramadori G, Sipe JD, Dinarello CA, et al. Pretranslational modulation of acute phase hepatic protein synthesis by murine recombinant interleukin 1 (IL-1) and purified human IL-1. J Exp Med. 1985;162(3):930–942. doi: 10.1084/jem.162.3.930
- Sandri S, Rodriguez D, Gomes E, et al. Is serum amyloid a an endogenous TLR4 agonist? J Leukocyte Biol. 2008;83(5):1174–1180. doi: 10.1189/jlb.0407203
- Yan SD, Zhu H, Zhu A, et al. Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis [see comments]. Nat Med. 2000;6(6):643–651. doi: 10.1038/76216
- Li W, Bao G, Chen W, et al. Connexin 43 hemichannel as a novel mediator of sterile and infectious inflammatory diseases. Sci Rep. 2018;8(1):166–18452. doi: 10.1038/s41598-017-18452-1
- Chen W, Zhu S, Wang Y, et al. Enhanced macrophage pannexin 1 expression and hemichannel activation exacerbates lethal experimental sepsis. Sci Rep. 2019;9(1):160–37232. doi: 10.1038/s41598-018-37232-z
- Zhu S, Wang Y, Chen W, et al. High-Density Lipoprotein (HDL) Counter-Regulates Serum Amyloid a (SAA)-induced sPLA2-IIE and sPLA2-V expression in macrophages. Plos One. 2016;11(11):e0167468. doi: 10.1371/journal.pone.0167468
- Li W, Zhu S, Li J, et al. Serum amyloid a stimulates PKR expression and HMGB1 release possibly through TLR4/RAGE receptors. Mol Med. 2015;21(1):515–525. doi: 10.2119/molmed.2015.00109
- Zhu CS, Qiang X, Chen W, et al. Identification of procathepsin L (Pcts-L)-neutralizing monoclonal antibodies to treat potentially lethal sepsis. Sci Adv. 2023;9(5):eadf4313. doi: 10.1126/sciadv.adf4313
- Johns EW. History,definitions and problems. In: Johns E, editor. The HMG chromosomal proteins. London: Academic Press Inc. (London) Ltd; 1982. p. 1–8. doi: 10.1016/B978-0-12-386050-7.50006-X.
- Bustin M. Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem Sci. 2001;26(3):152–153. doi: 10.1016/S0968-0004(00)01777-1
- Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol. 1999;19(8):5237–5246. doi: 10.1128/MCB.19.8.5237
- Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–251. doi: 10.1126/science.285.5425.248.
- Yang K, Fan M, Wang X, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022;29(1):133–146. doi: 10.1038/s41418-021-00841-9
- Lu B, Antoine DJ, Kwan K, et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci U S A. 2014;111(8):3068–3073. doi: 10.1073/pnas.1316925111
- Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. 2006;177(11):7889–7897. doi: 10.4049/jimmunol.177.11.7889
- Rendon-Mitchell B, Ochani M, Li J, et al. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-Dependent Mechanism. J Immunol. 2003;170(7):3890–3897. doi: 10.4049/jimmunol.170.7.3890
- Gardella S, Andrei C, Ferrera D, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3(10):955–1001. doi: 10.1093/embo-reports/kvf198
- Bonaldi T, Talamo F, Scaffidi P, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo J. 2003;22(20):5551–5560. doi: 10.1093/emboj/cdg516
- Garcia de Durango CR, Monteiro MN, Bijnsdorp IV, et al. Lipopolysaccharide-regulated secretion of soluble and vesicle-based proteins from a panel of colorectal cancer cell lines. Proteomics Clin Appl. 2021;15(2–3):e1900119. doi: 10.1002/prca.201900119
- Volchuk A, Ye A, Chi L, et al. Indirect regulation of HMGB1 release by gasdermin D. Nat Commun. 2020;11(1):4561–18443. doi: 10.1038/s41467-020-18443-3
- Hagar JA, Powell DA, Aachoui Y, et al. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341(6151):1250–1253. doi: 10.1126/science.1240988
- Lamkanfi M, Sarkar A, Vande WL, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185(7):4385–4392. doi: 10.4049/jimmunol.1000803
- Lu B, Nakamura T, Inouye K, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488(7413):670–674. doi: 10.1038/nature11290
- Vanaja SK, Russo AJ, Behl B, et al. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell. 2016;165(5):1106–1119. doi: 10.1016/j.cell.2016.04.015
- Rathinam VA, Vanaja SK, Waggoner L, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150(3):606–619. doi: 10.1016/j.cell.2012.07.007
- Aachoui Y, Kajiwara Y, Leaf IA, et al. Canonical inflammasomes drive IFN-Î3 to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe. 2015;18(3):320–332. doi: 10.1016/j.chom.2015.07.016
- Kim JH, Kim SJ, Lee IS, et al. Bacterial endotoxin induces the release of high mobility group box 1 via the IFN-beta signaling pathway. J Immunol. 2009;182(4):2458–2466. doi: 10.4049/jimmunol.0801364
- Yang X, Cheng X, Tang Y, et al. The role of type 1 interferons in coagulation induced by gram-negative bacteria. Blood. 2020;135(14):1087–1100. doi: 10.1182/blood.2019002282
- Niemi K, Teirila L, Lappalainen J, et al. Serum amyloid a activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol. 2011;186(11):6119–6128. doi: 10.4049/jimmunol.1002843
- Ather JL, Ckless K, Martin R, et al. Serum amyloid a activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol. 2011;187(1):64–73. doi: 10.4049/jimmunol.1100500
- Phulphagar K, Kühn LI, Ebner S, et al. Proteomics reveals distinct mechanisms regulating the release of cytokines and alarmins during pyroptosis. Cell Rep. 2021;34(10):108826. doi: 10.1016/j.celrep.2021.108826
- Yang H, Hreggvidsdottir HS, Palmblad K, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107(26):11942–11947. doi: 10.1073/pnas.1003893107
- Park JS, Gamboni-Robertson F, He Q, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290(3):C917–C924. doi: 10.1152/ajpcell.00401.2005
- Yu M, Wang H, Ding A, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26(2):174–179. doi: 10.1097/01.shk.0000225404.51320.82
- Zhu CS, Wang W, Qiang X, et al. Endogenous regulation and pharmacological modulation of sepsis-induced HMGB1 release and action: An updated review. Cells. 2021;10(9):2220. doi: 10.3390/cells10092220
- Zhu S, Ashok M, Li J, et al. Spermine protects mice against lethal sepsis partly by attenuating surrogate inflammatory markers. Mol Med. 2009;15(7–8):275–282. doi: 10.2119/molmed.2009.00062
- Fan J, Li Y, Levy RM, et al. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling. J Immunol. 2007;178(10):6573–6580. doi: 10.4049/jimmunol.178.10.6573
- Fiuza C, Bustin M, Talwar S, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101(7):2652–2660. doi: 10.1182/blood-2002-05-1300
- Ling Y, Yang ZY, Yin T, et al. Heparin changes the conformation of high-mobility group protein 1 and decreases its affinity toward receptor for advanced glycation endproducts in vitro. Int Immunopharmacol. 2011;11(2):187–193. doi: 10.1016/j.intimp.2010.11.014
- Hori O, Brett J, Slattery T, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270(43):25752–25761. doi: 10.1074/jbc.270.43.25752
- Deng M, Tang Y, Li W, et al. The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity. 2018;49(4):740–753. doi: 10.1016/j.immuni.2018.08.016
- Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–496. doi: 10.1038/ni1457.
- Yanai H, Ban T, Wang Z, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462(7269):99–103. doi: 10.1038/nature08512
- Ivanov S, Dragoi AM, Wang X, et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 2007;110(6):1970–1981. doi: 10.1182/blood-2006-09-044776
- Xu J, Jiang Y, Wang J, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21(8):1229–1239. doi: 10.1038/cdd.2014.40
- Yang D, Chen Q, Yang H, et al. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukocyte Biol. 2007;81(1):59–66. doi: 10.1189/jlb.0306180
- Dumitriu IE, Bianchi ME, Bacci M, et al. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukocyte Biol. 2007;81(1):84–91. doi: 10.1189/jlb.0306171
- Orlova VV, Choi EY, Xie C, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. Embo J. 2007;26(4):1129–1139. doi: 10.1038/sj.emboj.7601552
- Watanabe H, Son M. The immune tolerance role of the HMGB1-RAGE axis. Cells. 2021;10(3):564. doi: 10.3390/cells10030564
- Gregoire M, Tadie JM, Uhel F, et al. Frontline science: HMGB1 induces neutrophil dysfunction in experimental sepsis and in patients who survive septic shock. J Leukocyte Biol. 2017;101(6):1281–1287. doi: 10.1189/jlb.5HI0316-128RR
- Patel VS, Sitapara RA, Gore A, et al. HMGB1 mediates hyperoxia-induced impairment of pseudomonas aeruginosa clearance and inflammatory lung injury in mice. Am J Respir Cell Mol Biol. 2013;48(3):280–287. doi: 10.1165/rcmb.2012-0279OC
- Yang H, Ochani M, Li J, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101(1):296–301. doi: 10.1073/pnas.2434651100.
- Liaw PC, Ito T, Iba T, et al. DAMP and DIC: The role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev. 2016;30(4):257–261. doi: 10.1016/j.blre.2015.12.004
- Hatada T, Wada H, Nobori T, et al. Plasma concentrations and importaNCE OF HIGH MOBILITY GROUP BOx protein in the prognosis of organ failure in patients with disseminated intravascular coagulation. Thromb Haemost. 2005;94(5):975–979. doi: 10.1160/TH05-05-0316
- Machado FR, Cesar MS. Sepsis, coagulation and anticoagulants. Endocr Metab Immune Disord Drug Targets. 2010;10(3):204–213. doi: 10.2174/187153010791936892
- Ito T, Kawahara K, Nakamura T, et al. High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. J Thromb Haemost. 2007;5(1):109–116. doi: 10.1111/j.1538-7836.2006.02255.x
- Pawlinski R. Platelet HMGB1: the venous clot coordinator. Blood. 2016;128(20):2376–2378. doi: 10.1182/blood-2016-09-738740
- Lv B, Wang H, Tang Y, et al. High-mobility group box 1 protein induces tissue factor expression in vascular endothelial cells via activation of NF-kappaB and Egr-1. Thromb Haemost. 2009;102(2):352–359. doi: 10.1160/TH08-11-0759
- Vogel S, Bodenstein R, Chen Q, et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest. 2015;125(12):4638–4654. doi: 10.1172/JCI81660
- Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8(10):776–787. doi: 10.1038/nri2402
- Wang H, Yang H, Czura CJ, et al. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med. 2001;164(10):1768–1773. doi: 10.1164/ajrccm.164.10.2106117
- Qin S, Wang H, Yuan R, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006;203(7):1637–1642. doi: 10.1084/jem.20052203
- Wang H, Yang H, Tracey KJ. Extracellular role of HMGB1 in inflammation and sepsis. J Intern Med. 2004;255(3):320–331. doi: 10.1111/j.1365-2796.2003.01302.x
- Wang H, Zhu S, Zhou R, et al. Therapeutic potential of HMGB1-targeting agents in sepsis. Expert Rev Mol Med. 2008;10:e32. DOI:10.1017/S1462399408000884
- Wang H, Ward MF, Sama AE. Novel HMGB1-inhibiting therapeutic agents for experimental sepsis. Shock. 2009;32(4):348–357. doi: 10.1097/SHK.0b013e3181a551bd
- Abeyama K, Stern DM, Ito Y, et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115(5):1267–1274. doi: 10.1172/JCI22782.
- Yang H, Wang H, Levine YA, et al. Identification of CD163 as an antiinflammatory receptor for HMGB1-haptoglobin complexes. JCI Insight. 2016;1(7):e85375. doi: 10.1172/jci.insight.85375.
- Son M, Porat A, He M, et al. C1q and HMGB1 reciprocally regulate human macrophage polarization. Blood. 2016;128(18):2218–2228. doi: 10.1182/blood-2016-05-719757.
- Chen W, Qiang X, Wang Y, et al. Identification of tetranectin-targeting monoclonal antibodies to treat potentially lethal sepsis. Sci Transl Med. 2020;12(539):12/539. doi: 10.1126/scitranslmed.aaz3833.
- Herzog C, Lorenz A, Gillmann HJ, et al. Thrombomodulin’s lectin-like domain reduces myocardial damage by interfering with HMGB1-mediated TLR2 signalling. Cardiovasc Res. 2014;101(3):400–410. doi: 10.1093/cvr/cvt275
- Mollica L, De Marchis F, Spitaleri A, et al. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem Biol. 2007;14(4):431–441. doi: 10.1016/j.chembiol.2007.03.007
- Okuma Y, Liu K, Wake H, et al. Glycyrrhizin inhibits traumatic brain injury by reducing HMGB1-RAGE interaction. Neuropharmacology. 2014;85:18–26. DOI:10.1016/j.neuropharm.2014.05.007
- Li W, Li J, Sama AE, et al. Carbenoxolone blocks endotoxin-induced Protein Kinase R (PKR) activation and High Mobility Group Box 1 (HMGB1) release. Mol Med. 2013;19(1):203–211. doi: 10.2119/molmed.2013.00064
- Li W, Li J, Ashok M, et al. A cardiovascular drug rescues mice from lethal sepsis by selectively attenuating a late-acting proinflammatory mediator, high mobility group box 1. J Immunol. 2007;178(6):3856–3864. doi: 10.4049/jimmunol.178.6.3856
- Zhang Y, Li W, Zhu S, et al. Tanshinone IIA sodium sulfonate facilitates endocytic HMGB1 uptake. Biochem Pharmacol. 2012;84(11):1492–1500. doi: 10.1016/j.bcp.2012.09.015
- Takehara K, Murakami T, Kuwahara-Arai K, et al. Evaluation of the effect of recombinant thrombomodulin on a lipopolysaccharide-induced murine sepsis model. Exp Ther Med. 2017;13(6):2969–2974. doi: 10.3892/etm.2017.4308
- Yang H, Wang H, Andersson U. Targeting Inflammation Driven by HMGB1. Front Immunol. 2020;11:484. doi: 10.3389/fimmu.2020.00484
- Liu T, Xiang A, Peng T, et al. HMGB1-C1q complexes regulate macrophage function by switching between leukotriene and specialized proresolving mediator biosynthesis. Proc Natl Acad Sci U S A. 2019;116(46):23254–23263. doi: 10.1073/pnas.1907490116
- Li J, Bao G, Wang H. Time to develop therapeutic antibodies against harmless proteins colluding with sepsis mediators? Immunotargets Ther. 2020;9:157–166. doi: 10.2147/ITT.S262605
- Paterson CW, Ford ML, Coopersmith CM. Breaking the bond between tetranectin and HMGB1 in sepsis. Sci Transl Med. 2020;12(539):12/539. doi: 10.1126/scitranslmed.abb2575
- Crunkhorn S. Antibody intervention rescues mice from sepsis. Nat Rev Drug Discov. 2020;19(6):385–00077. doi: 10.1038/d41573-020-00077-1
- Wang W, Zhao F, Fang Y, et al. Glycyrrhizin protects against porcine endotoxemia through modulation of systemic inflammatory response. Crit Care. 2013;17(2):R44. doi: 10.1186/cc12558
- Kuester D, Lippert H, Roessner A, et al. The cathepsin family and their role in colorectal cancer. Pathol Res Pract. 2008;204(7):491–500. doi: 10.1016/j.prp.2008.04.010
- Joseph LJ, Chang LC, Stamenkovich D, et al. Complete nucleotide and deduced amino acid sequences of human and murine preprocathepsin L. An abundant transcript induced by transformation of fibroblasts. J Clin Invest. 1988;81(5):1621–1629. doi: 10.1172/JCI113497.
- Chauhan SS, Popescu NC, Ray D, et al. Cloning, genomic organization, and chromosomal localization of human cathepsin L. J Biol Chem. 1993;268(2):1039–1045. doi: 10.1016/S0021-9258(18)54038-2
- McCoy KL, Schwartz RH. The role of intracellular acidification in antigen processing. Immunol Rev. 1988;106(1):129–147. doi: 10.1111/j.1600-065X.1988.tb00777.x
- Hsieh CS, deRoos P, Honey K, et al. A role for cathepsin L and cathepsin S in peptide generation for MHC class II presentation. J Immunol. 2002;168(6):2618–2625. doi: 10.4049/jimmunol.168.6.2618
- Ishidoh K, Kominami E. Gene regulation and extracellular functions of procathepsin L. Biol Chem. 1998;379(2):131–135. doi: 10.1515/bchm.1998.379.2.131
- Fiebiger E, Maehr R, Villadangos J, et al. Invariant chain controls the activity of extracellular cathepsin L. J Exp Med. 2002;196(9):1263–1269. doi: 10.1084/jem.20020762.
- Lemaire R, Huet G, Zerimech F, et al. Selective induction of the secretion of cathepsins B and L by cytokines in synovial fibroblast-like cells. Br J Rheumatol. 1997;36(7):735–743. doi: 10.1093/rheumatology/36.7.735
- Lah TT, Hawley M, Rock KL, et al. Gamma-interferon causes a selective induction of the lysosomal proteases, cathepsins B and L, in macrophages. FEBS Lett. 1995;363(1–2):85–89. doi: 10.1016/0014-5793(95)00287-J.
- Kharbanda KK, McVicker DL, Zetterman RK, et al. Ethanol consumption alters trafficking of lysosomal enzymes and affects the processing of procathepsin L in rat liver. Biochim Biophys Acta. 1996;1291(1):45–52. doi: 10.1016/0304-4165(96)00043-8.
- Takahashi H, Ishidoh K, Muno D, et al. Cathepsin L activity is increased in alveolar macrophages and bronchoalveolar lavage fluid of smokers. Am Rev Respir Dis. 1993;147(6 Pt 1):1562–1568. doi: 10.1164/ajrccm/147.6_Pt_1.1562.
- Klose A, Wilbrand-Hennes A, Brinckmann J, et al. Alternate trafficking of cathepsin L in dermal fibroblasts induced by UVA radiation. Exp Dermatol. 2010;19(8):e117–e123. doi: 10.1111/j.1600-0625.2009.01014.x
- Kitamoto S, Sukhova GK, Sun J, et al. Cathepsin L deficiency reduces diet-induced atherosclerosis in low-density lipoprotein receptor-knockout mice. Circulation. 2007;115(15):2065–2075. doi: 10.1161/CIRCULATIONAHA.107.688523.
- Schurigt U, Eilenstein R, Gajda M, et al. Decreased arthritis severity in cathepsin L-deficient mice is attributed to an impaired T helper cell compartment. Inflamm Res. 2012;61(9):1021–1029. doi: 10.1007/s00011-012-0495-x.
- Bauer C, Duewell P, Mayer C, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59(9):1192–1199. doi: 10.1136/gut.2009.197822
- Wartmann T, Mayerle J, Kahne T, et al. Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology. 2010;138(2):726–737. doi: 10.1053/j.gastro.2009.10.048.
- Cai J, Zhong H, Wu J, et al. Cathepsin L promotes vascular intimal hyperplasia after arterial injury. Mol Med. 2017;23(1):92–100. doi: 10.2119/molmed.2016.00222.
- Kane SE, Gottesman MM. The role of cathepsin L in malignant transformation. Semin Cancer Biol. 1990;1(2):127–136.
- Heuer JG, Sharma GR, Gerlitz B, et al. Evaluation of protein C and other biomarkers as predictors of mortality in a rat cecal ligation and puncture model of sepsis. Crit Care Med. 2004;32(7):1570–1578. doi: 10.1097/01.CCM.0000129488.54282.1A
- Li W, Ashok M, Li J, et al. A major ingredient of green tea rescues mice from lethal sepsis partly by inhibiting HMGB1. Plos One. 2007;2(11):e1153. doi: 10.1371/journal.pone.0001153
- Bozza FA, Salluh JI, Japiassu AM, et al. Cytokine profiles as markers of disease severity in sepsis: a multiplex analysis. Crit Care. 2007;11(2):R49. doi: 10.1186/cc5783
- Zhou B, Liu J, Zeng L, et al. Extracellular SQSTM1 mediates bacterial septic death in mice through insulin receptor signalling. Nat Microbiol. 2020;5(12):1576–1587. doi: 10.1038/s41564-020-00795-7
- Machlus KR, Cardenas JC, Church FC, et al. Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice. Blood. 2011;117(18):4953–4963. doi: 10.1182/blood-2010-11-316885
- Robker RL, Russell DL, Espey LL, et al. Progesterone-regulated genes in the ovulation process: ADAMTS-1 and cathepsin L proteases. Proc Natl Acad Sci U S A. 2000;97(9):4689–4694. doi: 10.1073/pnas.080073497
- Yanai H, Matsuda A, An J, et al. Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci U S A. 2013;110(51):20699–20704. doi: 10.1073/pnas.1320808110
- Kang R, Zhang Q, Hou W, et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology. 2014;146(4):1097–1107. doi: 10.1053/j.gastro.2013.12.015
- Kang R, Chen R, Zhang Q, et al. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116.
- Chen W, Zhu CS, Qiang X, et al. Development of Procathepsin L (Pcts-L)-inhibiting lanosterol-carrying liposome nanoparticles to treat lethal sepsis. Int J Mol Sci. 2023;24(10):8649. doi: 10.3390/ijms24108649.
- Buchman TG, Simpson SQ, Sciarretta KL, et al. Sepsis among medicare beneficiaries: 1. The burdens of sepsis, 2012-2018. Crit Care Med. 2020;48(3):276–288. doi: 10.1097/CCM.0000000000004224
- Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358(2):111–124. doi: 10.1056/NEJMoa071366
- Ziegler EJ, Fisher CJ Jr., Sprung CL, et al. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. The HA-1A sepsis study group. N Engl J Med. 1991;324(7):429–436. doi: 10.1056/NEJM199102143240701
- Ziegler EJ, McCutchan JA, Fierer J, et al. Treatment of gram-negative bacteremia and shock with human antiserum to a mutant Escherichia coli. N Engl J Med. 1982;307(20):1225–1230. doi: 10.1056/NEJM198211113072001
- Abraham E, Wunderink R, Silverman H, et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb sepsis study group. JAMA. 1995;273(12):934–941. doi: 10.1001/jama.1995.03520360048038
- Dinarello CA, Gelfand JA, Wolff SM. Anticytokine strategies in the treatment of the systemic inflammatory response syndrome. JAMA. 1993;269(14):1829–1835. doi: 10.1001/jama.1993.03500140081040
- Opal SM, Laterre PF, Francois B, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309(11):1154–1162. doi: 10.1001/jama.2013.2194
- Fisher CJ Jr., Dhainaut JF, Opal SM, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra sepsis syndrome study group. JAMA. 1994;271(23):1836–1843. doi: 10.1001/jama.1994.03510470040032
- Dellinger RP, Levy MM, Carlet JM, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36(1):296–327. doi: 10.1097/01.CCM.0000298158.12101.41
- Cohen J, Vincent JL, Adhikari NK, et al. Sepsis: a roadmap for future research. Lancet Infect Dis. 2015;15(5):581–614. doi: 10.1016/S1473-3099(15)70112-X
- Song Y, Shi Y, Ao LH, et al. TLR4 mediates LPS-induced HO-1 expression in mouse liver: role of TNF-alpha and IL-1beta. World J Gastroenterol. 2003;9(8):1799–1803. doi: 10.3748/wjg.v9.i8.1799
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368–1377. doi: 10.1056/NEJMoa010307
- Feldmann M, Maini RN. Anti-TNFα therapy of rheumatoid arthritis: What have we learned? Annu Rev Immunol. 2001;19(1):163–196. doi: 10.1146/annurev.immunol.19.1.163
- Wang H, Ward MF, Sama AE. Targeting HMGB1 in the treatment of sepsis. Expert Opin Ther Targets. 2014;18(3):257–268. doi: 10.1517/14728222.2014.863876
- Vicentino ARR, da Silva Fraga-Junior V, Palazzo M, et al. High mobility group box 1, ATP, lipid mediators, and tissue factor are elevated in COVID-19 patients: HMGB1 as a biomarker of worst prognosis. Clin Transl Sci. 2023;16(4):631–646. doi: 10.1111/cts.13475
- Chen L, Long X, Xu Q, et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell Mol Immunol. 2020;17(9):992–994. doi: 10.1038/s41423-020-0492-x
- Andersson U, Ottestad W, Tracey KJ. Extracellular HMGB1: a therapeutic target in severe pulmonary inflammation including COVID-19? Mol Med. 2020;26(1):42. doi: 10.1186/s10020-020-00172-4
- Wei J, Alfajaro MM, DeWeirdt PC, et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell. 2021;184(1):76–91.e13. doi: 10.1016/j.cell.2020.10.028
- Chen R, Huang Y, Quan J, et al. HMGB1 as a potential biomarker and therapeutic target for severe COVID-19. Heliyon. 2020;6(12):e05672. doi: 10.1016/j.heliyon.2020.e05672
- Qiang X, Zhu S, Li J, et al. Monoclonal antibodies capable of binding SARS-CoV-2 spike protein receptor-binding motif specifically prevent GM-CSF induction. J Leukocyte Biol. 2022;22(1):261–267. doi: 10.1002/JLB.3COVCRA0920-628RR
- Muus C, Luecken MD, Eraslan G, et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat Med. 2021;27(3):546–559. doi: 10.1038/s41591-020-01227-z
- Zhao MM, Yang WL, Yang FY, et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct Target Ther. 2021;6(1):134. doi: 10.1038/s41392-021-00558-8